Identification and Evaluation of Inhibitors of Lipase from Malassezia restricta using Virtual High-Throughput Screening and Molecular Dynamics Studies

 ,
,  ,
,  ,
,
Abstract
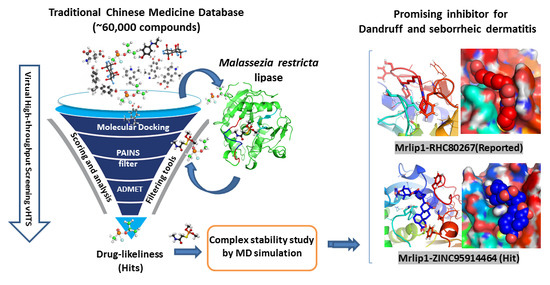
1. Introduction
2. Results and Discussion
2.1. vHTS: Molecular Docking
2.2. Hit Selection and Drug-Ability Assessment
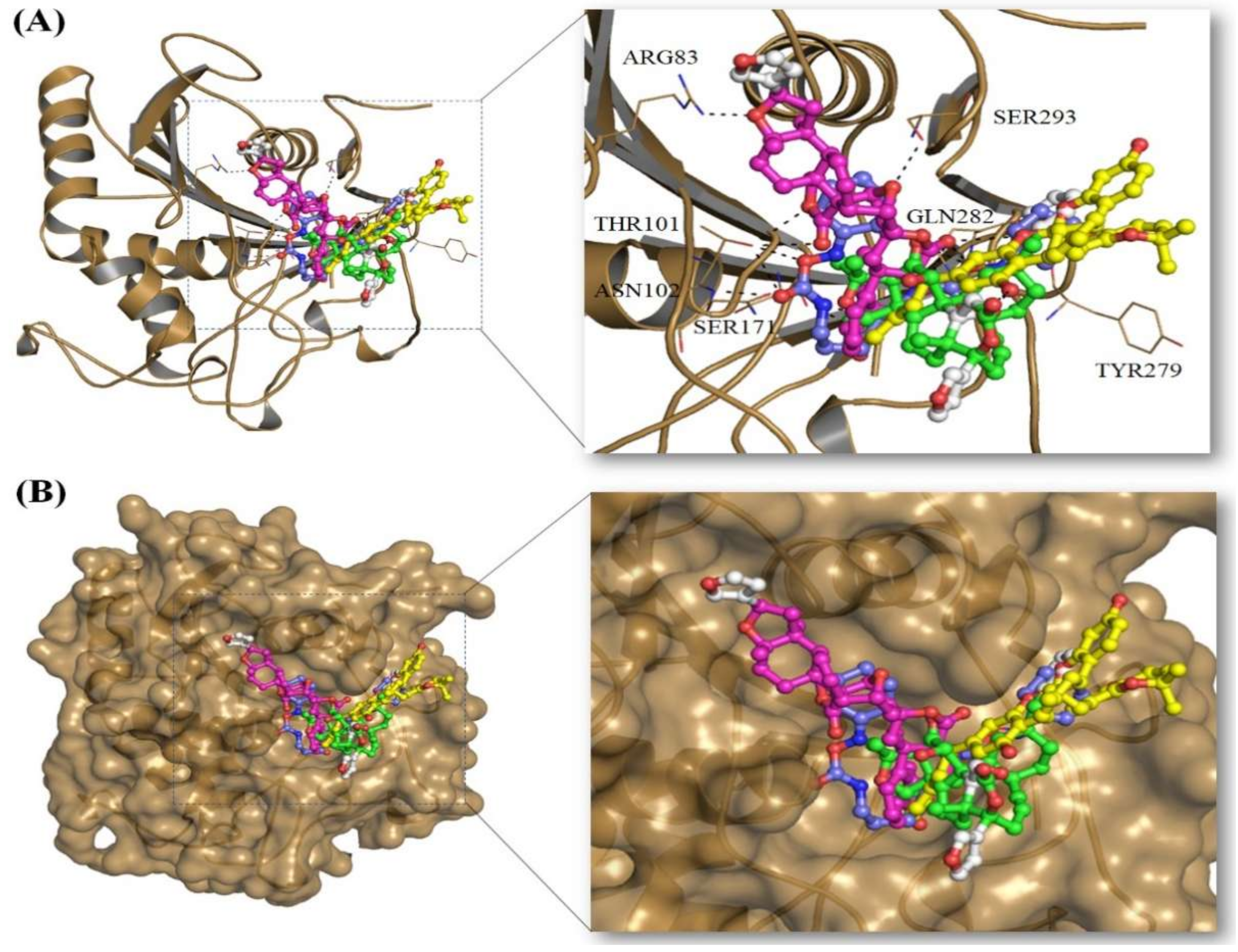
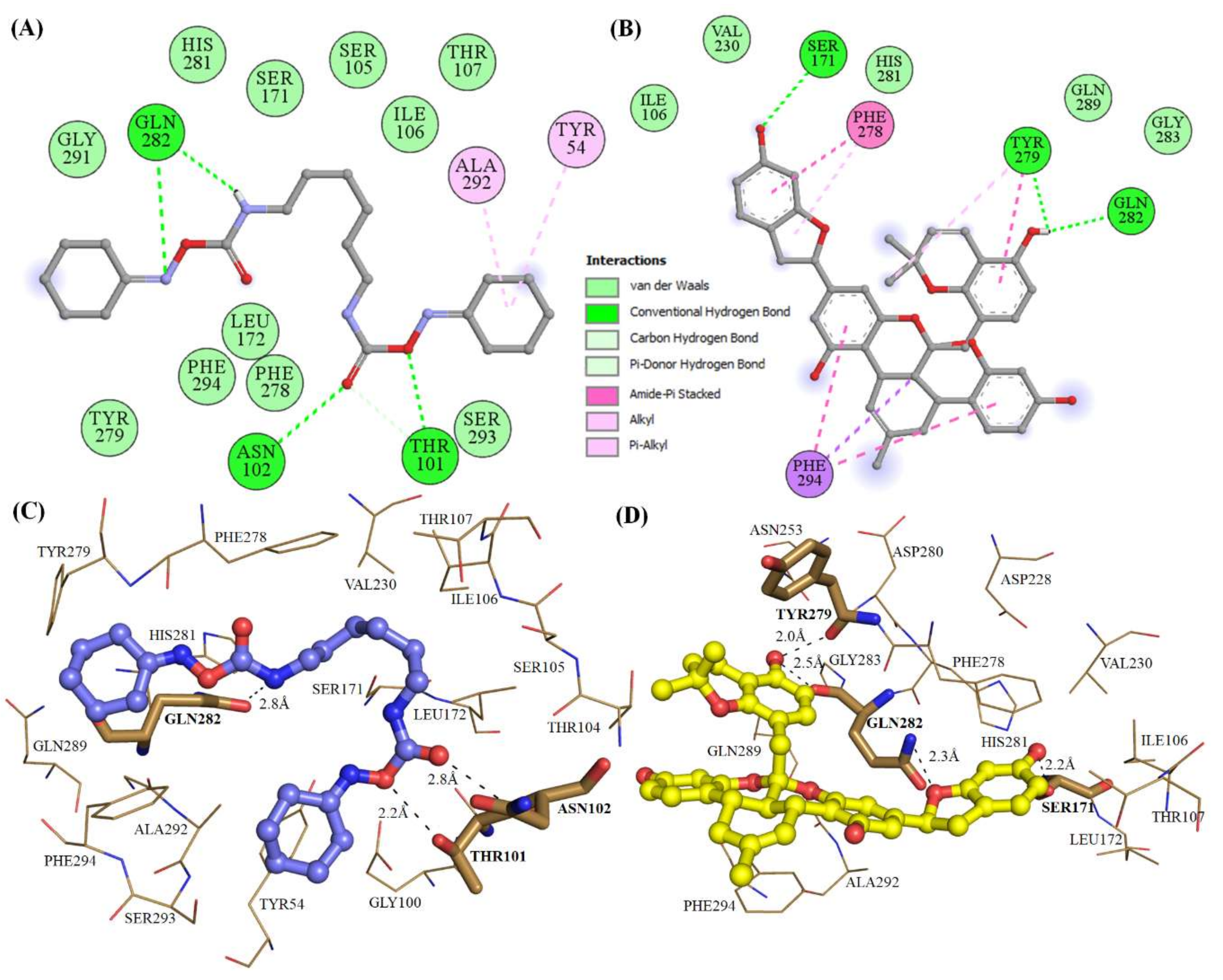
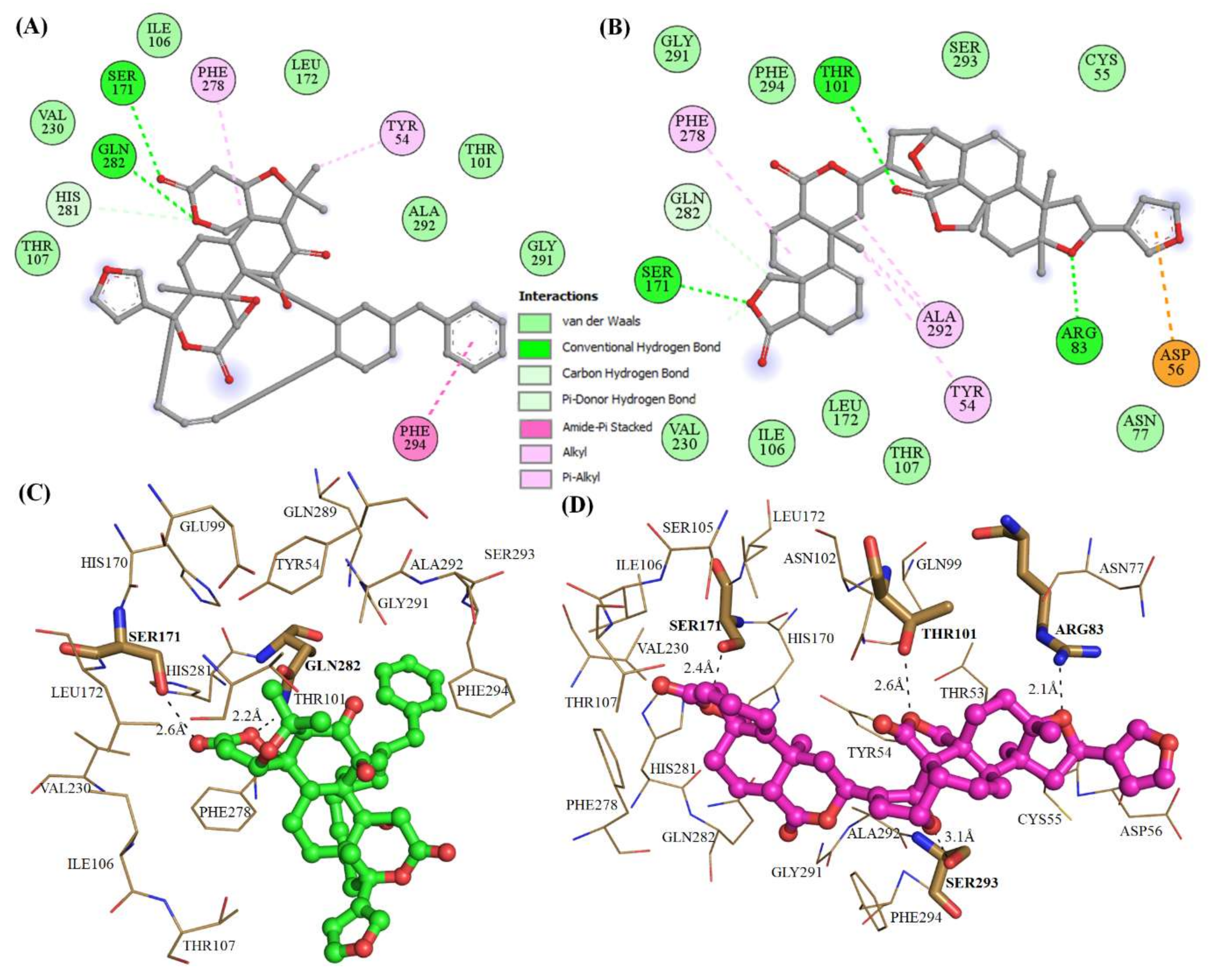
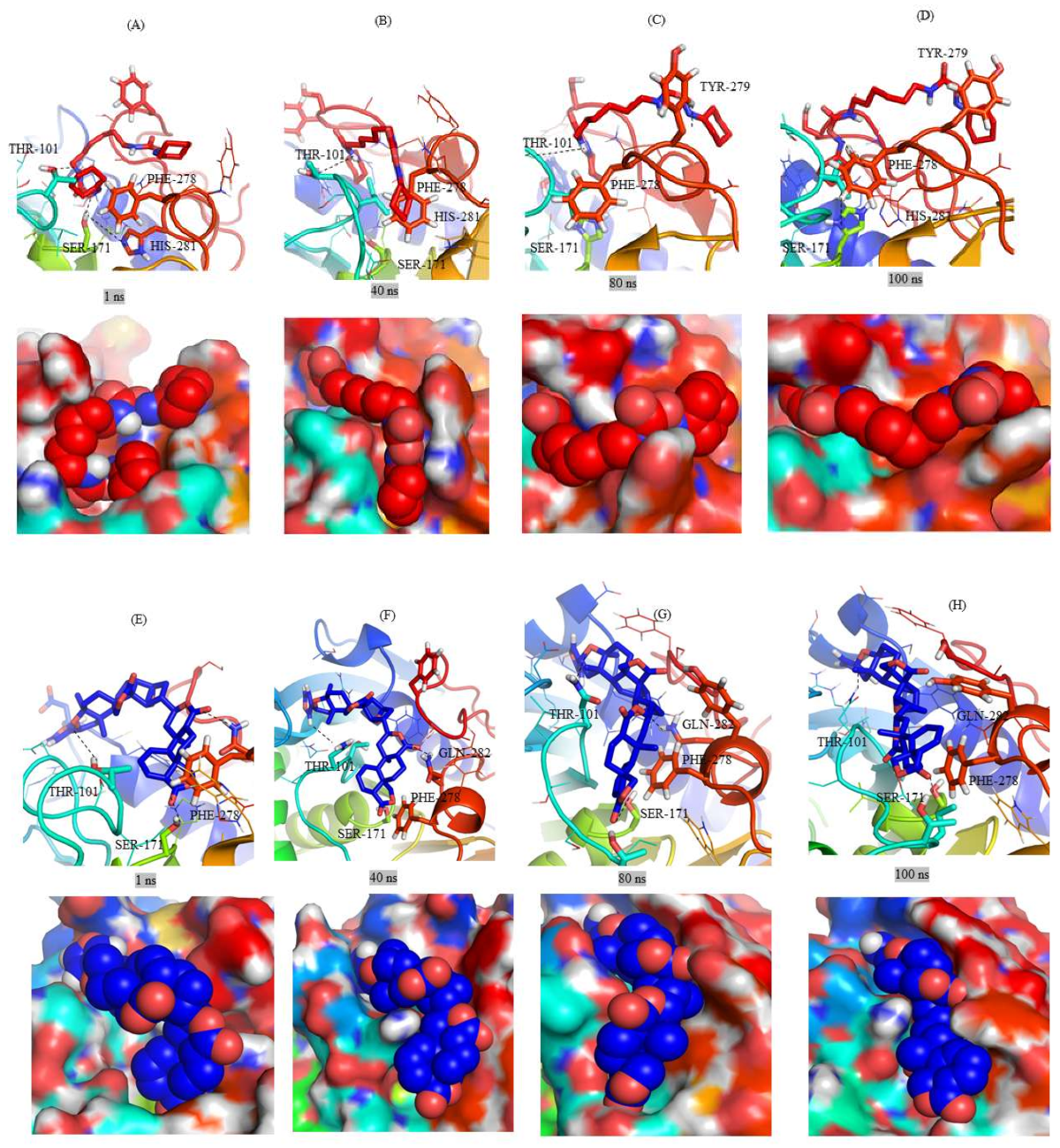
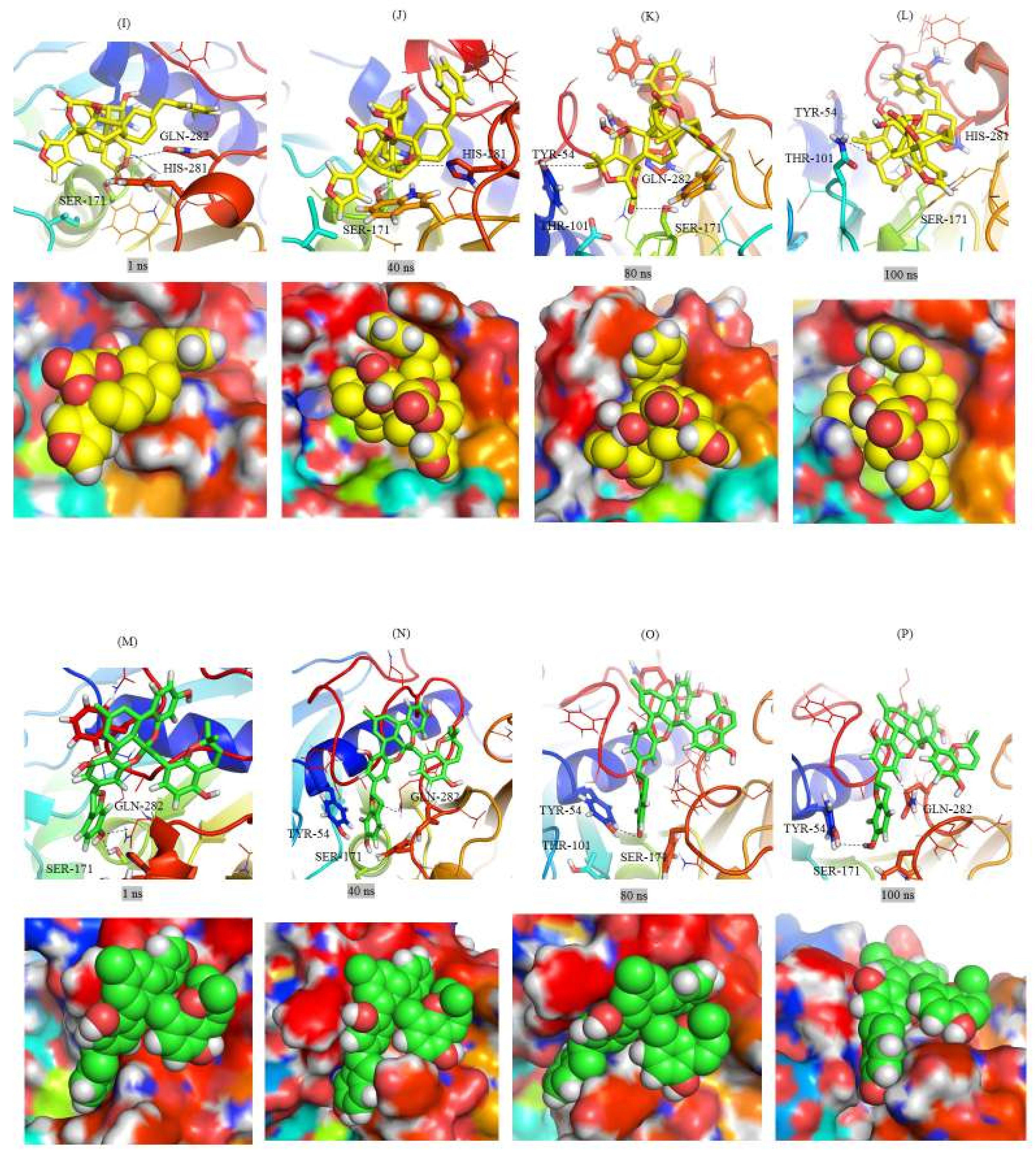
2.3. Structural Analysis of Mrlip1 Complexes
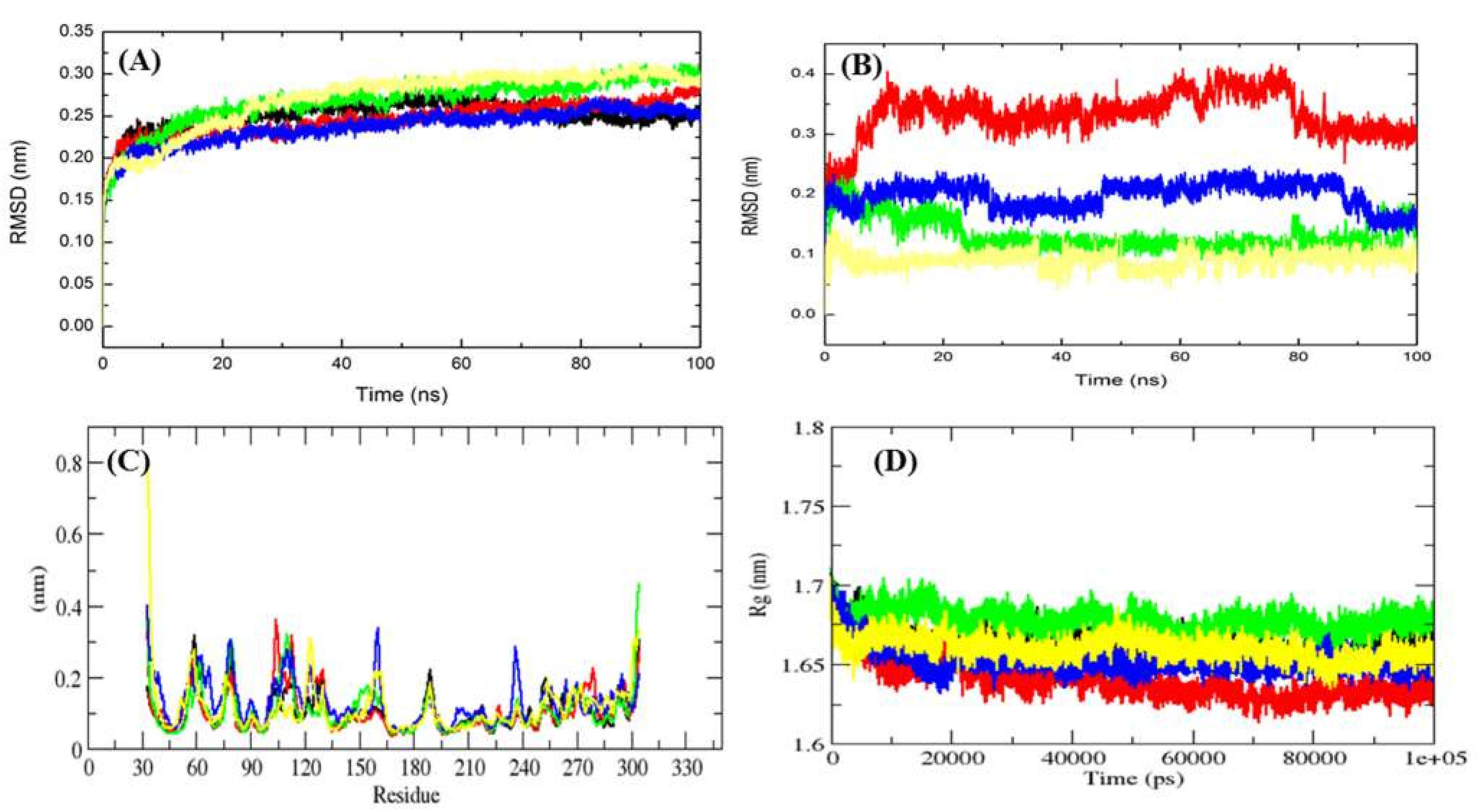
2.4. MD Simulation Data Analysis
2.4.1. Average Potential Energy of Complex Systems
2.4.2. Conformational of Mrlip1
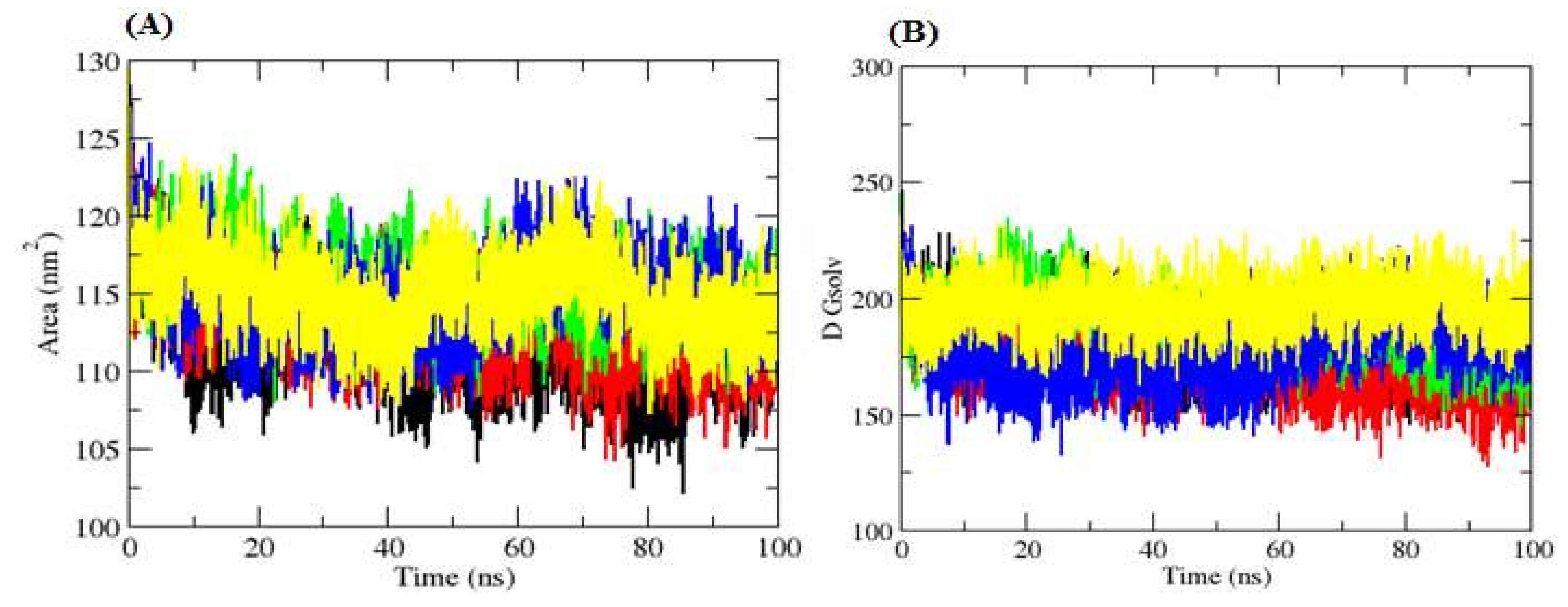
2.4.3. Solvent Accessible Surface Area
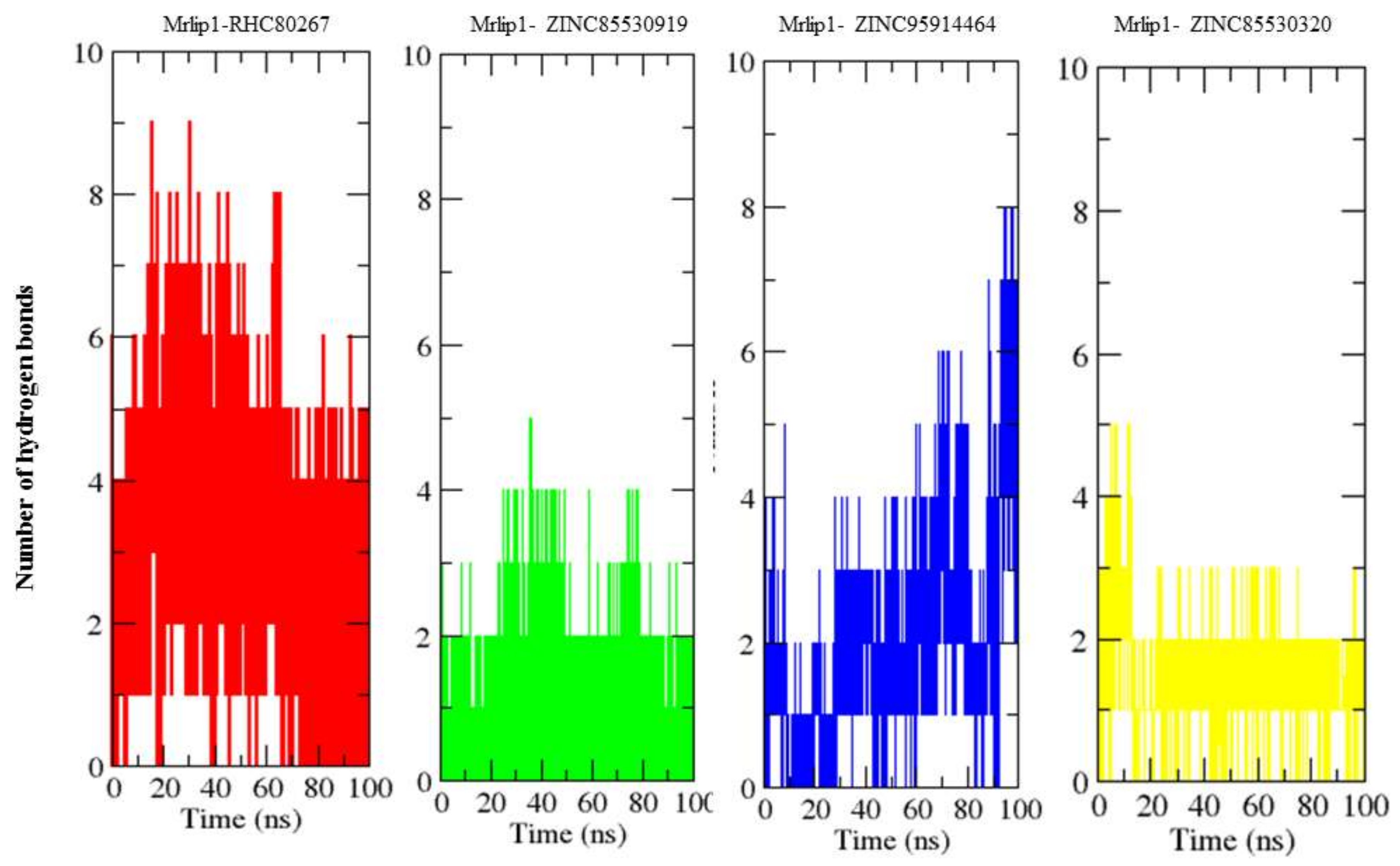
2.4.4. Hydrogen Bonds Analysis
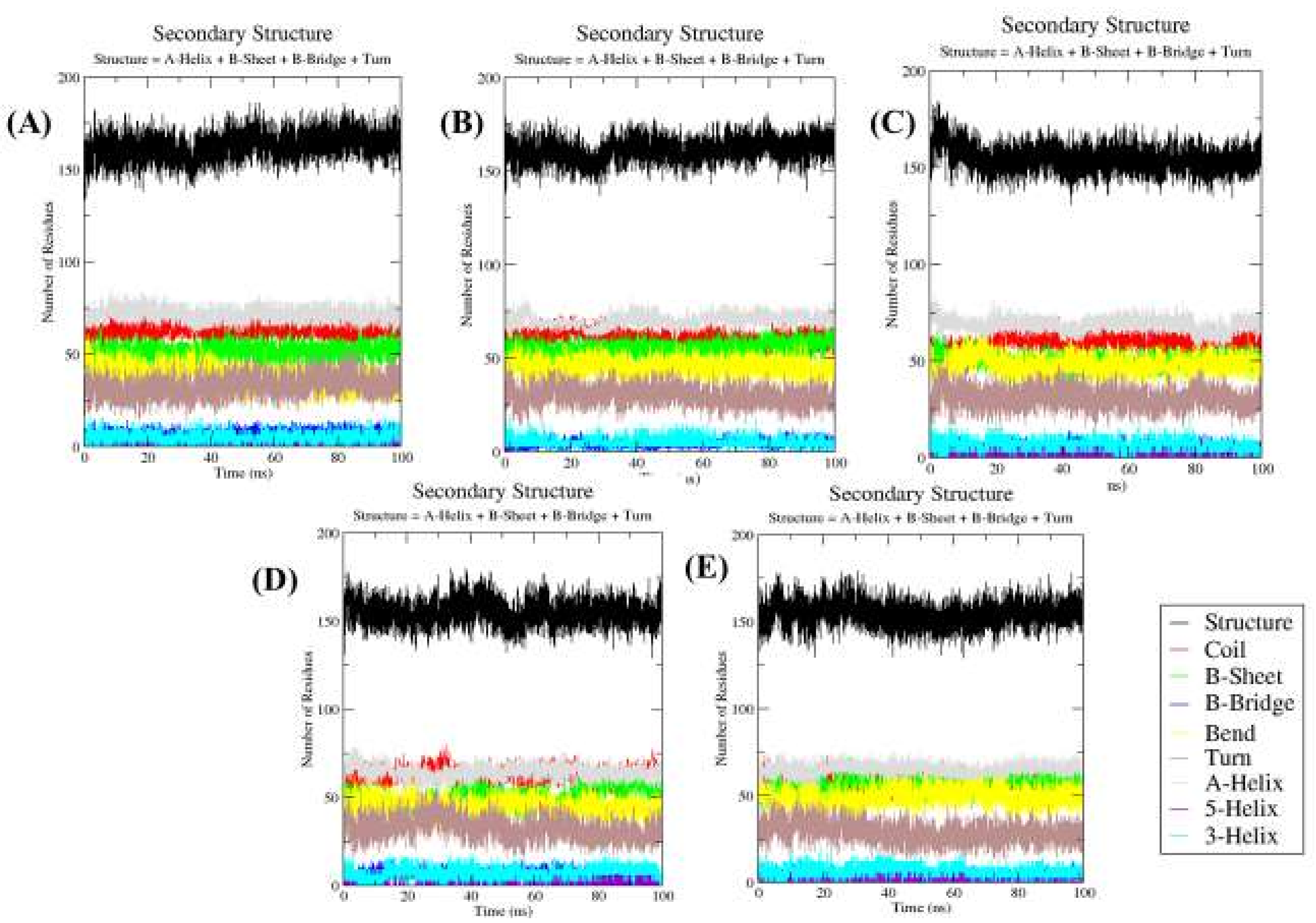
2.4.5. Secondary Structure Changes upon Ligands Binding
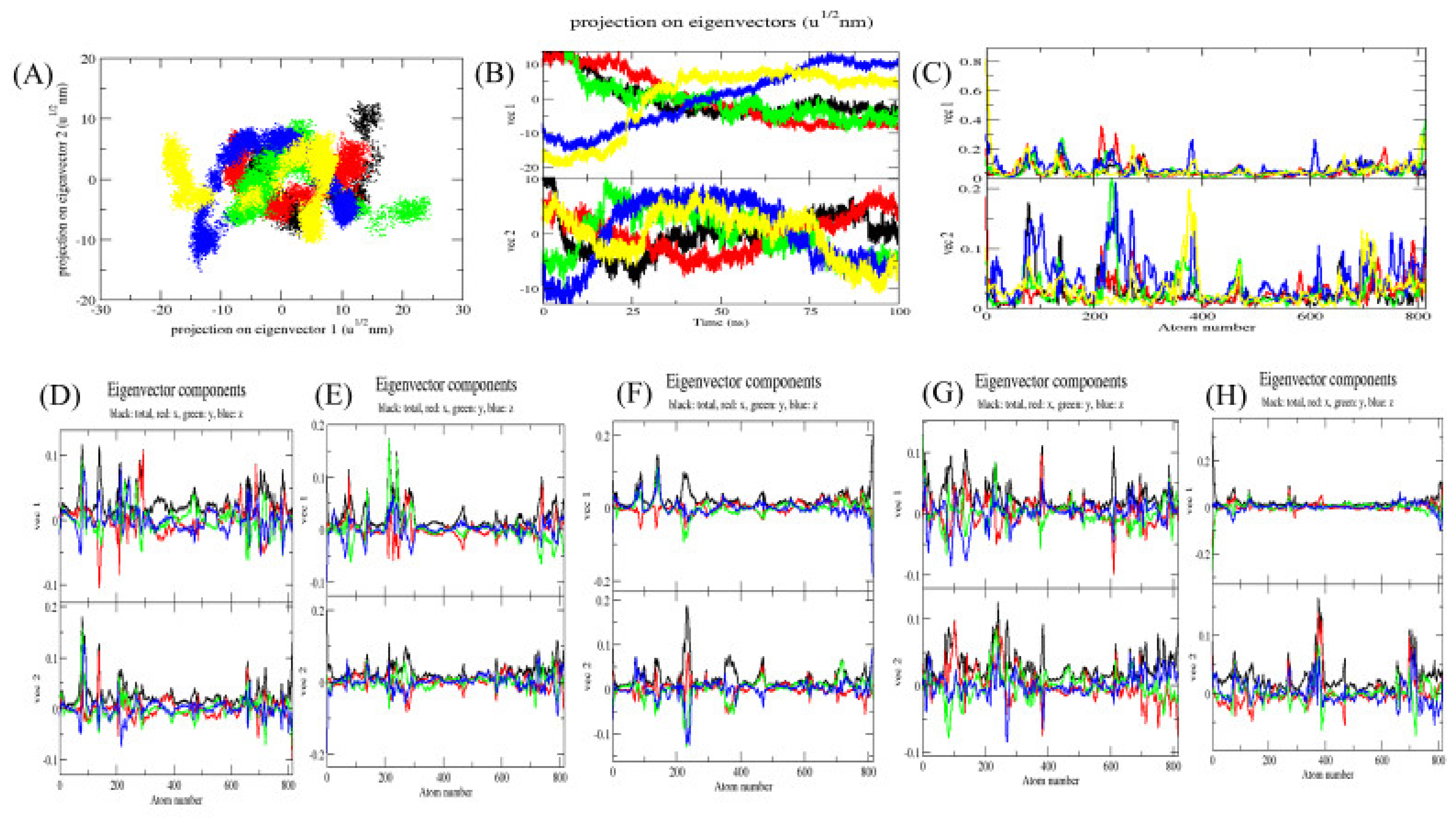
2.5. Principal Component Analysis
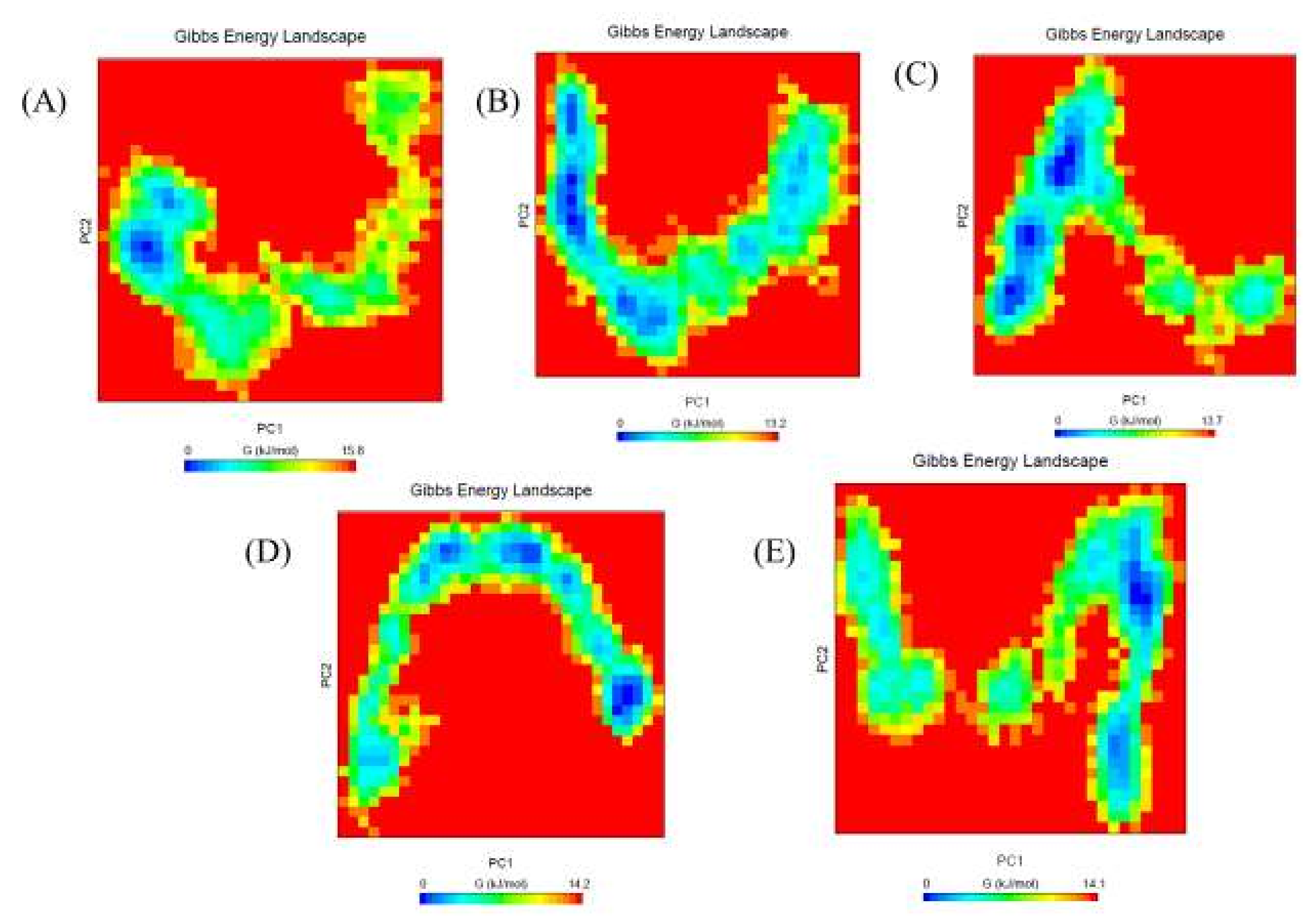
2.6. Gibbs Free Energy Landscape
2.7. MMPBSA Binding Energy Analysis
3. Materials and Methods
3.1. Preparation of Target Protein and Natural Compounds Library
3.2. Hit Selection and Drug-Ability Evaluation
3.3. Mrlip1 Structural Analysis: Visualization and Evaluation
3.4. MD Simulations
3.5. MMPBSA Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DAG | Diacyl glyceride |
| MMPBSA | Molecular mechanics Poisson Boltzmann surface area |
| TCM | Traditional Chinese Medicine |
| vTHS | Virtual High-throughput Screening |
References
- Cao, M.; Fonseca, L.M.; Schoenfuss, T.C.; Rankin, S.A. Homogenization and lipase treatment of milk and resulting methyl ketone generation in blue cheese. J. Agric. Food Chem. 2014, 62, 5726–5733. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Sun, L.; Chen, H.; Lan, D.; Wang, Y.; Yang, B. Enzymatic synthesis of diacylglycerols enriched with conjugated linoleic acid by a novel lipase from Malassezia globosa. J. Am. Oil Chem. Soc. 2012, 89, 1259–1266. [Google Scholar] [CrossRef]
