Hippocampal LMNA Gene Expression is Increased in Late-Stage Alzheimer’s Disease


Abstract
1. Introduction
2. Results
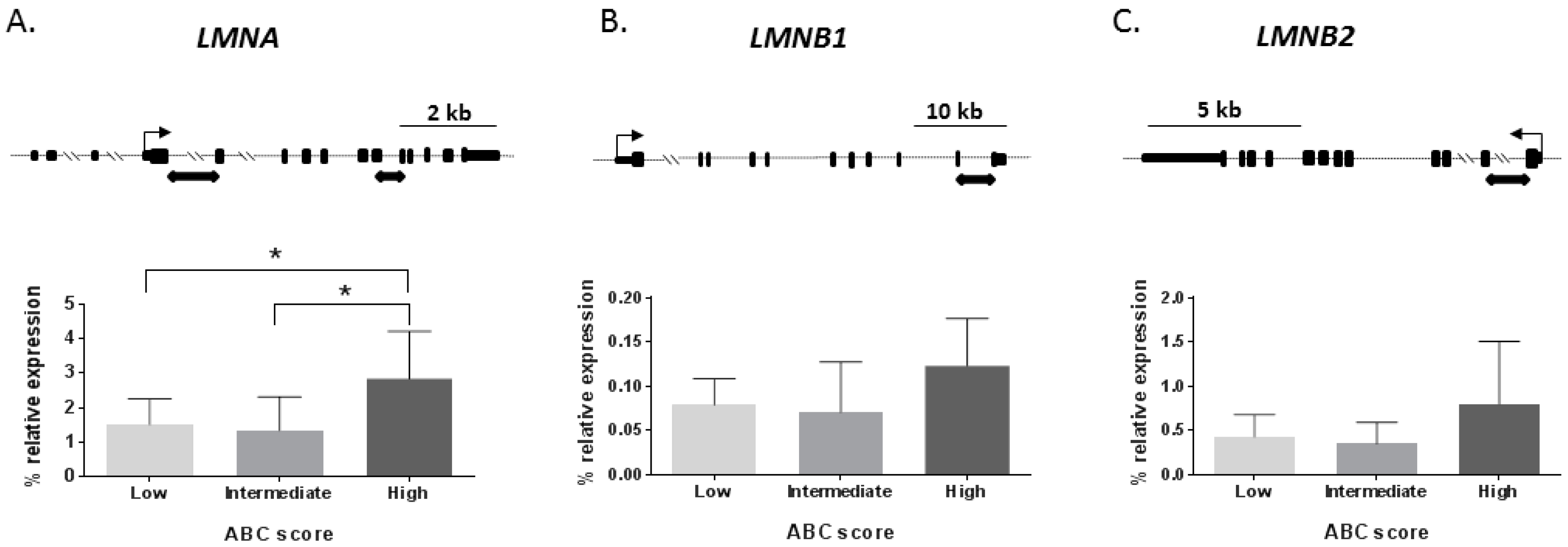
2.1. Lamin mRNA Levels across Alzheimer’s Disease Stages in the Human Hippocampus
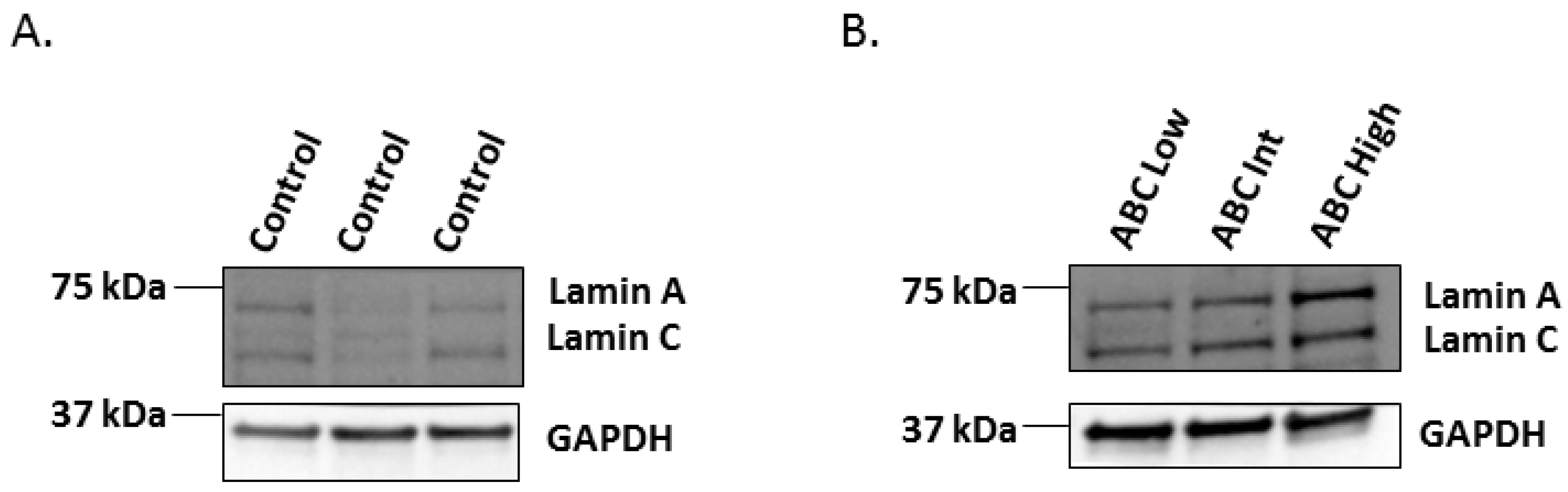
2.2. Western-Blot of Lamin A/C in the Human Hippocampus Affected by AD
2.3. Correlation between Lamin Genes mRNA Levels and p-Tau Burden
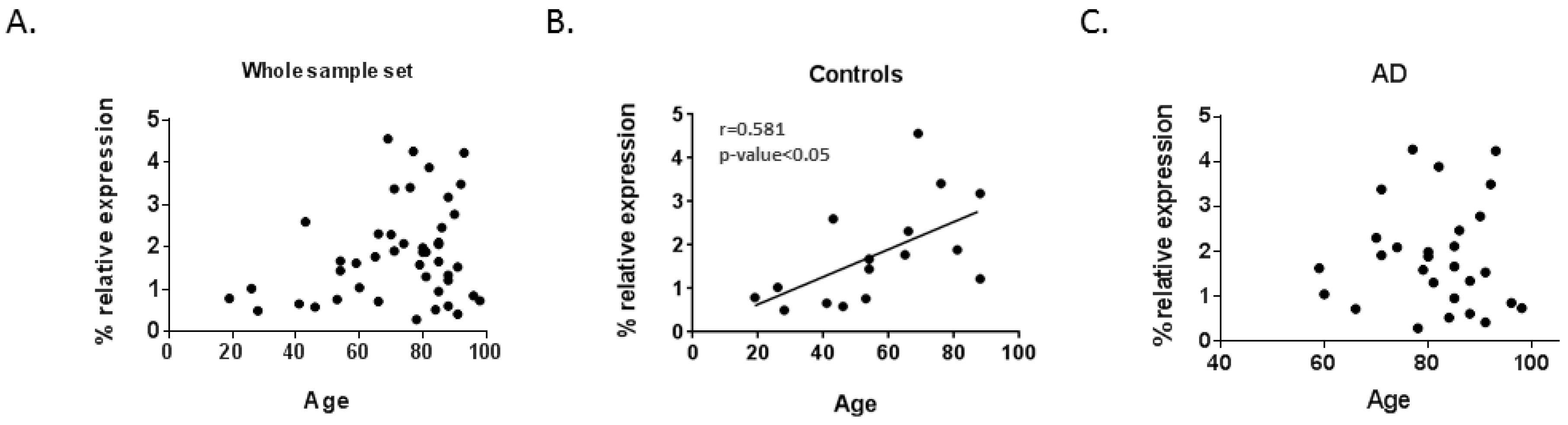
2.4. Correlation between Lamin Genes mRNA Levels and Age
3. Discussion
4. Materials and Methods
4.1. Human Hippocampal Samples and Neuropathological Examination
4.2. Lamin mRNA Expression Analysis by RT-qPCR
4.3. Progerin mRNA Expression Analysis by RT-PCR
4.4. LMNA Protein Expression Analysis by Western Blot
4.5. Quantitative Assessment of P-Tau Deposits in Hippocampal Samples
4.6. Statistical Data Analysis
4.7. Ethics Approval and Consent to Participate
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Mendez-Lopez, I.; Worman, H.J. Inner nuclear membrane proteins: Impact on human disease. Chromosoma 2012, 121, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Padiath, Q.S.; Saigoh, K.; Schiffmann, R.; Asahara, H.; Yamada, T.; Koeppen, A.; Hogan, K.; Ptáček, L.J.; Fu, Y.H. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 2006, 38, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.A.; Afawi, Z.; Bahlo, M.; Mauermann, M.; Misk, A.; Arsov, T.; Oliver, K.L.; Dahl, H.H.; Shearer, A.E.; Smith, R.J.; et al. Mutation of the nuclear lamin gene LMNB2 in progressive myoclonus epilepsy with early ataxia. Hum. Mol. Genet. 2015, 24, 4483–4490. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Cao, H.; Liu, D.M.; Costain, G.A.; Charlton-Menys, V.; Wilson Rodger, N.; Durrington, P.N. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am. J. Hum. Genet. 2006, 79, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Coppede, F. The epidemiology of premature aging and associated comorbidities. Clin. Interv. Aging 2013, 8, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Isaev, N.K.; Genrikhs, E.E.; Oborina, M.V.; Stelmashook, E.V. Accelerated aging and aging process in the brain. Rev. Neurosci. 2018, 29, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.J.; Gordon, L.B. Hutchinson-Gilford progeria syndrome. Handb. Clin. Neurol. 2015, 132, 249–264. [Google Scholar] [PubMed]
