1. Introduction
The genus
Allium (Amaryllidaceae) includes more than 800 species, making it one of the largest monocotyledonous genera. For the purpose of this study we selected the two species which, according to the Food and Agriculture Organization of the United Nations (FAO), make the largest contribution to food production,
A. cepa and
A. sativum, (onion and garlic), and we also included one distant relative,
A. ursinum (wild garlic), which is of only marginal economic importance, but is interesting for several other reasons. Firstly, it grows relatively abundantly and is native to Europe and Asia. Secondly,
A. ursinum belongs to the oldest evolutionary clade in
Allium. The other two selected species,
A. cepa and
A. sativum, belong to the youngest evolutionary line [
1].
Allium genomes belong to the group of giant plant genomes:
A. cepa has 1C=16.75 pg [
2],
A. sativum has more or less the same genome size 1C = 16.25 pg [
3], but
A. ursinum has almost twice as large a genome 1C = 31.45 pg in comparison with the previous two [
4].
A. cepa and
A. sativum represent members of the genus with the most common chromosome number
x = 8, but other numbers (e.g.,
x = 7, 9, 10, 11) and variability in ploidy also occur (reviewed in [
1]).
A. ursinum is an example with the chromosome number
x = 7.
The
Allium genome assembly has not been released so far. There are several genomic strategies on how to prepare an informative repeatome of an as yet unassembled genome, but most of these need some reference genome of a closely related species or general repeat database at least, and NGS data with high genomic coverage. On the other hand, identification of major repeats
de novo using RepeatExplorer, TAREAN and Tandem repeats finder is independent on any reference sequence [
5,
6,
7].
Among repetitive elements, special attention is paid to ribosomal RNAs (rRNAs) and their genes (rDNAs) due to the essential importance of rRNA in proteosynthesis. These remnants of the RNA-world are relatively highly conserved and their genes are present in every cellular genome (reviewed in [
8]). However, the structure and organization of rDNA may differ [
9]. 45S rDNA codes for 18S, 5.8S and 25S rRNA, while 5S rDNA codes for 5S rRNA. Both loci may exist either separately as tandemly repeated units, or in a linked arrangement where a single unit of 5S rDNA is inside the intergenic spacer between 45S rDNA units. Physical linkage between 45S and 5S rDNA in plants has been described in some early diverging taxa of mosses, algae and ferns [
10,
11], while a separate arrangement is typical for most land plants. Exceptions were discovered, e.g., in Asteraceae [
12] and gymnosperms [
13]. The number of loci possessing 45S and 5S varies from a single one to several tens per haploid genome [
14]. Their number is usually species-specific, however, exceptions with hypervariability and polymorphisms in the rDNA signals were described in
Anacyclus (Asteraceae) [
15].
Most
Allium species studied have a low number of rDNA clusters per genome, except for
A. ursinum, where amplification of 45S rDNA was observed [
16]. One strong and one very weak signal of 45S rDNA per haploid genome were detected in
A. fistulosum (
n = 8), while two strong signals of 45S rDNA clusters per haploid genome were described in
A. cepa [
17]. In
A. wakegi (
n = 8), a natural allodiploid between
A. cepa and
A. fistulosum, three signals per haploid genome were detected [
18]. Genbank contains numerous accessions with short partial 45S rDNA sequences from
Allium, e.g., partial gene sequences and intergenic spacers from
A. ursinum (HF934582; 456 nt, KM103427; 1174 nt, and KX167936; 470 nt). Two notably long sequences represent clones containing either complete 45S rDNA unit sequences (KM117265; 10,621 nt) or partial sequences of 25S and 18S rRNA genes and the spacer between them from
A. cepa (EU256494; 6117 nt). Regarding 5S rDNA, one locus was observed e.g., in
A. fistulosum, two distinct loci of 5S rDNA in
A. cepa, and three in
A. sativum [
19,
20]. Numerous 5S rDNA sequences are publicly available for several
Allium species, e.g., for
A. cepa (Genbank AF101244; 352 nt and KM117264; 522 nt) and
A. sativum (AF101249; 603 nt).
Centromeres are nucleoprotein structures responsible for proper segregation of chromosomes in mitosis and meiosis, and they are usually species-specific in their sequences. The characterization of centromere elements
de novo is well described in [
21]. The method is based on Chip-Seq using antibodies specific for the centromere histone variant CenH3. The only centromere chromatin immunoprecipitated sequences from
Allium are from
A. fistulosum (GenBank AB735740, AB735741, AB735743, AB735742, AB735744; 144–376 nt) [
22].
Telomeres are chromatin structures forming the ends of linear chromosomes. Their function is to protect the natural ends and distinguish them from DNA breaks. The telomere sequence is usually formed by a minisatellite with a motif that can be summarized as (T
xA
yG
z)
n. Recently we demonstrated that an unusual telomere repeat (TTATGGGCTCGG)
n forms telomeres in 11
Allium species [
16], suggesting that the sequence is common across the genus.
Long tandem repeats were among the first characterized in
A. cepa. It was shown that approximately 4% of the
A. cepa genome is composed of a satellite with a unit of 375 bp and it localizes near all chromosomal termini except the ends occupied by nucleolus organizer regions (NORs). Three variants of this satellite were published, ACSAT1, ACSAT2, and ACSAT3 (GenBank X02572; 372 nt), differing in several single nucleotide positions and short insertions and deletions [
23]. Much higher variability was found in more detailed studies, when sequence and length variants were described, including, e.g., a 314 bp long repeat called pAc074 (GenBank AF227152; 314 nt), [
24]. This showed a similar terminal localization as ACSATs. Similar satellites were detected and characterized in closely related
Allium species, such as
A. fistulosum (AFISAT), which is 378–380 bp long and it has 82% homology to ACSAT. Furthermore, from
A. roylei, AROSAT (GenBank: Z69035),
A. oschaninii, AOSAT (GenBank: Z69034),
A. altaicum, AALSAT (GenBank: Z69033), and from
A. galanthum, AGASAT were described (GenBank: Z69036) [
25]. It was estimated that 2.8 million copies of AFISAT exist per haploid
A. fistulosum genome, which represents a genomic proportion of 4.5% [
26]. An original model of a simple tandem array built only from satellites has been updated with information about inversions of its repeat units, interruptions by microsatellites, and Ty1/copia elements [
27].
