Low Endotoxin Recovery—Masking of Naturally Occurring Endotoxin

 ,
,
Abstract
1. Introduction
2. Results
2.1. Preparation and Characterization of Naturally Occurring Endotoxin (NOE)
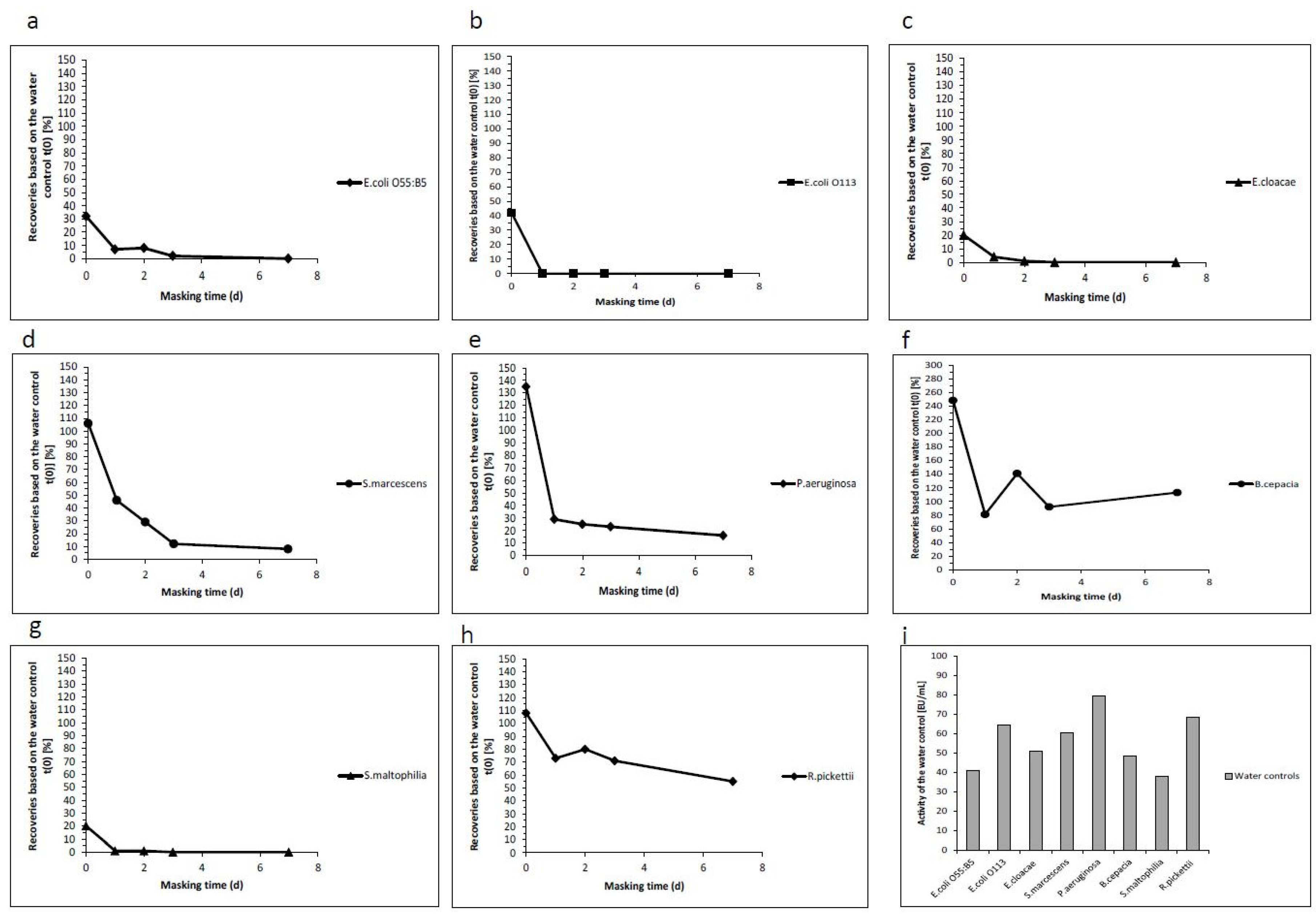
2.2. Analysis of the Masking Ability of Different NOE
2.3. Analysis of the Masking Ability of Different NOEs Depending on Their Growth Conditions
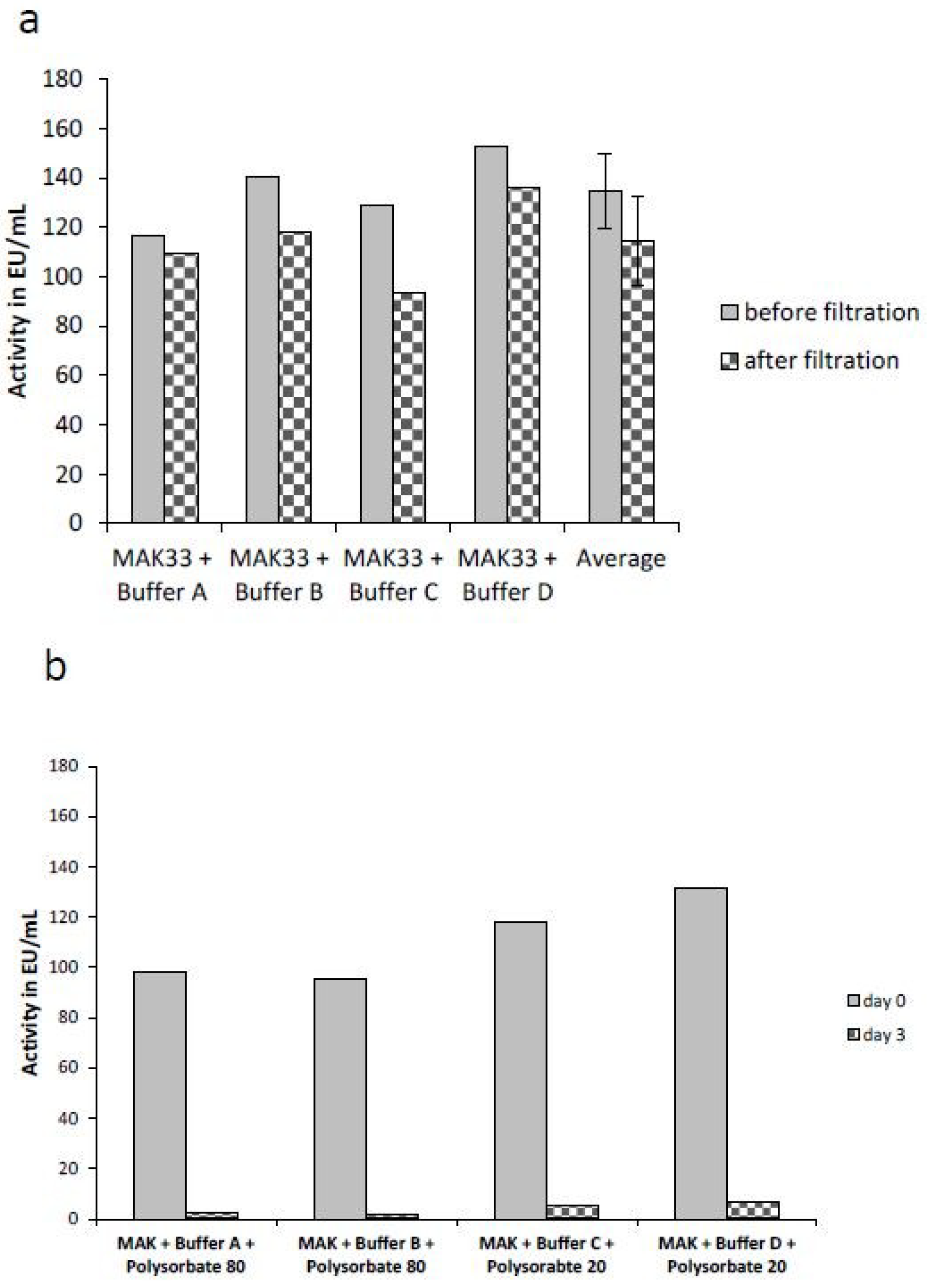
2.4. Comparison of the Masking Behavior of Purified Lipopolysaccharides (LPS) and NOE
2.5. Masking Behavior of an Endogenous Endotoxin Contamination
3. Discussion
4. Material and Methods
4.1. Chemicals and Materials
4.2. Endotoxins and Bacteria
4.3. Preparation of Crude Endotoxin Extracts (Naturally Occurring Endotoxins, NOEs) (Prep a)
4.4. Preparation of NOEs from Bacteria Grown under Low-Nutrition Conditions (Prep b)
4.5. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Silver Staining
4.6. Sample Masking Preparation for Masking Kinetics
4.7. Sample Preparation using an Endogenous Endotoxin Contamination
4.8. Endotoxin Detection
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rietschel, E.T.; Kirikae, T.; Schade, F.U.; Mamat, U.; Schmidt, G.; Loppnow, H.; Ulmer, A.J.; Zähringer, U.; Seydel, U.; Di Padova, F. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994, 8, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Rietschel, E.T.; Cavaillon, J.-M. Richard Pfeiffer and Alexandre Besredka: Creators of the concept of endotoxin and anti-endotoxin. Microbes Infect. 2003, 5, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.F.; Sá-Correia, I.; Valvano, M.A. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 2016, 40, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, H.; Gornicec, J.; Neuper, T.; Parigiani, M.A.; Wallner, M.; Duschl, A.; Horejs-Hoeck, J. Biological Activity of Masked Endotoxin. Sci. Rep. 2017, 7, 44750. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.C. Endotoxemia: Methods of detection and clinical correlates. Clin. Microbiol. Rev. 1995, 8, 268–292. [Google Scholar] [CrossRef] [PubMed]
