The Role of Osteoprotegerin and Its Ligands in Vascular Function

 ,
,
Abstract
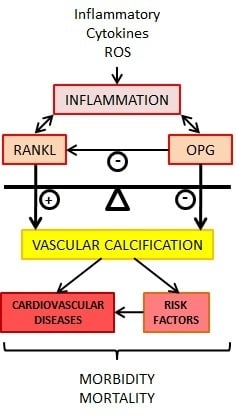
{kind=link}
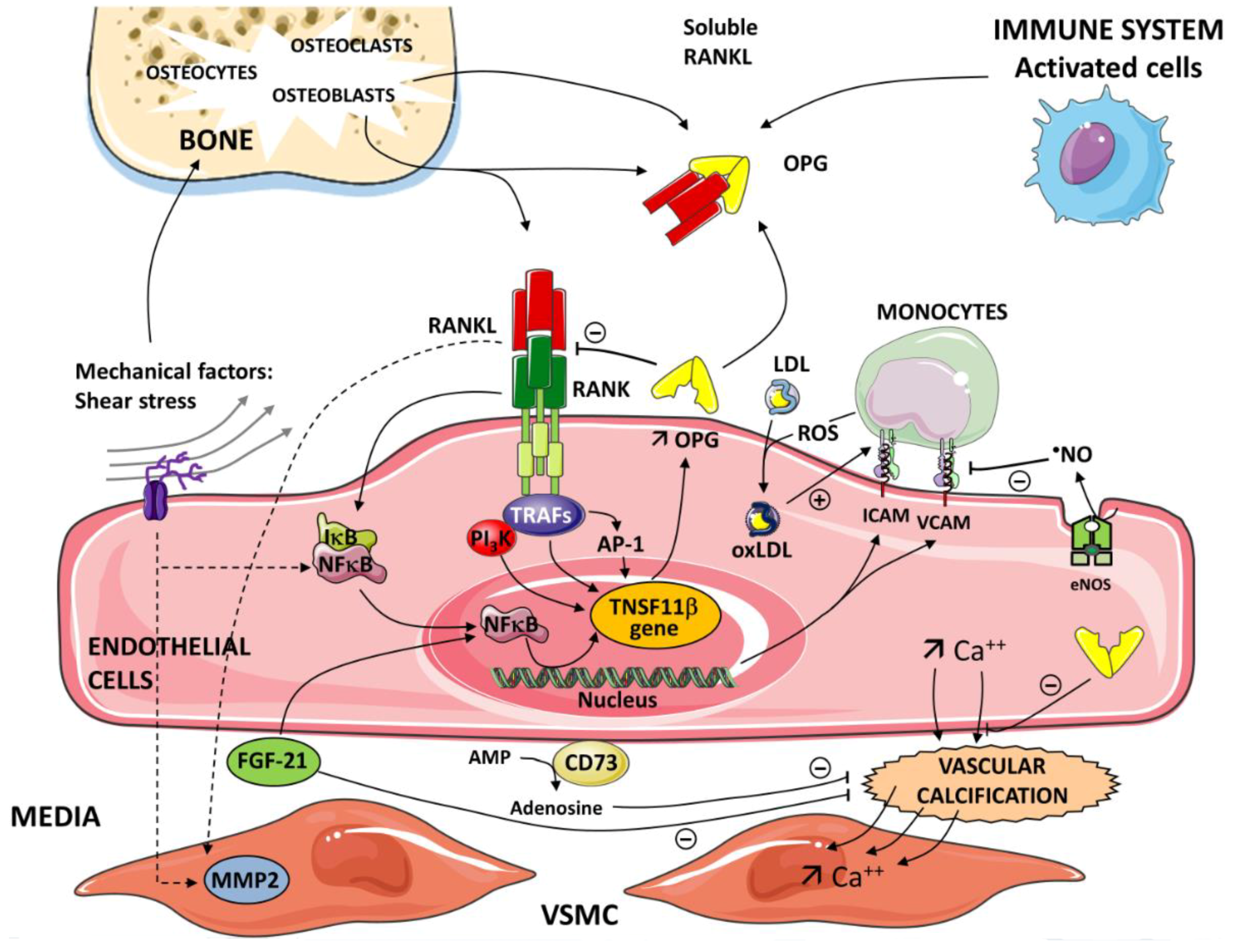{kind=link}
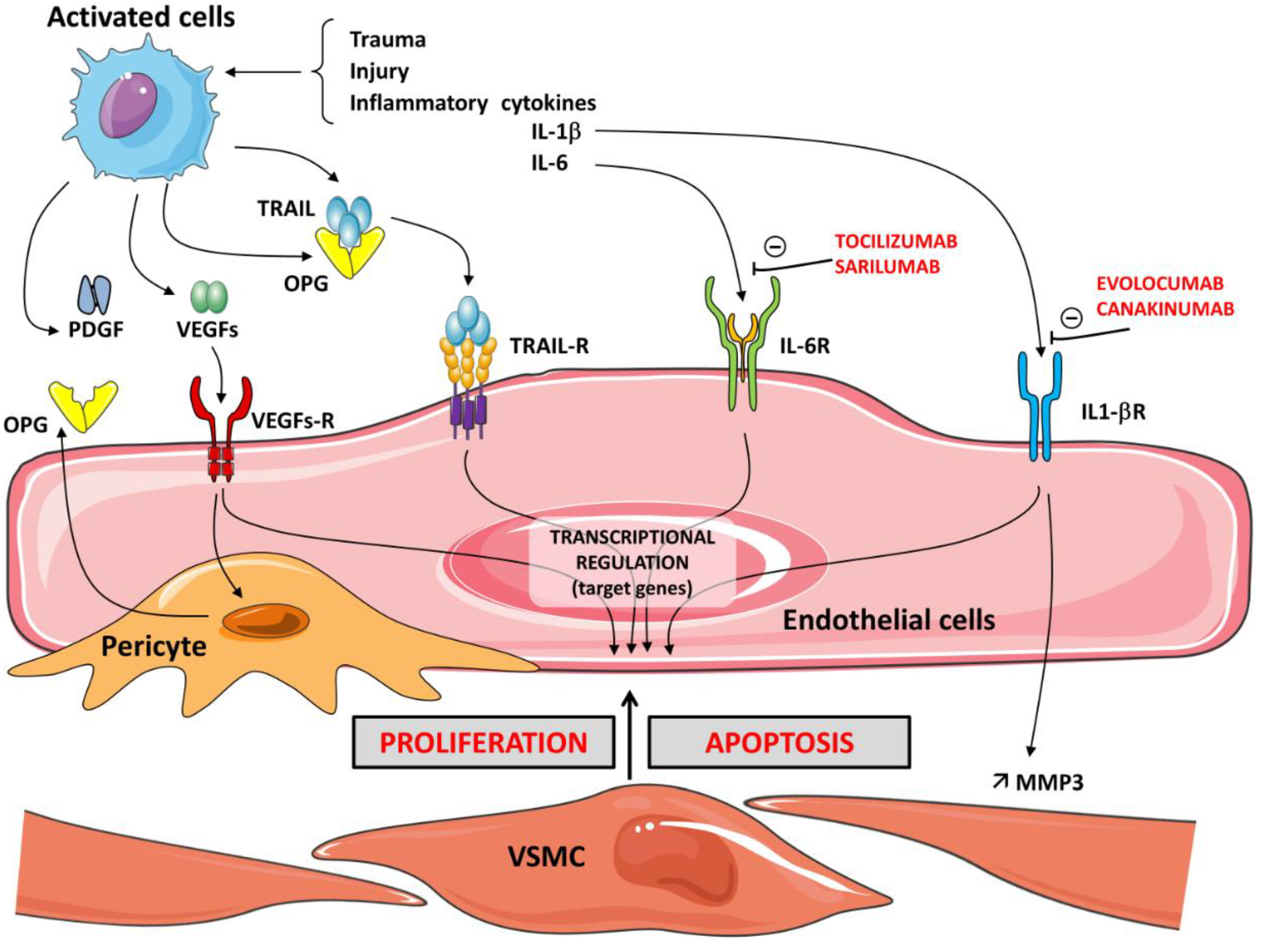{kind=link}
1. Introduction
2. The OPG/RANKL/RANK/TRAIL System: Structures, Localization, and Characterization
3. Interactions between OPG/RANKL/RANK and Endogenous Factors in the Heart: Incidences on Metabolism and Functions of Endothelial Cells.