- Grupe, A.; Abraham, R.; Li, Y.; Rowland, C.; Hollingworth, P.; Morgan, A.; Jehu, L.; Segurado, R.; Stone, D.; Schadt, E.; et al. Evidence for novel susceptibility genes for late-onset Alzheimer’s disease from a genome-wide association study of putative functional variants. Hum. Mol. Genet. 2007, 16, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Belbin, O.; Carrasquillo, M.M.; Crump, M.; Culley, O.J.; Hunter, T.A.; Ma, L.; Bisceglio, G.; Zou, F.; Allen, M.; Dickson, D.W.; et al. Investigation of 15 of the top candidate genes for late-onset Alzheimer’s disease. Hum. Genet. 2011, 129, 273–282. [Google Scholar] [CrossRef]
- Yeh, H.L.; Hou, S.J.; Yen, F.C.; Hong, C.J.; Liou, Y.J.; Yang, A.C.; Liu, M.E.; Tsai, S.J. Polymorphisms in LMNA and near a SERPINA13 gene are not associated with cognitive performance in Chinese elderly males without dementia. Neurosci. Lett. 2011, 504, 32–34. [Google Scholar] [CrossRef]
- Schjeide, B.M.; McQueen, M.B.; Mullin, K.; DiVito, J.; Hogan, M.F.; Parkinson, M.; Hooli, B.; Lange, C.; Blacker, D.; Tanzi, R.E.; et al. Assessment of Alzheimer’s disease case-control associations using family-based methods. Neurogenetics 2009, 10, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Black, D.S.; Cole, S.W.; Irwin, M.R.; Breen, E.; St Cyr, N.M.; Nazarian, N.; Khalsa, D.S.; Lavretsky, H. Yogic meditation reverses NF-κB and IRF-related transcriptome dynamics in leukocytes of family dementia caregivers in a randomized controlled trial. Psychoneuroendocrinology 2013, 38, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Bardai, F.H.; Feany, M.B. Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr. Biol. 2016, 26, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Cornelison, G.L.; Levy, S.A.; Jenson, T.; Frost, B. Tau-induced nuclear envelope invagination causes a toxic accumulation of mRNA in Drosophila. Aging Cell 2018, e12847. [Google Scholar] [CrossRef] [PubMed]
- Lace, G.; Savva, G.M.; Forster, G.; de Silva, R.; Brayne, C.; Matthews, F.E.; Barclay, J.J.; Dakin, L.; Ince, P.G.; Wharton, S.B. MRC-CFAS, Hippocampal tau pathology is related to neuroanatomical connections: An ageing population-based study. Brain 2009, 132, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef]