- Khan, F.I.; Nizami, B.; Anwer, R.; Gu, K.R.; Bisetty, K.; Hassan, M.I.; Wei, D.Q. Structure prediction and functional analyses of a thermostable lipase obtained from Shewanella putrefaciens. J. Biomol. Struct. Dyn. 2017, 35, 2123–2135. [Google Scholar] [CrossRef] [PubMed]
- Sommer, B.; Overy, D.; Kerr, R.G. Identification and characterization of lipases from Malassezia restricta, a causative agent of dandruff. FEMS Yeast Res. 2015, 15, fov078. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Jung, W.H.; Han, S.H.; Lee, Y.H.; Lee, Y.W. Characterisation and Expression Analysis of MrLip1, a Class 3 Family Lipase of Malassezia restricta. Mycoses 2015, 58, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Z.; Yuan, C.; Liu, X.; Yang, F.; Wang, T.; Wang, J.; Manabe, K.; Qin, O.; Wang, X.; et al. Dandruff is associated with the conjoined interactions between host and microorganisms. Sci. Rep. 2016, 6, 24877. [Google Scholar] [CrossRef] [PubMed]
- Gemmer, C.M.; DeAngelis, Y.M.; Theelen, B.; Boekhout, T.; Dawson, T.L., Jr. Fast, noninvasive method for molecular detection and differentiation of Malassezia yeast species on human skin and application of the method to dandruff microbiology. J. Clin. Microbiol. 2002, 40, 3350–3357. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, Y.M.; Gemmer, C.M.; Kaczvinsky, J.R.; Kenneally, D.C.; Schwartz, J.R.; Dawson, T.L., Jr. Three etiologic facets of dandruff and seborrheic dermatitis: Malassezia fungi, sebaceous lipids, and individual sensitivity. J. Investig. Dermatol. Symp. Proc. 2005, 10, 295–297. [Google Scholar] [CrossRef]
- Schofield, D.A.; Westwater, C.; Warner, T.; Balish, E. Differential Candida albicans lipase gene expression during alimentary tract colonization and infection. FEMS Microbiol. Lett. 2005, 244, 359–365. [Google Scholar] [CrossRef]