Recently, two new tandem repeats with specific chromosomal positions were described in
A. fistulosum. The first one was called “Heterochromatin-associated
Allium tandem repeat from CL58” (HAT58, KX137121; 65 nt). Species-specific HAT58 has a unit 65 nt long and it localizes to chromosomes 6, 7, and 8. It occurs as a polymorphic region on chromosome 7. The second repeat, “periCentromeric
Allium tandem repeat from CL36” (CAT36, KX137122; 197 nt) localizes to pericentromeres of chromosomes 5 and 6. These two repeats represent 0.25% of the
A. fistulosum genome. It was estimated that there are 160,000 copies of HAT58 and 93,000 copies of CAT36 per haploid genome. [
28]
Mobile elements represent another important class of repeats in
Allium genomes. Heterogeneous retrotransposons belonging to the Ty1/copia group were estimated to represent roughly 3% of the
A. cepa genome, (100,000–200,000 copies per haploid genome). Ty1/copia elements were shown to be distributed all over the
A. cepa chromosomes, but they were enriched in the terminal heterochromatin. A similar finding was obtained in the case of EnSpm-like DNA transposons [
29,
30]. An enrichment of Ty3/gypsy retrotransposons in the centromeres was observed in
A. cepa and
A. fistulosum, using a probe derived from a genomic clone (GenBank ET645811; 910 nt). The total number of these centromere-enriched retrotransposons was estimated at 26,000 copies per haploid genome, i.e., 0.89% in terms of genomic proportion. The authors of this study hypothesized that centromeric chromatin is a safe environment due to its reduced rate of recombination and thus a lower risk of removal of the element from the genome [
31].
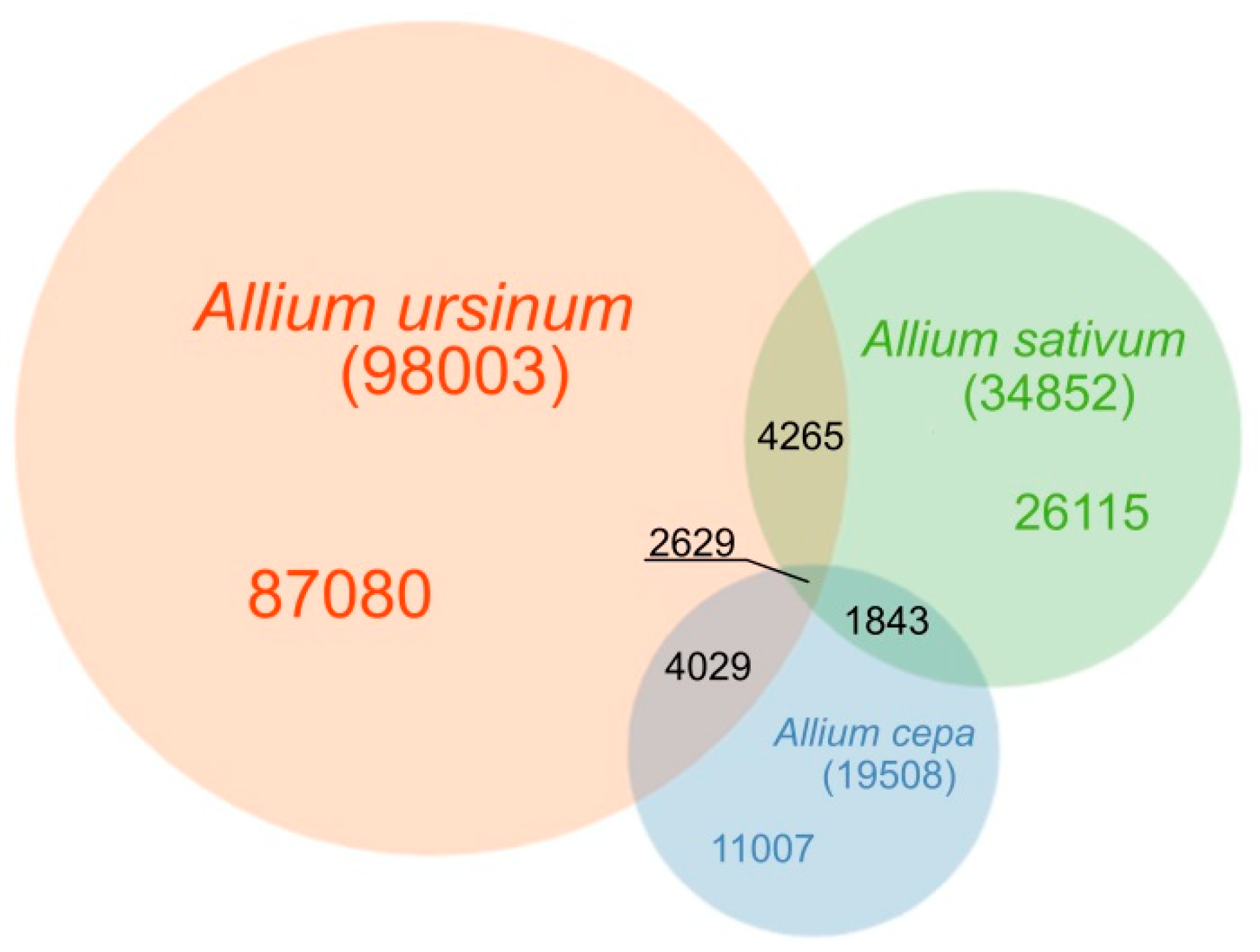
Overall, patterns of diverse types of repeats may reflect processes involved in speciation and karyotype evolution. Furthermore, knowledge of repeatomes can be applied to breeding strategies in respective crop species. In this study, we perform a comparative description of repeats in three Allium species using a genome assembly-independent approach, and provide a repeat database with information on major short and long tandems, mobile elements, and plastid DNA.