- Luo, Y.B.; Mitrpant, C.; Johnsen, R.D.; Fabian, V.A.; Fletcher, S.; Mastaglia, F.L.; Wilton, S.D. Investigation of age-related changes in LMNA splicing and expression of progerin in human skeletal muscles. Int. J. Clin. Exp. Pathol. 2013, 6, 2778–2786. [Google Scholar]
- Messner, M.; Ghadge, S.K.; Goetsch, V.; Wimmer, A.; Dorler, J.; Polzl, G.; Zaruba, M.M. Upregulation of the aging related LMNA splice variant progerin in dilated cardiomyopathy. PloS ONE 2018, 13, e0196739. [Google Scholar] [CrossRef] [PubMed]
- Harhouri, K.; Navarro, C.; Baquerre, C.; Da Silva, N.; Bartoli, C.; Casey, F.; Mawuse, G.K.; Doubaj, Y.; Levy, N.; De Sandre-Giovannoli, A. Antisense-Based Progerin Downregulation in HGPS-Like Patients’ Cells. Cells 2016, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Frost, B. Alzheimer’s disease: An acquired neurodegenerative laminopathy. Nucleus 2016, 7, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Celarain, N.; Sánchez-Ruiz de Gordoa, J.; Zelaya, M.V.; Roldán, M.; Larumbe, R.; Pulido, L.; Echavarri, C.; Mendioroz, M. TREM2 upregulation correlates with 5-hydroxymethycytosine enrichment in Alzheimer’s disease hippocampus. Clin. Epigenet. 2016, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Ragnauth, C.D.; Warren, D.T.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C.M. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, P.; Riva, A.; Montano, M.; Pham, P.; Torkamani, A.; Scherba, E.; Benson, G.; Milton, J.N.; Baldwin, C.T.; Andersen, S.; et al. Whole genome sequences of a male and female supercentenarian, ages greater than 114 years. Front. Genet. 2011, 2, 90. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, P.; Solovieff, N.; Dewan, A.T.; Walsh, K.M.; Puca, A.; Hartley, S.W.; Melista, E.; Andersen, S.; Dworkis, D.A.; Wilk, J.B.; et al. Genetic signatures of exceptional longevity in humans. PloS ONE 2012, 7, e29848. [Google Scholar] [CrossRef]
- Worman, H.J.; Schirmer, E.C. Nuclear membrane diversity: Underlying tissue-specific pathologies in disease? Curr. Opin. Cell Biol. 2015, 34, 101–112. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef]
- Young, S.G.; Jung, H.J.; Coffinier, C.; Fong, L.G. Understanding the roles of nuclear A- and B-type lamins in brain development. J. Biol. Chem. 2012, 287, 16103–16110. [Google Scholar] [CrossRef] [PubMed]