- Hort, E.C.; Penfold, W.J. Microorganisms and their Relation to Fever: Preliminary Communication. J. Hyg. 1912, 12, 361–390. [Google Scholar] [CrossRef] [PubMed]
- Greisman, S.E.; Hornick, R.B. Comparative pyrogenic reactivity of rabbit and man to bacterial endotoxin. Proc. Soc. Exp. Biol. Med. 1969, 131, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Fennrich, S.; Fischer, M.; Hartung, T.; Lexa, P.; Montag-Lessing, T.; Sonntag, H.G.; Weigandt, M.; Wendel, A. Evaluation and further development of a pyrogenicity assay based on human whole blood. ALTEX 1998, 15, 123–128. [Google Scholar] [PubMed]
- Fennrich, S.; Fischer, M.; Hartung, T.; Lexa, P.; Montag-Lessing, T.; Sonntag, H.G.; Weigandt, M.; Wendel, A. Detection of endotoxins and other pyrogens using human whole blood. Dev. Biol. Stand. 1999, 101, 131–139. [Google Scholar] [PubMed]
- Schindler, S.; von Aulock, S.; Daneshian, M.; Hartung, T. Development, validation and applications of the monocyte activation test for pyrogens based on human whole blood. ALTEX 2009, 26, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Bang, F.B. A bacterial disease of Limulus Polyphemus. Bull. Johns Hopkins Hosp. 1956, 98, 325–351. [Google Scholar] [PubMed]
- Levin, J.; Bang, F.B. Clottable protein in Limulus; Its localization and kinetics of its coagulation by endotoxin. Thromb. Diath. Haemorrh. 1968, 19, 186–197. [Google Scholar] [PubMed]
- Levin, J.; Bang, F.B. The role of endotoxin in the extracellular coagulation of limulus blood. Bull. Johns Hopkins Hosp. 1964, 115, 265–274. [Google Scholar] [PubMed]
- Ding, J.L.; Ho, B. Endotoxin Detection—From Limulus Amebocyte Lysate to Recombinant Factor C. In Endotoxins: Structure, Function and Recognition; Springer: Dordrecht, The Netherlands, 2010; pp. 187–208. [Google Scholar]
- McCullough, K.Z.; Parenteral Drug Association. The Bacterial Endotoxins Test: A Practical Approach; River Grove: Bethesda, MD, USA, 2011. [Google Scholar]
- Williams, K.L.; Roberts, K.; Nnalue, N.A. Endotoxins Pyrogens, LAL Testing and Depyrogenation, 2nd ed.; Dekker: New York, NY, USA, 2001. [Google Scholar]