3. Discussion
We have performed a comprehensive pilot study of the repeatome in three
Allium species,
A. cepa,
A. ursinum and
A. sativum. All our results are based on incomplete and low genome coverage approaches (
Table 4). Previously, similar analyzes were done in
A. fistulosum [
28,
35] and
A. cepa [
31] but not in
A. sativum,
A. ursinum or any other species from the old
Allium evolutionary line. Our selection of species for comparative analysis covered several aspects.
A. cepa and
A. sativum, two economically valuable crop species from the same young evolutionary line, are compared to each other and to the wild growing species from the old evolutionary line,
A. ursinum. Our data are based on slightly higher genome coverage than in previous studies. For example, we used NGS data corresponding to 0.004× in comparative and 0.01× in separate TAREAN and RepeatExplorer analyses, and 0.277× in TRFi-TRM in the case of
A. cepa. But it is not the coverage what makes the data specific. Considering their sensible volume, the data were produced in a way that minimized amplification bias (PCR-free libraries), and ensured at least minimal control of sequencing reliability (3× parallel libraries per species from the same DNA extract).
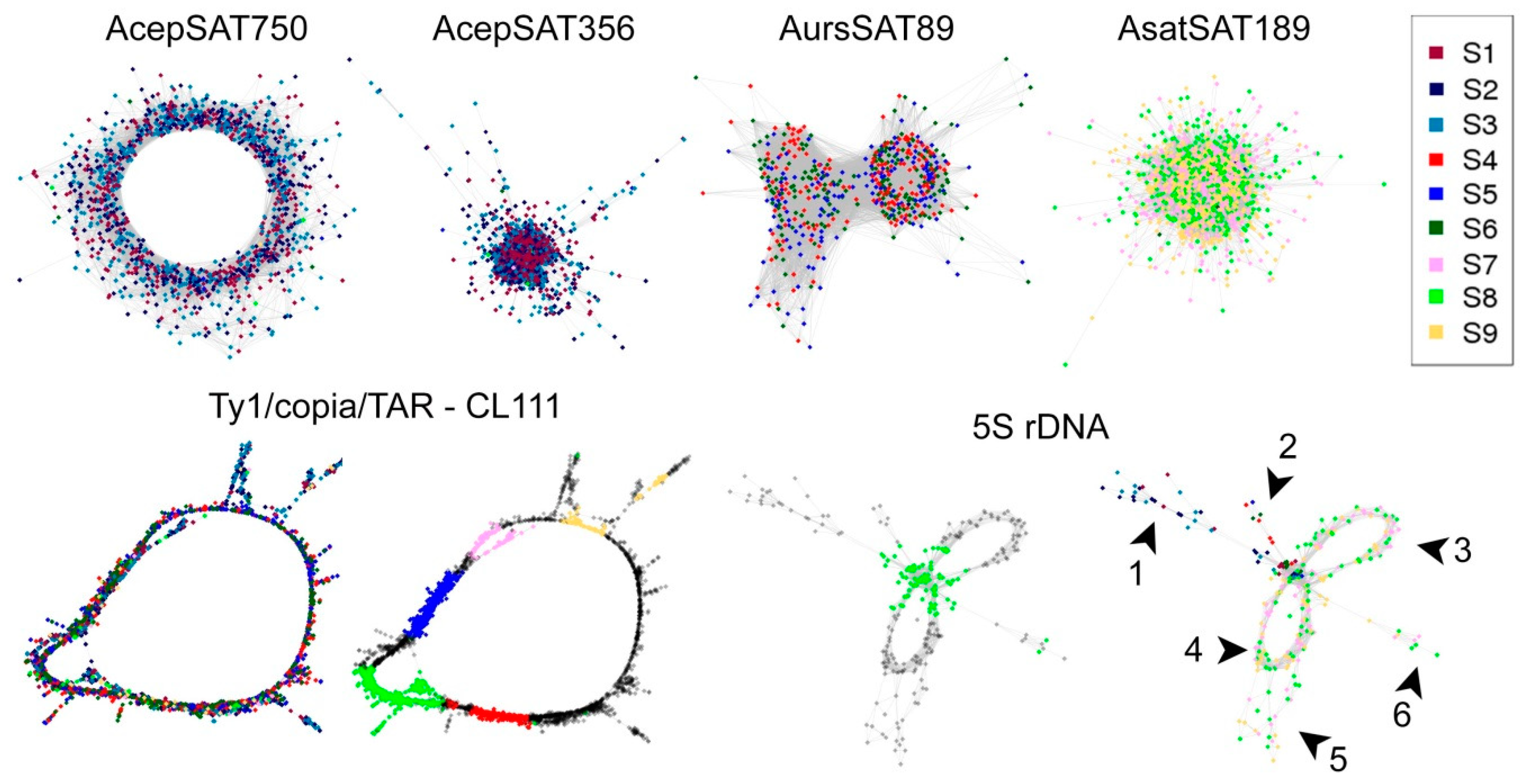
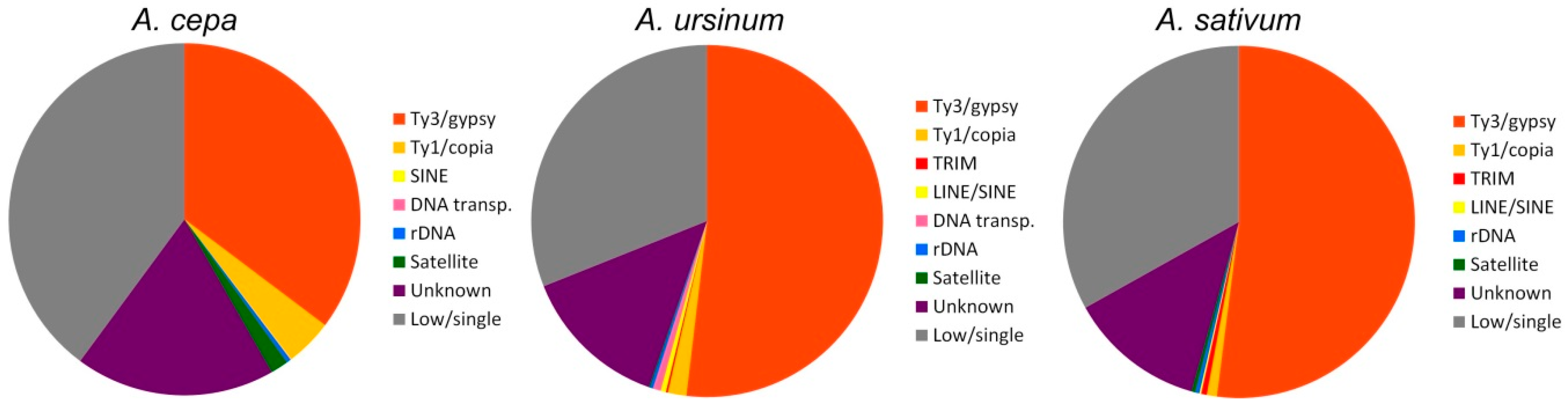