- Stehr, F.; Felk, A.; Gácser, A.; Kretschmar, M.; Mähnss, B.; Neuber, K.; Hube, B.; Schäfer, W. Expression analysis of the Candida albicans lipase gene family during experimental infections and in patient samples. FEMS Yeast Res. 2004, 4, 401–408. [Google Scholar] [CrossRef]
- Brunke, S.; Hube, B. MfLIP1, a gene encoding an extracellular lipase of the lipid-dependent fungus Malassezia furfur. Microbiology 2006, 152, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.C.; Zani, M.B.; Arruda, A.C.; Arruda, L.H.; Paulino, L.C. Malassezia intra-specific diversity and potentially new species in the skin microbiota from Brazilian healthy subjects and seborrheic dermatitis patients. PLoS ONE 2015, 10, e0117921. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.I.; Lan, D.; Durrani, R.; Huan, W.; Zhao, Z.; Wang, Y. The Lid Domain in Lipases: Structural and Functional Determinant of Enzymatic Properties. Front. Bioeng. Biotechnol. 2017, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Monhemi, H.; Housaindokht, M.R.; Moosavi-Movahedi, A.A.; Bozorgmehr, M.R. How a protein can remain stable in a solvent with high content of urea: Insights from molecular dynamics simulation of Candida antarctica lipase B in urea: Choline chloride deep eutectic solvent. Phys. Chem. Chem. Phys. 2014, 16, 14882–14893. [Google Scholar] [CrossRef] [PubMed]
- Knapp, B.; Ospina, L.; Deane, C.M. Avoiding False Positive Conclusions in Molecular Simulation: The Importance of Replicas. J. Chem. Theory Comput. 2018, 14, 6127–6138. [Google Scholar] [CrossRef] [PubMed]
- Gramany, V.; Khan, F.I.; Govender, A.; Bisetty, K.; Singh, S.; Permaul, K. Cloning, expression, and molecular dynamics simulations of a xylosidase obtained from Thermomyces lanuginosus. J. Biomol. Struct. Dyn. 2016, 34, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.I.; Shahbaaz, M.; Bisetty, K.; Waheed, A.; Sly, W.S.; Ahmad, F.; Hassan, M.I. Large scale analysis of the