- Chen, J.; Vinther, A. Low endotoxin recovery in common biologics products. In Proceedings of the PDA Annual Meeting, Orlando, FL, USA, 15–17 April 2013. [Google Scholar]
- Bolden, J.S.; Claerbout, M.E.; Miner, M.K.; Murphy, M.A.; Smith, K.R.; Warburton, R.E. Evidence Against a Bacterial Endotoxin Masking Effect in Biologic Drug Products by Limulus Amebocyte Lysate Detection. PDA J. Pharm. Sci. Technol. 2014, 68, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.; Knight, M.; Stockman, S.; Omokoko, B. Results of a harmonized endotoxin recovery study protocol evaluation by 14 BioPhorum Operations Group (BPOG) member companies. Biologicals 2017, 48, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Reich, J.; Lang, P.; Grallert, H.; Motschmann, H. Masking of endotoxin in surfactant samples: Effects on Limulus-based detection systems. Biologicals 2016, 44, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Reich, J.; Tamura, H.; Nagaoka, I.; Motschmann, H. Investigation of the kinetics and mechanism of low endotoxin recovery in a matrix for biopharmaceutical drug products. Biologicals 2018, 53, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M. Factors Affecting Reduction of Reference Endotoxin Standard Activity Caused by Chelating Agent/Detergent Matrices: Kinetic Analysis of Low Endotoxin Recovery. PDA J. Pharm. Sci. Technol. 2017, 71, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Rudbach, J.A.; Akiya, F.I.; Elin, R.J.; Hochstein, H.D.; Luoma, M.K.; Milner, E.C.; Milner, K.C.; Thomas, K.R. Preparation and properties of a national reference endotoxin. J. Clin. Microbiol. 1976, 3, 21–25. [Google Scholar] [PubMed]
- Cooper, J.F. LER: Microbiology’s Hottest Urban Myth. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/181837-LER-Microbiology-s-Hottest-Urban-Myth/ (accessed on 30 November 2015).
- Platco, C.; Kave, J.; Tidswell, E. Resolution of Low Endotoxin/Lipopolysaccharide Recovery (LER/LLR) Testing. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/190994-Resolution-of-Low-Endotoxin-Lipopolysaccharide-Recovery-LER-LLR-Testing/ (accessed on 30 July 2016).
- Erridge, C.; Bennett-Guerrero, E.; Poxton, I.R. Structure and function of lipopolysaccharides. Microbes Infect. 2002, 4, 837–851. [Google Scholar] [CrossRef]