We used two bioinformatic approaches. The first approach included Tandem Repeats Finder and the Tandem Repeats Merger tool to identify short tandem repeats. The second approach consisted of TAREAN and RepeatExplorer analyses. The estimated genomic proportion of repeats identified by all approaches together in all three species was roughly 63%. A list of identified short tandem repeats from TRFi-TRM is in the
supplementary tables (Tables S1–4). In our estimation, these STMs represent 0.9–2.4% of the genome in the species studied. Repeats identified in RepeatExplorer represented ca. 60% of the genome in the three species (
Figure 4). Clusters representing a genomic proportion of at least 0.01% were automatically analyzed in more detail. This analysis was focused on the similarity to already known repetitive elements using BLAST. Separate reads in clusters were assembled to contigs, and the clusters were depicted in graphs according to overlaps and information from pair-end mates. We were able to annotate most of them thanks to the automatic prediction in RepeatExplorer, TAREAN pipeline, and graphs shape (
Tables S5–8). Only up to 4% of clusters analyzed in detail remain unannotated (
Table 1 and
Table 2). Nevertheless, a relatively large proportion of repeats remain unclassified (violet sector in
Figure 6), most of these are clusters representing less than 0.01% of each genome. Very low- or single-copy sequences remained as non-clustered reads (grey sectors in
Figure 6). Higher genomic coverage might uncover further repeats from this fraction as we hypothesize further in the discussion. The sequences from all, annotated and unannotated clusters can be found in