mutational landscape in beta-glucuronidase: A major player of mucopolysaccharidosis type VII. Gene 2016, 576, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.I.; Wei, D.Q.; Gu, K.R.; Hassan, M.I.; Tabrez, S. Current updates on computer aided protein modeling and designing. Int. J. Biol. Macromol. 2016, 85, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Mazola, Y.; Guirola, O.; Palomares, S.; Chinea, G.; Menéndez, C.; Hernández, L.; Musacchio, A. A comparative molecular dynamics study of thermophilic and mesophilic beta-fructosidase enzymes. J. Mol. Model. 2015, 21, 2772. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.E.; Kamran Haider, M. Hydrogen Bonds in Proteins: Role and Strength; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2001. [Google Scholar]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Principal component analysis for protein folding dynamics. J. Mol. Biol. 2009, 385, 312–329. [Google Scholar] [PubMed]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [PubMed]
- Tiana, G.; Simona, F.; De Mori, G.M.; Broglia, R.A.; Colombo, G. Understanding the determinants of stability and folding of small globular proteins from their energetics. Protein Sci. 2004, 13, 113–124. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Warren, D. The PyMOL Molecular Graphics System. 2002. Available online: http://pymol.sourceforge.net/overview/index.htm (accessed on 10 July 2018).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2015. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Zhang, L.; Ai, H.; Chen, W.; Yin, Z.; Hu, H.; Zhu, J.; Zhao, J.; Zhao, Q.; Liu, H. CarcinoPred-EL: Novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci. Rep. 2017, 7, 2118. [Google Scholar] [CrossRef]