4. OPG/RANKL/RANK and Vascular Signaling
5. OPG/RANKL/RANK and Regulation of Angiogenesis
6. OPG/RANKL/RANK and Inflammation
7. OPG/RANKL/RANK and the Proteasome
8. OPG/RANKL/RANK and Cellular Senescence
9. OPG/RANKL/RANK and Vascular Calcification
10. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Amino acid |
| ALP | Alkaline phosphatase |
| Ang | Angiotensin |
| AP-1 | Activator protein-1 |
| ASCs | Adipose-stromal cells |
| CAD | Coronary artery disease |
| CIMT | Carotid intima-media thickness |
| CKD | Chronic kidney disease |
| CVD | Cardiovascular disease; |
| DR | Death receptors; |
| EC | Endothelial cell; |
| ECFC | Endothelial colony-forming cell; |
| EndMT | Endothelial to mesenchymal transition |
| EPC | Endothelial progenitor cell |
| FA | Fatty acid |
| FAO | Fatty acid–beta-oxidation |
| FGFs | Fibroblast growth factors |
| GDF-11 | Growth differentiation factor-11 |
| GFR | Glomerular Filtration Rate |
| GLUT | Glucose transporter |
| HDL | High Density Lipoproteins |
| HSPGs | Heparan sulfate proteoglycans |
| ICAM | Intercellular adhesion molecule |
| IL | Interleukin |
| kD | kiloDalton |
| MMP | Matrix metalloprotease |
| Nf-κB | Nuclear factor κB |
| NO | Nitric oxide |
| NOS | NO synthetase |
| NOX | NADPH oxidase |
| Nrf2 | Nuclear factor-E2-related factor 2 |
| OCIF | Osteoclastogenesis inhibitory factor |
| OPG | Osteoprotegerin |
| OxLDL | Oxidized low density lipoprotein |
| PPARs | Peroxisome proliferator-activated receptors |
| RA | Rheumatoid arthritis |
| RANK | Receptor activator of nuclear factor κ B |
| RANKL | Receptor activator of nuclear factor κ B ligand |
| ROS | Reactive oxygen species |
| SMPC | Smooth muscle progenitor cells |
| TGF | Transforming growth factor |
| TNF | Tumor necrosis factor |
| TNFR | Tumor necrosis factor receptor |
| TNFRS | Tumor necrosis factor receptor superfamily |
| TRAF | TNFR-associated factor |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| TSP-1 | Thrombospondin-1 |
| VCAM | Vascular adhesion molecule |
| VEGF | Vascular endothelial growth factor |
| VSMC | Vascular smooth muscle cells |
| vWF | von Willebrand factor |
| WPB | Weibel-Palade bodies |
References
- Walsh, M.C.; Choi, Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Front. Immunol. 2014, 5, 511. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Meloux, A.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. The role of osteoprotegerin in the crosstalk between vessels and bone: Its potential utility as a marker of cardiometabolic diseases. Pharm. Ther. 2018, 182, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Barbu, C.G.; Arsene, A.L.; Florea, S.; Albu, A.; Sirbu, A.; Martin, S.; Nicolae, A.C.; Burcea-Dragomiroiu, G.T.A.; Popa, D.E.; Velescu, B.S.; et al. Cardiovascular risk assessment in osteoporotic patients using osteoprotegerin as a reliable predictive biochemical marker. Mol. Med. Rep. 2017, 16, 6059–6067. [Google Scholar] [CrossRef] [PubMed]