Supplemetary Files S4 and S8–10. In total, the annotated fraction represents roughly 40–55% of the genomes studied (
Table 1 and
Table 2;
Figure 6).
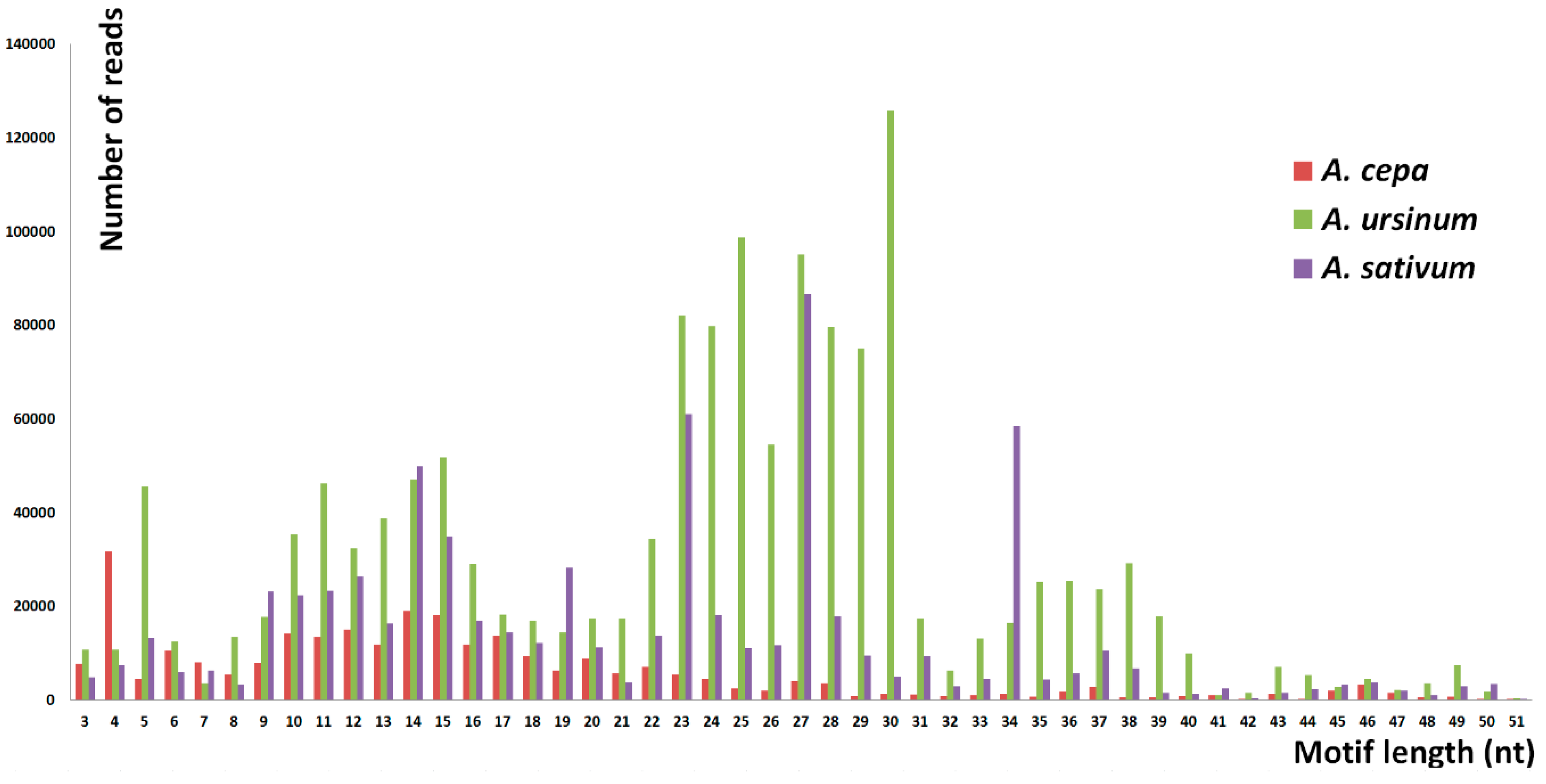
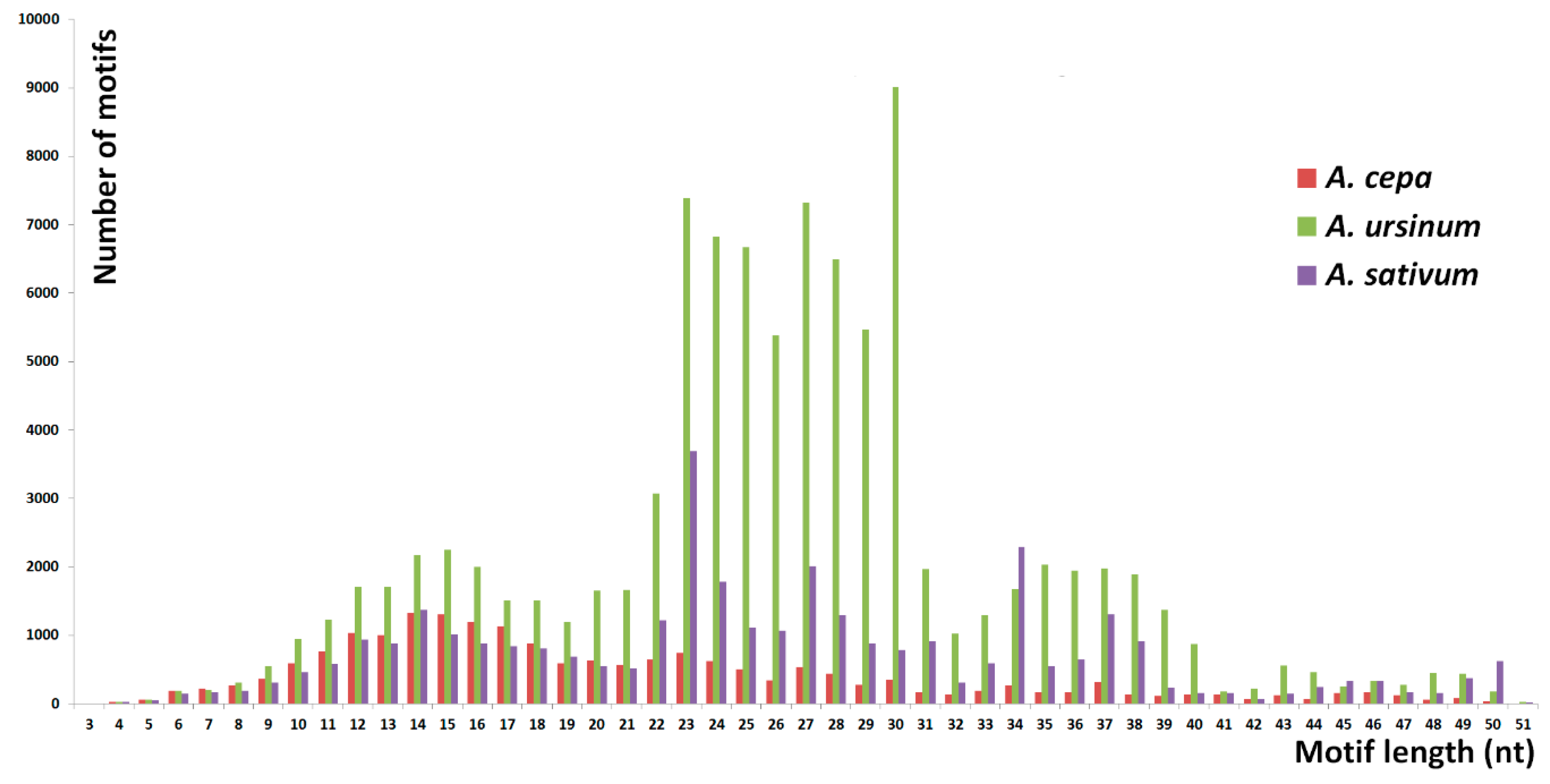
Short tandem repeats with monomer lengths between three and 52 nucleotides, considering three adjacent monomers as the shortest arrangement, were analyzed in Tandem Repeats Finder and a set of custom made scripts called Tandem Repeat Merger [
7,
36]. In this way, we identified 136,968 tandem motifs in a sample of 40 million reads (10 million reads from
A. cepa and
A. sativum each, and 20 million reads from
A. ursinum). We used a relatively high number of reads per species, yet the enormous sizes of the genomes studied means that even repeats detected in fewer reads represent a considerable DNA content. This fact is evident from recalculation demonstrated in
Table 5. For example, numbers of reads with vertebrate-like and
Arabidopsis-like telomere sequences in our TRFi-TRM result represent a genomic proportion corresponding to a short gene in the
Allium genome context. Most of abundant tandem motifs in
A. cepa belong to the category of genomic proportion corresponding to 160 kb–16 Mb, e.g., the telomere sequence corresponds to 406 kb in total in
A. cepa (red bold frame) which also corresponds to the experimental results [
16]. All detected motifs and estimations of their genomic proportions are shown in
supplementary tables (Supplementary Tables S1–S4).