- Ali, S.; Khan, F.I.; Chen, W.; Rahaman, A.; Wang, Y. Open and closed states of Mrlip1 DAG lipase revealed by molecular dynamics simulation. Mol. Simul. 2018, 44, 1520–1528. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Khan, F.I.; Mohammad, T.; Khan, P.; Hasan, G.M.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. Exploring molecular insights into the interaction mechanism of cholesterol derivatives with the Mce4A: A combined spectroscopic and molecular dynamic simulation studies. Int. J. Biol. Macromol. 2018, 111, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.B.; Khan, F.I.; Khan, S.H.; Srivastava, S.; Hasan, G.M.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. Mechanistic insights into the urea-induced denaturation of kinase domain of human integrin linked kinase. Int. J. Biol. Macromol. 2018, 111, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.I.; Aamir, M.; Wei, D.Q.; Ahmad, F.; Hassan, M.I. Molecular mechanism of Ras-related protein Rab-5A and effect of mutations in the catalytically active phosphate-binding loop. J. Biomol. Struct. Dyn. 2017, 35, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Anwer, K.; Sonani, R.; Madamwar, D.; Singh, P.; Khan, F.; Bisetty, K.; Ahmad, F.; Hassan, M.I. Role of N-terminal residues on folding and stability of C-phycoerythrin: Simulation and urea-induced denaturation studies. J. Biomol. Struct. Dyn. 2015, 33, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Stephens, D.E.; Khan, F.I.; Singh, P.; Bisetty, K.; Singh, S.; Permaul, K. Creation of thermostable and alkaline stable xylanase variants by DNA shuffling. J. Biotechnol. 2014, 187, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Khan, F.I.; Xu, Q.; Wei, D.Q. Recent Studies of Mitochondrial SLC25, Integration of Experimental and Computational Approaches. Curr. Protein Pept. Sci. 2018, 19, 507–522. [Google Scholar] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Pol-Fachin, L.; Fernandes, C.L.; Verli, H. GROMOS96 43a1 performance on the characterization of glycoprotein conformational ensembles through molecular dynamics simulations. Carbohydr. Res. 2009, 344, 491–500. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Norberto de Souza, O.; Ornstein, R.L. Molecular dynamics simulations of a protein-protein dimer: Particle-mesh Ewald electrostatic model yields far superior results to standard cutoff model. J. Biomol. Struct. Dyn. 1999, 16, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Compound ID | MW | LogP | HBD | HBA | rBonds |
|---|---|---|---|---|---|---|
| 1. | RHC80267© | 394.51 | 5.42 | 2 | 8 | 13 |
| 2. | ZINC85627948 | 721.99 | 4.18 | 7 | 8 | 2 |
| 3. | ZINC85530320 | 682.8 | 4.20 | 1 | 9 | 3 |
| 4. | ZINC85569100 | 676.82 | 4.89 | 7 | 7 | 9 |
| 5. | ZINC95914464 | 670.79 | 4.35 | 0 | 9 | 2 |
| 6. | ZINC95914661 | 690.92 | 4.64 | 4 | 8 | 3 |
| 7. | ZINC85545357 | 597.87 | 3.96 | 3 | 7 | 1 |
| 8. | ZINC95914660 | 691.93 | 5.03 | 5 | 7 | 3 |
| 9. | ZINC85542923 | 552.85 | 4.90 | 4 | 3 | 0 |
| 10. | ZINC85632555 | 701.87 | 2.56 | 5 | 9 | 4 |
| 11. | ZINC85545857 | 662.94 | 4.22 | 5 | 6 | 2 |
| 12. | ZINC85546208 | 618.8 | 4.53 | 3 | 5 | 3 |
| 13. | ZINC85569586 | 710.94 | 3.77 | 3 | 7 | 1 |
| 14. | ZINC85632546 | 715.9 | 2.37 | 5 | 9 | 4 |
| 15. | ZINC85569094 | 720.91 | 5.48 | 7 | 7 | 8 |
| 16. | ZINC85531411 | 688.8 | 4.09 | 1 | 8 | 4 |
| 17. | ZINC85569130 | 674.84 | 5.25 | 6 | 6 | 8 |
| 18. | ZINC85546197 | 670.88 | 4.39 | 3 | 5 | 3 |
| 19. | ZINC85570604 | 684.99 | 5.00 | 4 | 5 | 2 |
| 20. | ZINC85530916 | 644.71 | 3.98 | 4 | 8 | 3 |
| 21. | ZINC85530919 | 644.71 | 4.05 | 4 | 8 | 3 |
| 22. | ZINC85542736 | 691.02 | 6.17 | 4 | 3 | 2 |
| 23. | ZINC85570874 | 723.98 | 4.74 | 4 | 7 | 7 |
| 24. | ZINC85570863 | 750.02 | 4.76 | 4 | 7 | 7 |
| 25. | ZINC85542639 | 715.04 | 6.46 | 4 | 3 | 3 |
| S. No. | Molecule | BBB Perme Ant | PPB | HIA | TPSA | Log S | Skin Permea Bility | CYP2D6 Inhibitor | Carcinogenicity |
|---|---|---|---|---|---|---|---|---|---|
| 1. | RHC80267© | No | 88.69 | 89.79 | 101.38 | −5.27 | −3.40 | No | NC |
| 2. | ZINC85530916 | No | 100.00 | 94.70 | 117.84 | −9.52 | −2.49 | No | NC |
| 3. | ZINC85530919 | No | 100.00 | 94.70 | 117.84 | −9.52 | −2.49 | No | NC |