- Harper, E.; Forde, H.; Davenport, C.; Rochfort, K.D.; Smith, D.; Cummins, P.M. Vascular calcification in type-2 diabetes and cardiovascular disease: Integrative roles for OPG, RANKL and TRAIL. Vasc. Pharm. 2016, 82, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Matsuo, K. Molecular mechanisms of triggering, amplifying and targeting RANK signaling in osteoclasts. World J. Orthop. 2012, 3, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Gyurkovska, V.; Ivanovska, N. Distinct roles of TNF-related apoptosis-inducing ligand (TRAIL) in viral and bacterial infections: From pathogenesis to pathogen clearance. Inflamm. Res. 2016, 65, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Forde, H.; Harper, E.; Davenport, C.; Rochfort, K.D.; Wallace, R.; Murphy, R.P.; Smith, D.; Cummins, P.M. The beneficial pleiotropic effects of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) within the vasculature: A review of the evidence. Atherosclerosis 2016, 247, 87–96. [Google Scholar] [CrossRef]
- D’Auria, F.; Centurione, L.; Centurione, M.A.; Angelini, A.; Di Pietro, R. Tumor Necrosis Factor Related Apoptosis Inducing Ligand (Trail) in endothelial response to biomechanical and biochemical stresses in arteries. J. Cell. Biochem. 2015, 116, 2427–2434. [Google Scholar] [CrossRef]
- Lang, I.; Fullsack, S.; Wyzgol, A.; Fick, A.; Trebing, J.; Arana, J.A.; Schafer, V.; Weisenberger, D.; Wajant, H. Binding Studies of TNF Receptor Superfamily (TNFRSF) Receptors on Intact Cells. J. Biol. Chem. 2016, 291, 5022–5037. [Google Scholar] [CrossRef]
- Hur, J.; Ghosh, A.; Kim, K.; Ta, H.M.; Kim, H.; Kim, N.; Hwang, H.Y.; Kim, K.K. Design of a RANK-Mimetic Peptide Inhibitor of Osteoclastogenesis with Enhanced RANKL-Binding Affinity. Mol. Cells 2016, 39, 316–321. [Google Scholar]
- Munasinghe, A.; Lin, P.; Colina, C.M. Unraveling Binding Interactions between Human RANKL and Its Decoy Receptor Osteoprotegerin. J. Phys. Chem. B 2017, 121, 9141–9148. [Google Scholar] [CrossRef] [PubMed]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Schoppet, M.; Henser, S.; Ruppert, V.; Stubig, T.; Al-Fakhri, N.; Maisch, B.; Hofbauer, L.C. Osteoprotegerin expression in dendritic cells increases with maturation and is NF-kappaB-dependent. J. Cell. Biochem. 2007, 100, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.A.; Cito, M.; Carlsson, P.O.; Vanderwinden, J.M.; Molkentin, J.D.; Bugliani, M.; Marchetti, P.; Eizirik, D.L.; Cnop, M. Thrombospondin 1 protects pancreatic beta-cells from lipotoxicity via the PERK-NRF2 pathway. Cell Death Differ. 2016, 23, 1995–2006. [Google Scholar] [CrossRef] [PubMed]
- Milanova, V.; Ivanovska, N.; Dimitrova, P. TLR2 elicits IL-17-mediated RANKL expression, IL-17, and OPG production in neutrophils from arthritic mice. Mediat. Inflamm. 2014, 2014, 643406. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.L.; Yeh, C.C.; Wu, J.Y.; Lin, H.C.; Wang, Y.F.; Kuo, Y.Y.; Hsieh, Y.T.; Hsu, Y.J.; Kuo, C.C. TLR2 Promotes Vascular Smooth Muscle Cell Chondrogenic Differentiation and Consequent Calcification Via the Concerted Actions of Osteoprotegerin Suppression and IL-6-Mediated RANKL Induction. Arter. Thromb. Vasc. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Schaalan, M.; Mohamed, W. Predictive ability of circulating osteoprotegerin as a novel biomarker for early detection of acute kidney injury induced by sepsis. Eur. Cytokine Netw. 2017, 28, 52–62. [Google Scholar]