- IMattsby-Baltzer; Mielniczuk, Z.; Larsson, L.; Lindgren, K.; Goodwin, S. Lipid A in Helicobacter pylori. Infect. Immun. 1992, 60, 4383–4387. [Google Scholar]
- Steimle, A.; Autenrieth, I.B.; Frick, J.-S. Structure and function: Lipid A modifications in commensals and pathogens. Int. J. Med. Microbiol. 2016, 306, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.A.; Munford, R.S. Dephosphorylation of the lipid A moiety of Escherichia coli lipopolysaccharide by mouse macrophages. Infect. Immun. 1987, 55, 974–978. [Google Scholar] [PubMed]
- Bowers, K.; Lynn, T. Creation of an in-house naturally occurring endotoxin preparation for use in endotoxin spiking studies and LAL sample hold time analysis. Am. Pharm. Rev. 2011, 14, 92–97. [Google Scholar]
- Dubczak, J. The Great LER Debate. Available online: https://www.outsourcedpharma.com/doc/the-great-ler-debate-0001 (accessed on 28 January 2016).
- Lüderitz, O.; Staub, A.M.; Westphal, O. Immunochemistry of O and R antigens of Salmonella and related Enterobacteriaceae. Bacteriol. Rev. 1966, 30, 192–255. [Google Scholar] [PubMed]
- Bradley, S.G. Cellular and molecular mechanisms of action of bacterial endotoxins. Annu. Rev. Microbiol. 1979, 33, 67–94. [Google Scholar] [CrossRef]
- Yan, A.; Guan, Z.; Raetz, C.R.H. An undecaprenyl phosphate-aminoarabinose flippase required for polymyxin resistance in Escherichia coli. J. Biol. Chem. 2007, 282, 36077–36089. [Google Scholar] [CrossRef] [PubMed]
- Correa, W.; Brandenburg, K.; Zähringer, U.; Ravuri, K.; Khan, T.; von Wintzingerode, F. Biophysical Analysis of Lipopolysaccharide Formulations for an Understanding of the Low Endotoxin Recovery (LER) Phenomenon. Int. J. Mol. Sci. 2017, 18, 2737. [Google Scholar] [CrossRef] [PubMed]
- Department of Health and Human Services Food and Drug Administration; Center for Drug Evaluation and Research (CDER) Center for Veterinary Medicine (CVM). Bioanalytical Method Validation Guidance for Industry. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm070107.pdf (accessed on 24 May 2018).