An estimated total genomic proportion of short tandem repeats was 0.9% in
A. cepa, 2.4% in
A. ursinum, and 2.3% in
A. sativum. However, the total number of identified motifs is probably overestimated as even the abundant motifs have rare mutant and degenerate variants that do not pass the TRFi-TRM threshold of tolerance for variability in the motif pattern to which they belong. There is also a strict limitation in the estimation of genomic proportion for tandems with monomer lengths approaching 50 nt. Such tandems have only three units in a read (151 nt), from which their consensus of the motif is calculated in TRFi. This fact itself probably decreases the success rate of tandem identification in the case of degenerate repeats. Moreover, the more degenerate the 50 nt tandem repeat is, the more incongruent are TRFi consensus sequences produced from distinct reads carrying this tandem. In the case of very short monomer lengths, e.g., 3 nt, the consensus produced was more representative and probably congruent for most reads with this tandem. Briefly, the shorter the motifs are, the more accurate the estimation of genomic proportion in terms of the number of motifs detected and their abundancy was. From this point of view, our results from degenerate tandems and tandems with motif lengths approaching 50 nt detected in TRFi-TRM are only semiquantitative and their value is mainly in qualitative information on their presence in the three
Allium species and their comparison. In the case of conserved tandems such as telomere sequences, approaching the lower limit of motif lengths detected (minisatellites and microsatellites), the estimated genomic proportion is not burdened by the error of low motif representation in reads and their coverage corresponds to the abundancy. We estimate here that an average telomere length is 25.389 kb in
A. cepa, 50.285 kb in
A. sativum, and 90.998 kb in
A. ursinum. Although the genome proportion of telomere sequence in tested species was very low (0.002–0.005%), the estimated average telomere length in several tens of kilobase pairs ranks
Allium among plants with rather long telomeres. For example, the longest terminal fragments in plants were detected in
Nicotiana tabacum (160 kb) [
37] and
Nicotiana sylvestris (200 kb) [
38] while most ecotypes of
Arabidopsis thaliana range between 3.5–9 kb [
39,
40].
Interestingly, we also detected vertebrate-like and
Arabidopsis-like telomere sequences in genomic DNA of all three species. The detected numbers of reads mostly represent a very small DNA content that could be a trace of an ancient telomere sequence in the
Allium ancestor (the number of
Arabidopsis-like reads in library S2, 1843 (
Table 3), was neglected as an outlier or experimental artifact). This actually exemplifies the reason why three parallel libraries were used for each species. If the other two libraries were not prepared (the number of
Arabidopsis-like reads in library S1–8 reads and S3–6 reads (
Table 3)), we would not be able to recognize the value of this repeat of interest in S2 library as irrelevant. In the case of the vertebrate-like sequence in
A. sativum and the
Arabidopsis-like sequence in
A. ursinum, the number of reads detected corresponds to several kb (
Table 5). This finding corresponds to the previously published occurrence of both repeats at non-terminal positions in
Allium species [
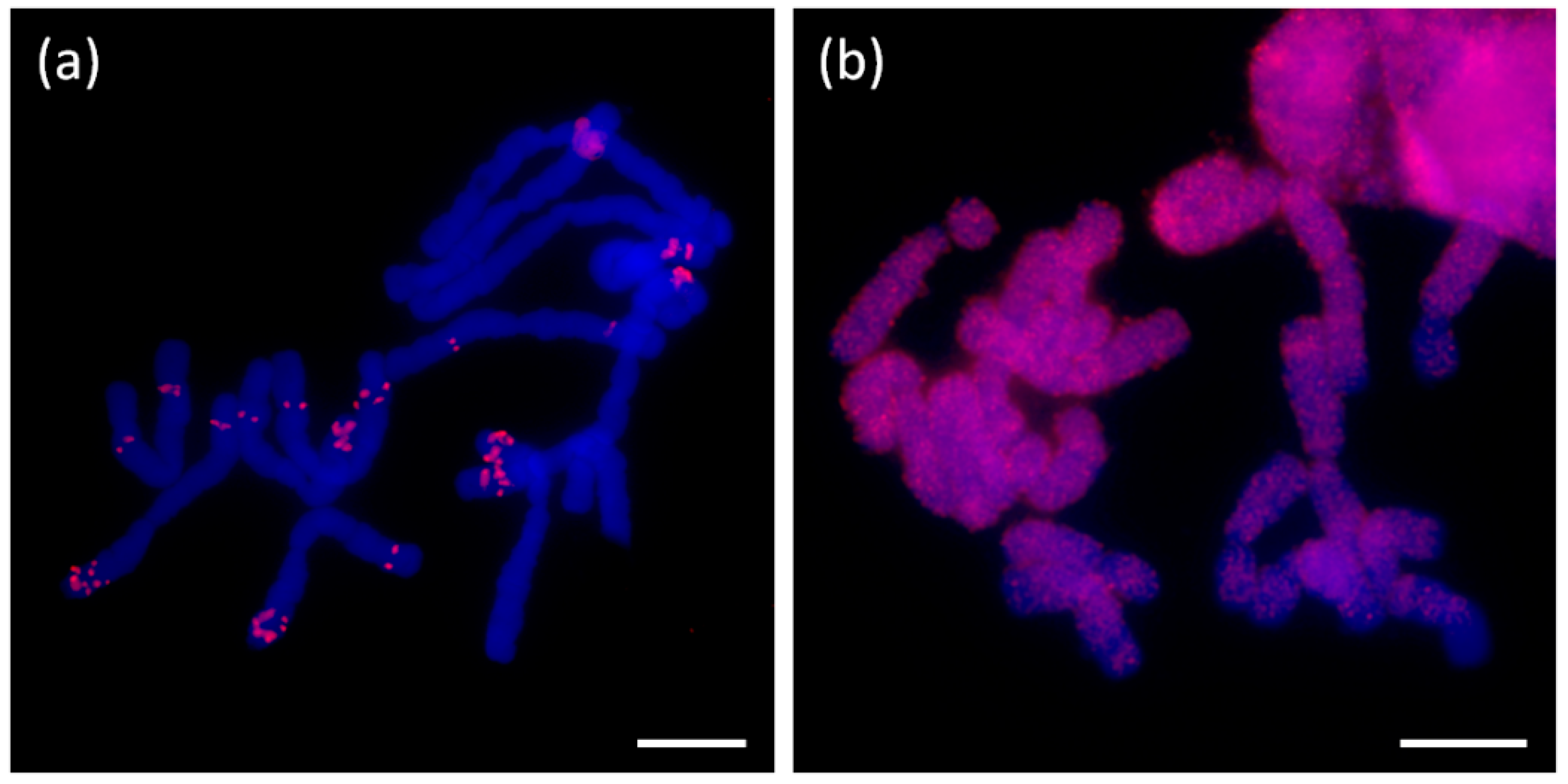
32]. Other short tandem repeats selected for FISH localization did not provide any specific signal. In all cases, the FISH signal was interspersed throughout the genome with diverse intensity, which was congruent with the number of reads detected per tandem. The only specific terminal localization was detected in the case of telomere short tandem repeats (CTCGGTATTGGG)
n in
A. sativum,
A. cepa, and
A. ursinum (
Figure 8 and [
16]) and (TCGGATCGGT)
n in
A. ursinum (at interstitial and terminal loci; see
Figure 9a). Neither interstitial nor terminal signals were detected in
Arabidopsis- or vertebrate-like telomere sequences.