- Bonati, U.; Bechtel, N.; Heinimann, K.; Rutz, E.; Schneider, J.; Frank, S.; Weber, P.; Fischer, D. Congenital muscular dystrophy with dropped head phenotype and cognitive impairment due to a novel mutation in the LMNA gene. Neuromuscul. Disord. 2014, 24, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Komaki, H.; Kawatani, M.; Sakuma, H.; Saito, Y.; Nakagawa, E.; Sugai, K.; Sasaki, M.; Hayashi, Y.K.; Nonaka, I.; et al. A novel mutation in the LMNA gene causes congenital muscular dystrophy with dropped head and brain involvement. Neuromuscul. Disord. 2012, 22, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Nissan, X.; Blondel, S.; Navarro, C.; Maury, Y.; Denis, C.; Girard, M.; Martinat, C.; De Sandre-Giovannoli, A.; Levy, N.; Peschanski, M. Unique preservation of neural cells in Hutchinson- Gilford progeria syndrome is due to the expression of the neural-specific miR-9 microRNA. Cell Rep. 2012, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Coffinier, C.; Choe, Y.; Beigneux, A.P.; Davies, B.S.; Yang, S.H.; Barnes, R.H., 2nd; Hong, J.; Sun, T.; Pleasure, S.J.; et al. Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. USA 2012, 109, E423–E431. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Schmidt, E.; Viceconte, N.; Strandgren, C.; Pernold, K.; Richard, T.J.; Van Leeuwen, F.W.; Dantuma, N.P.; Damberg, P.; Hultenby, K.; et al. Expression of progerin in aging mouse brains reveals structural nuclear abnormalities without detectible significant alterations in gene expression, hippocampal stem cells or behavior. Hum. Mol. Genet. 2015, 24, 1305–1321. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef]
- Pedersen, K.M.; Finsen, B.; Celis, J.E.; Jensen, N.A. Expression of a novel murine phospholipase D homolog coincides with late neuronal development in the forebrain. J. Biol. Chem. 1998, 273, 31494–31504. [Google Scholar] [CrossRef]
- Wegner, L.; Anthonsen, S.; Bork-Jensen, J.; Dalgaard, L.; Hansen, T.; Pedersen, O.; Poulsen, P.; Vaag, A. LMNA rs4641 and the muscle lamin A and C isoforms in twins—Metabolic implications and transcriptional regulation. J. Clin. Endocrinol. Metab. 2010, 95, 3884–3892. [Google Scholar] [CrossRef]