- Caroff, M.; Karibian, D. Structure of bacterial lipopolysaccharides. Carbohydr. Res. 2003, 338, 2431–2447. [Google Scholar] [CrossRef] [PubMed]
- Survarna, K.; Lolas, A.; Hughes, P.; Friedman, R.L. Case Studies of Microbial Contamination in Biologic Product Manufacturing. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/36755-Case-Studies-of-Microbial-Contamination-in-Biologic-Product-Manufacturing/ (accessed on 1 January 2011).
- Platco, C. Low Lipopolysaccharide Recovery versus Low Endotoxin Recovery in Common Biological Product Matrices. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/167451-Low-Lipopolysaccharide-Recovery-versus-Low-Endotoxin-Recovery-in-Common-Biological-Product-Matrices/ (accessed on 1 September 2014).
- Tirumalai, R. Naturally Occurring Endotoxin: A New Reference Material Proposed By the US Pharmacopeia. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/190860-Naturally-Occurring-Endotoxin-A-New-Reference-Material-Proposed-By-the-US-Pharmacopeia/ (accessed on 29 July 2016).
- Groisman, E.A.; Kayser, J.; Soncini, F.C. Regulation of polymyxin resistance and adaptation to low-Mg2+ environments. J. Bacteriol. 1997, 179, 7040–7045. [Google Scholar] [CrossRef]
- Silipo, A. Full structural characterization of the lipid A components from the Agrobacterium tumefaciens strain C58 lipopolysaccharide fraction. Glycobiology 2004, 14, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Vinion-Dubiel, A.D.; Goldberg, J.B. Lipopolysaccharide of Burkholderia cepacian complex. J. Endotoxin Res. 2003, 9, 201–213. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Absorbance | Activity |
|---|---|---|
| (600 nm) | (EU/mL) | |
| E. coli O55:B5 | 1.9 | 146,174 |
| E. coli O113 | 1.7 | 402,789 |
| E. cloacae | 1.7 | 189,103 |
| S. marcescens | 1.4 | 116,175 |
| P. aeruginosa | 0.9 | 8595 |
| B. cepacia | 0.7 | 357 |
| S. maltophilia | 0.3 | 4557 |
| R. pickettii | 0.8 | 77,815 |
| Masking Time in Days | Endotoxin Recoveries Based on the Water Control t(0) (%) | Activity of the Water Control (EU/mL) | ||||
|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 7 | ||
| E. coli O55:B5 | 32 | 7 | 8 | 2 | 0 | 40.9 |
| E. coli O113 | 42 | 0 | 0 | 0 | 0 | 64.7 |
| E. cloacae | 20 | 4 | 1 | 0 | 0 | 50.9 |
| S. marcescens | 106 | 46 | 29 | 12 | 8 | 60.5 |
| P. aeruginosa | 135 | 29 | 25 | 23 | 16 | 79.6 |
| B. cepacia | 248 | 81 | 141 | 92 | 113 | 48.5 |
| S. maltophilia | 20 | 1 | 1 | 0 | 0 | 37.9 |
| R. pickettii | 108 | 73 | 80 | 71 | 55 | 68.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reich, J.; Weyer, F.A.; Tamura, H.; Nagaoka, I.; Motschmann, H. Low Endotoxin Recovery—Masking of Naturally Occurring Endotoxin. Int. J. Mol. Sci. 2019, 20, 838. https://doi.org/10.3390/ijms20040838
Reich J, Weyer FA, Tamura H, Nagaoka I, Motschmann H. Low Endotoxin Recovery—Masking of Naturally Occurring Endotoxin. International Journal of Molecular Sciences. 2019; 20(4):838. https://doi.org/10.3390/ijms20040838
Chicago/Turabian StyleReich, Johannes, Felix Alexander Weyer, Hiroshi Tamura, Isao Nagaoka, and Hubert Motschmann. 2019. "Low Endotoxin Recovery—Masking of Naturally Occurring Endotoxin" International Journal of Molecular Sciences 20, no. 4: 838. https://doi.org/10.3390/ijms20040838
APA StyleReich, J., Weyer, F. A., Tamura, H., Nagaoka, I., & Motschmann, H. (2019). Low Endotoxin Recovery—Masking of Naturally Occurring Endotoxin. International Journal of Molecular Sciences, 20(4), 838. https://doi.org/10.3390/ijms20040838