| 4. | ZINC85531411 | No | 92.38 | 97.27 | 112.27 | −9.96 | −1.69 | No | NC |
| 5. | ZINC85546197 | No | 97.26 | 96.06 | 86.99 | −9.30 | −1.02 | No | NC |
| 6. | ZINC85546208 | No | 95.88 | 95.90 | 86.99 | −8.30 | −1.48 | No | NC |
| 7. | ZINC85545357 | No | 90.33 | 94.84 | 129.39 | −7.94 | −3.14 | No | NC |
| 8. | ZINC85545857 | No | 97.47 | 90.39 | 118.22 | −9.05 | −2.51 | No | NC |
| 9. | ZINC85530320 | No | 91.92 | 98.24 | 124.8 | −7.32 | −2.00 | No | NC |
| 10. | ZINC95914464 | No | 89.91 | 99.27 | 110.5 | −5.52 | −4.20 | No | NC |
| 11. | ZINC95914660 | No | 69.28 | 94.49 | 62.28 | −8.69 | −3.36 | No | NC |
| 12. | ZINC85569586 | No | 95.00 | 95.81 | 105.45 | −6.59 | −1.10 | No | NC |
| 13. | ZINC85570604 | No | 100.00 | 93.72 | 97.99 | −7.96 | −3.27 | No | NC |
| 14 | ZINC85570874 | No | 96.46 | 94.37 | 133.38 | −9.87 | −1.03 | No | NC |
| S. No | Compound IDs | Catalytic Pocket Interacting Residues | ||
|---|---|---|---|---|
| Hydrogen Bonds | π-π | π-Alkyl | ||
| 1. | RHC80267© | Ser101, Asn102, Gln282 | - | Tyr54, Ile106, Phe278, Tyr279 |
| 2. | ZINC85530919 | Ser171, Tyr279, Gln282 | Phe278, Phe294, Tyr279 | Phe278, Tyr279 |
| 3. | ZINC85530320 | Ser171, Gln282 | Phe294 | Tyr54, Phe278 |
| 4. | ZINC95914464 | Ser171, Arg83, Thr101, His281 | - | Ala292, Tyr54, Phe278 |
| Complexes | Average Potential Energy (kJ/mol) | Radius of Gyration (nm) | Average RMSD (nm) | Average SASA (Backbone, nm2) | Free Energy of Solvation (kJ/mol/nm2) |
|---|---|---|---|---|---|
| Mrlip1 | −642,080 | 1.67 | 0.25 | 132.15 | 181.83 |
| Mrlip1-RHC80267© | −630,167 | 1.64 | 0.21 | 134.81 | 172.17 |
| Mrlip1-ZINC85530919 | −629,866 | 1.67 | 0.27 | 133.69 | 187.16 |
| Mrlip1-ZINC95914464 | −629,362 | 1.65 | 0.20 | 134.06 | 177.10 |
| Mrlip1-ZINC85530320 | −632,500 | 1.67 | 0.28 | 132.77 | 196.83 |
| Complexes | Percentage of Protein Secondary Structure (SS%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Structure * | Coil | β-sheet | β-bridge | Bend | Turn | α-helix | 310-helix | |