- Bell, J.E.; Alafuzoff, I.; Al-Sarraj, S.; Arzberger, T.; Bogdanovic, N.; Budka, H.; Dexter, D.T.; Falkai, P.; Ferrer, I.; Gelpi, E.; et al. Management of a twenty-first century brain bank: Experience in the BrainNet Europe consortium. Acta Neuropathol. 2008, 115, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Total Sample Set (n = 44) | Controls (n = 16) | AD Patients (n = 28) | ||||
|---|---|---|---|---|---|---|
| Gene | r Coefficient | p-Value | r Coefficient | p-Value | r Coefficient | p-Value |
| LMNA | 0.254 | 0.092 | 0.581* | 0.018 | 0.002 | 0.990 |
| LMNB1 | 0.211 | 0.164 | 0.149 | 0.582 | 0.358 | 0.061 |
| LMNB2 | 0.222 | 0.143 | 0.396 | 0.129 | 0.165 | 0.401 |
| ABC Score Staging | Controls (n = 16) | ABC Low (n = 14) | ABC Intermediate (n = 6) | ABC High (n = 8) |
|---|---|---|---|---|
| Age at death (years) | 56.1 ± 21.6 | 78.6 ± 9.4 | 88.3 ± 6.6 | 81.2 ± 12 |
| Gender (Female) % | 43.8 | 64.3 | 66.7 | 62.5 |
| PMI (h) | 7.5 ± 4.6 | 8.3 ± 8.6 | 4.9 ± 2.9 | 6.3 ± 4.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Méndez-López, I.; Blanco-Luquin, I.; Sánchez-Ruiz de Gordoa, J.; Urdánoz-Casado, A.; Roldán, M.; Acha, B.; Echavarri, C.; Zelaya, V.; Jericó, I.; Mendioroz, M. Hippocampal LMNA Gene Expression is Increased in Late-Stage Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 878. https://doi.org/10.3390/ijms20040878
Méndez-López I, Blanco-Luquin I, Sánchez-Ruiz de Gordoa J, Urdánoz-Casado A, Roldán M, Acha B, Echavarri C, Zelaya V, Jericó I, Mendioroz M. Hippocampal LMNA Gene Expression is Increased in Late-Stage Alzheimer’s Disease. International Journal of Molecular Sciences. 2019; 20(4):878. https://doi.org/10.3390/ijms20040878
Chicago/Turabian StyleMéndez-López, Iván, Idoia Blanco-Luquin, Javier Sánchez-Ruiz de Gordoa, Amaya Urdánoz-Casado, Miren Roldán, Blanca Acha, Carmen Echavarri, Victoria Zelaya, Ivonne Jericó, and Maite Mendioroz. 2019. "Hippocampal LMNA Gene Expression is Increased in Late-Stage Alzheimer’s Disease" International Journal of Molecular Sciences 20, no. 4: 878. https://doi.org/10.3390/ijms20040878
APA StyleMéndez-López, I., Blanco-Luquin, I., Sánchez-Ruiz de Gordoa, J., Urdánoz-Casado, A., Roldán, M., Acha, B., Echavarri, C., Zelaya, V., Jericó, I., & Mendioroz, M. (2019). Hippocampal LMNA Gene Expression is Increased in Late-Stage Alzheimer’s Disease. International Journal of Molecular Sciences, 20(4), 878. https://doi.org/10.3390/ijms20040878