Our results with slightly higher content of TTTAGGG than TTAGGG in
A. cepa and
A. ursinum, and comparable numbers of both in
A. sativum raise the question of ancestral telomere sequences in the
Allium genus. It is not clear whether the ancestor’s telomerase synthesized purely (TTAGGG)
n or a mixed array of (TTAGGG/TTTAGGG)
n as was shown for some Asparagales [
33].
Major repeats, particularly mobile elements, rDNA and satellites, were characterized in the RepeatExplorer analysis (
Supplementary Tables S5–8 and Files S8–10). DNA transposons (mobile elements class II) represented only a tiny genome proportion (up to 0.07%). It is consistent with the “cut and paste” mode of mobilization which does not result in an expansion comparable to retrotransposons (mobile element class I). Generally, we conclude that the main contribution to all three genomes is by an abundancy of the Ty3/gypsy element Tekay (ca. 30% in
A. cepa, 45% in
A. ursinum, and almost 50% in
A. sativum). It is not clear whether the whole captured genomic proportion of such large repeats like retrotransposons correspond to fully functional variants. It is probable that incomplete, truncated and other derived variants were designated with the annotation of the full repeat, which may represent only a part of the specific cluster or supercluster. Only if the short version formed a very specific and considerably abundant population of copies in the genome, we would be able to annotate it as a separate element, such as in the case of TRIMs (
Table 1 and
Table 2). Nevertheless, a vast majority of detected retrotransposons, including TRIM elements, were identified in species-specific clusters regardless of their classification. Similar situation has already been studied in the comparative analysis of several
Fritillaria (Liliaceae) species with giant genomes. The study indicated, that the genome expansion occurred independently via accumulation of variable repeats with the lack of DNA removal [
41]. The repeatome profiles of obese
Fritillaria genomes were shown to be dramatically different, particularly in the presence of the species-specific satellites [
42]. A different observation was made in the case of
Allium in rather rare but conserved Ty1/copia/TAR element (CL111 in the
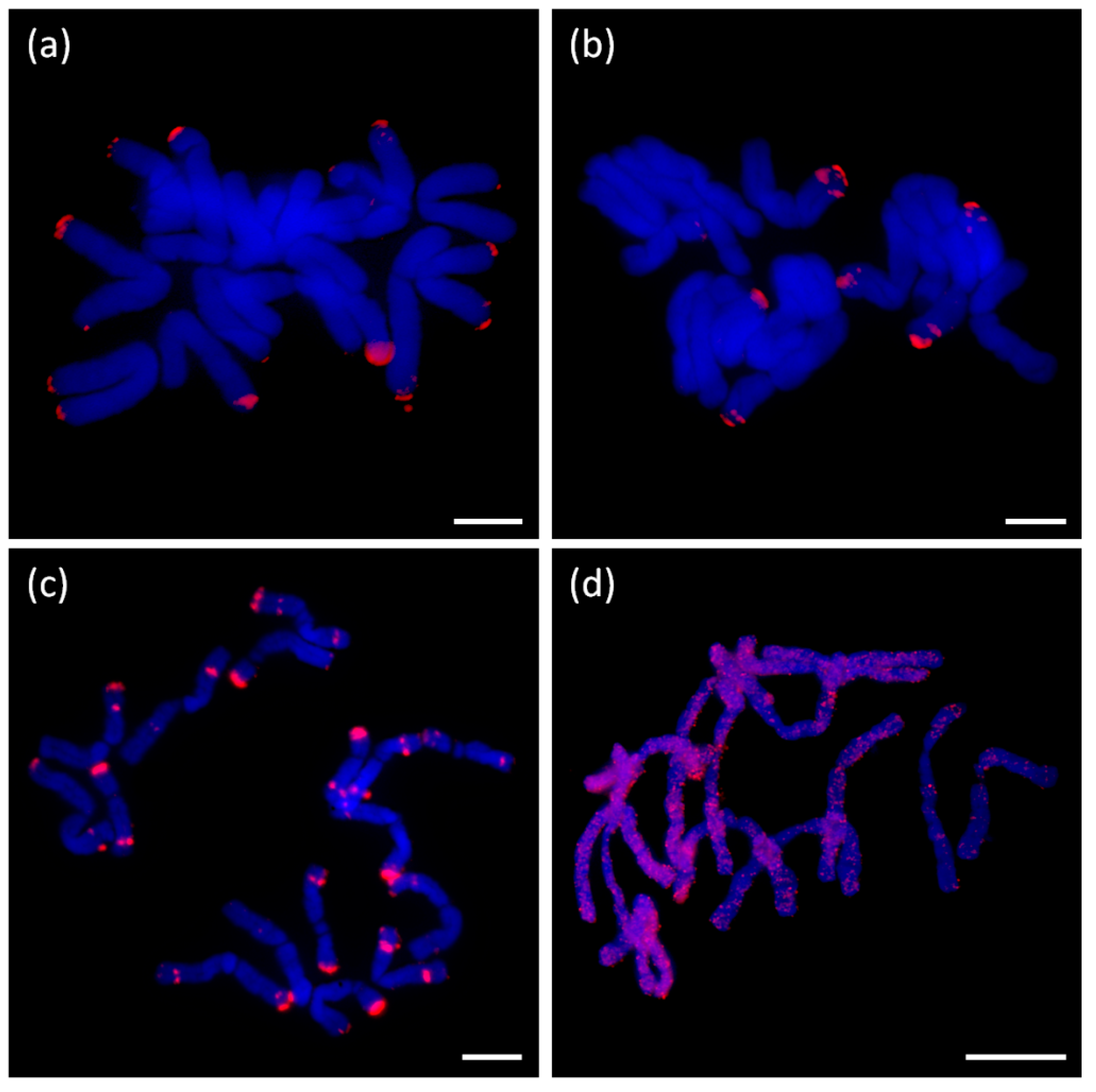
Supplementary File S2, Figure S3 shows mapping of contigs from CL111 to annotated hypothetical copy (5417 nt) as a reference sequence). Conservation of this repeat gave an impression that it could be connected to some specific loci, like centromeres or subtelomeres. Unexpectedly, using FISH, we did not find any enrichment of this element in any specific locus (
Figure 10d).