| Mrlip1 | 60 | 23 | 19 | 2 | 14 | 12 | 26 | 2 |
| Mrlip1-RHC80267© | 59 | 22 | 21 | 2 | 17 | 11 | 26 | 2 |
| Mrlip1-ZINC85530919 | 57 | 22 | 18 | 2 | 18 | 11 | 25 | 3 |
| Mrlip1-ZINC95914464 | 57 | 23 | 19 | 2 | 16 | 12 | 24 | 3 |
| Mrlip1-ZINC85530320 | 57 | 21 | 21 | 1 | 18 | 11 | 24 | 3 |
| S. No. | Complexes | Average Binding Energy (kJ/mol) |
|---|---|---|
| 1. | Mrlip1-RHC80267© | −98.51 ± 12.97 |
| 2. | Mrlip1-ZINC85530919 | −232.28 ± 16.27 |
| 3. | Mrlip1-ZINC95914464 | −183.17 ± 10.98 |
| 4. | Mrlip1-ZINC85530320 | −85.71 ± 12.53 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, S.; Khan, F.I.; Mohammad, T.; Lan, D.; Hassan, M.I.; Wang, Y. Identification and Evaluation of Inhibitors of Lipase from Malassezia restricta using Virtual High-Throughput Screening and Molecular Dynamics Studies. Int. J. Mol. Sci. 2019, 20, 884. https://doi.org/10.3390/ijms20040884
Ali S, Khan FI, Mohammad T, Lan D, Hassan MI, Wang Y. Identification and Evaluation of Inhibitors of Lipase from Malassezia restricta using Virtual High-Throughput Screening and Molecular Dynamics Studies. International Journal of Molecular Sciences. 2019; 20(4):884. https://doi.org/10.3390/ijms20040884
Chicago/Turabian StyleAli, Shahid, Faez Iqbal Khan, Taj Mohammad, Dongming Lan, Md. Imtaiyaz Hassan, and Yonghua Wang. 2019. "Identification and Evaluation of Inhibitors of Lipase from Malassezia restricta using Virtual High-Throughput Screening and Molecular Dynamics Studies" International Journal of Molecular Sciences 20, no. 4: 884. https://doi.org/10.3390/ijms20040884
APA StyleAli, S., Khan, F. I., Mohammad, T., Lan, D., Hassan, M. I., & Wang, Y. (2019). Identification and Evaluation of Inhibitors of Lipase from Malassezia restricta using Virtual High-Throughput Screening and Molecular Dynamics Studies. International Journal of Molecular Sciences, 20(4), 884. https://doi.org/10.3390/ijms20040884