Surprisingly, the genomic proportion of detected repeats is more similar in
A. sativum and
A. ursinum than between more closely related
A. cepa and
A. sativum (
Figure 6). The representativeness of the profiles should be guaranteed by technical triplication of NGS libraries from each species and PCR-free library preparation.
Contrary to most retrotransposons and long satellites, rDNA sequences from all three species were clustered in the same group, as expected according to their conserved character. We made several interesting observations in these rDNA clusters. In the case of 5S rDNA, a complete circular cluster shape was detected in
A. cepa and
A. sativum. In
A. ursinum, the part corresponding to a spacer between 5S rRNA genes was not completely assembled. It is possible that the variability of its sequence makes it impossible to assemble the spacer variants under the coverage used in RepeatExplorer. Remarkably, more reads from 5S rDNA were detected in
A. sativum (
Figure 5). The shape of clusters indicates the presence of four variants in
A. sativum and two intergenic spacer variants in
A. cepa (
Figure 7). Notably,
A. sativum has additional 5S loci compared to
A. cepa [
19,
20]. Further analysis is needed to localize these variants and to show whether they occupy the same or distinct chromosomal loci. In 45S rDNA clusters,
A. sativum exhibited a single circular cluster of the long tandem array (
Supplementary Figure S4a).
A. cepa 45S rDNA was assembled into two clusters that form one circular supercluster through mate pair reads. Therefore, they have been merged to obtain consensus and its variants (
Figure S4c,d; File S17). Interestingly, the assembled 45S rDNA from
A. cepa cultivar Všetana is more similar to a partial clone from
A. cepa cultivar Ailsa Craig (GenBank EU256494.1), [
43] than to the complete onion 45S rDNA sequence published recently (GenBank KM117265), (
Figure S5). Unexpectedly, in
A. ursinum, the graph of the 45S rDNA cluster was not enclosed in the circular shape, which is typical for tandem arrangement and stayed linear,
Figure S4B. It happened probably due to the presence of a long array of GC rich short tandem repeats in its spacer region, which can be a difficult substrate for enzymes used in NGS.
We identified new satellite sequences in
A. cepa,
A. ursinum and
A. sativum. We named these according to previous practice (similar to ACSAT from [
25]) according to species and monomer length. Since we expect more NGS data from other
Allium species to be published soon we suggest using a longer prefix, such as Acep for
A. cepa, not to be confused, e.g., with
A. cernuum. Further modification and unification of nomenclature will be probably needed once the other species will be analyzed (e.g.,
A. cepiforme). The newly identified satellites are AcepSAT750 and AcepSAT2500 from
A. cepa, and AsatSAT377, AsatSAT 189, AsatSAT 74, and AsatSAT 604 from
A. sativum. A putative satellite was detected in
A. ursinum and was termed AursSAT89. AcepSAT750, AcepSAT356, and AsatSAT189 satellites have specific chromosomal locations and potentially, these can be used as chromosomal markers. In comparison to similar previously described satellites from
A. cepa, e.g., pAc074 and ACSAT, which are 314–378 nt long [
23,
24,
25,
27,
30], we identified a consensus with a length of 356 nt and termed it AcepSAT356. This consensus, which actually stands for one major variant, was examined by FISH. We obtained a terminally positioned signal. However we observed a lack of signal at more than just one chromosomal arm (
Figure 10a). The satellite was previously estimated to constitute up to 4% of the genome in
A. cepa [
23]. Our NGS results show the proportion to be 0.28% in a comparative analysis and 1.3% in separate clustering with higher genomic coverage. This difference can be interpreted either as a low precision of the previous analyses, or as a result of a low genomic coverage in our analyses and, consequently, its biased quantitative result (
Table 4). Increased genomic coverage would probably minimize the bias but the input used is at the limit of the server with the web based pipeline of RepeatExplorer and TAREAN.
Some Allium retrotransposon lineages such as Ty1/copia Bianca, Ikeros, Ale, and Angela are currently near the limit of detection by RepeatExplorer. They were not detected using a genomic coverage of 0.004× and only when the genomic coverage was increased (0.007–0.01×) did they become obvious in the dataset as well as in the DNA element MITE from A. cepa. It is possible that by further extending the amount of sequences analyzed, we will detect other lineages of repeats with tiny genomic proportions. This hidden variability may uncover a substantial part of unclassified and non-clustered sequences in our study, mainly those of low copy number and high sequence variability.
A. cepa,
A. ursinum, and
A. sativum, with our estimations (based on the result from FastQC output [
44] on raw whole genome shotgun sequencing data) of 33% GC content, were shown to be one of the AT- richest plant species [
45,
46]. On the other hand, experimental measurement (39% GC) shows
Allium as rather an average plant genome rather than any extreme, with a low GC content [
47]. In this light, our results rather reject the hypothesis that the low GC content in
Allium is caused by expansion of non-genic tandem repeats. Although short AT rich tandem motifs were much more frequent than GC rich ones, STM altogether constituted only a few percent of the genome in the species studied (up to 0.9% in
A. cepa). Long satellites were estimated to constitute slightly, yet not dramatically, larger genomic proportions (1.6–1.7% in
A. cepa). Moreover, the satellites differed in GC content and were rarely below 30%. In contrast,
A. cepa retrotransposons, e.g., Ty1/copia/TAR were richer in AT than the most prevalent long satellite, AcepSAT356 (
Table 6).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}