- Kim, J.Y.; Park, Y.J.; Kim, K.J.; Choi, J.J.; Kim, W.U.; Cho, C.S. Osteoprotegerin causes apoptosis of endothelial progenitor cells by induction of oxidative stress. Arthritis Rheum. 2013, 65, 2172–2182. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Lee, C.Y.; Seo, H.H.; Shin, S.; Choi, J.W.; Kim, S.W.; Park, J.C.; Lim, S.; Hwang, K.C. Adipose-derived stem cell-released osteoprotegerin protects cardiomyocytes from reactive oxygen species-induced cell death. Stem Cell Res. 2017, 8, 195. [Google Scholar] [CrossRef]
- Chung, C.P.; Solus, J.F.; Oeser, A.; Li, C.; Raggi, P.; Smith, J.R.; Stein, C.M. A variant in the osteoprotegerin gene is associated with coronary atherosclerosis in patients with rheumatoid arthritis: Results from a candidate gene study. Int. J. Mol. Sci. 2015, 16, 3885–3894. [Google Scholar] [CrossRef]
- Mishra, P.K.; Givvimani, S.; Chavali, V.; Tyagi, S.C. Cardiac matrix: A clue for future therapy. Biochim. Biophys. Acta 2013, 1832, 2271–2276. [Google Scholar] [CrossRef] [PubMed]
- Brutsaert, D.L. Cardiac endothelial-myocardial signaling: Its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev. 2003, 83, 59–115. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- Culic, O.; Gruwel, M.L.; Schrader, J. Energy turnover of vascular endothelial cells. Am. J. Physiol. 1997, 273, C205–C213. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Maeda, K.; Hanaoka, H.; Suga, T.; Goto, K.; Syamsunarno, M.R.; Hishiki, T.; Nagahata, Y.; Matsui, H.; Arai, M.; et al. Capillary endothelial fatty acid binding proteins 4 and 5 play a critical role in fatty acid uptake in heart and skeletal muscle. Arter. Thromb. Vasc. Biol. 2013, 33, 2549–2557. [Google Scholar] [CrossRef]
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 2010, 464, 917–921. [Google Scholar] [CrossRef]
- Ingwall, J.S. Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 2009, 81, 412–419. [Google Scholar] [CrossRef]
- Ashrafian, H.; Frenneaux, M.P.; Opie, L.H. Metabolic mechanisms in heart failure. Circulation 2007, 116, 434–448. [Google Scholar] [CrossRef]
- Evans, B.A.; Elford, C.; Pexa, A.; Francis, K.; Hughes, A.C.; Deussen, A.; Ham, J. Human osteoblast precursors produce extracellular adenosine, which modulates their secretion of IL-6 and osteoprotegerin. J. Bone Miner. Res. 2006, 21, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, M.; Vieira, T.S.; Martins, M.J.; Lucas, R.; Costa, L.; Pereira, J.G.; Ventura, F.; Martins, E. Myocardial Perfusion in Rheumatoid Arthritis Patients: Associations with Traditional Risk Factors and Novel Biomarkers. Biomed. Res. Int. 2017, 2017, 6509754. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Cohen, S. The A3 adenosine receptor (A3AR): Therapeutic target and predictive biological marker in rheumatoid arthritis. Clin. Rheumatol. 2016, 35, 2359–2362. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi-Sakamoto, M.; Hirose, K.; Isogai, E.; Chiba, I. NF-kappaB-dependent induction of osteoprotegerin by Porphyromonas gingivalis in endothelial cells. BioChem. Biophys. Res. Commun 2004, 315, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi-Sakamoto, M.; Isogai, E.; Hirose, K.; Chiba, I. Role of alphav integrin in osteoprotegerin-induced endothelial cell migration and proliferation. Microvasc. Res. 2008, 76, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Benslimane-Ahmim, Z.; Poirier, F.; Delomenie, C.; Lokajczyk, A.; Grelac, F.; Galy-Fauroux, I.; Mohamedi, A.; Fischer, A.M.; Heymann, D.; Lutomski, D.; et al. Mechanistic study of the proangiogenic effect of osteoprotegerin. Angiogenesis 2013, 16, 575–593. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V.; Katakam, S.K.; Urbanowitz, A.K.; Gotte, M. Heparan sulphate as a regulator of leukocyte recruitment in inflammation. Curr. Protein Pept. Sci. 2015, 16, 77–86. [Google Scholar] [CrossRef]
- Wylie-Sears, J.; Aikawa, E.; Levine, R.A.; Yang, J.H.; Bischoff, J. Mitral valve endothelial cells with osteogenic differentiation potential. Arter. Thromb. Vasc. Biol. 2011, 31, 598–607. [Google Scholar] [CrossRef]
- Songia, P.; Branchetti, E.; Parolari, A.; Myasoedova, V.; Ferrari, G.; Alamanni, F.; Tremoli, E.; Poggio, P. Mitral valve endothelial cells secrete osteoprotegerin during endothelial mesenchymal transition. J. Mol. Cell Cardiol. 2016, 98, 48–57. [Google Scholar] [CrossRef]
- Lommi, J.I.; Kovanen, P.T.; Jauhiainen, M.; Lee-Rueckert, M.; Kupari, M.; Helske, S. High-density lipoproteins (HDL) are present in stenotic aortic valves and may interfere with the mechanisms of valvular calcification. Atherosclerosis 2011, 219, 538–544. [Google Scholar] [CrossRef]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharm. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress and therapeutic strategies. Biochim. Biophys. Acta 2014, 1840, 2709–2729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fu, M.; Myles, D.; Zhu, X.; Du, J.; Cao, X.; Chen, Y.E. PDGF induces osteoprotegerin expression in vascular smooth muscle cells by multiple signal pathways. FEBS Lett. 2002, 521, 180–184. [Google Scholar] [CrossRef]
- Kleemann, R.; Bureeva, S.; Perlina, A.; Kaput, J.; Verschuren, L.; Wielinga, P.Y.; Hurt-Camejo, E.; Nikolsky, Y.; van Ommen, B.; Kooistra, T. A systems biology strategy for predicting similarities and differences of drug effects: Evidence for drug-specific modulation of inflammation in atherosclerosis. BMC Syst. Biol. 2011, 5, 125. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.W.; Sanders, J.M.; Bevard, M.; Coleman, E.; Sarembock, I.J.; Schwartz, M.A. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: A potential role in atherosclerosis. J. Cell Biol. 2005, 169, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Givens, C.; Tzima, E. Endothelial Mechanosignaling: Does One Sensor Fit All? Antioxid. Redox Signal. 2016, 25, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Cheng, T.; Wang, S.; Zhang, C.; Jin, L.; Yang, Y. Shear stress inhibits IL-17A-mediated induction of osteoclastogenesis via osteocyte pathways. Bone 2017, 101, 10–20. [Google Scholar] [CrossRef]
- Husain, K.; Hernandez, W.; Ansari, R.A.; Ferder, L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J. Biol. Chem. 2015, 6, 209–217. [Google Scholar] [CrossRef]
- Malyankar, U.M.; Scatena, M.; Suchland, K.L.; Yun, T.J.; Clark, E.A.; Giachelli, C.M. Osteoprotegerin is an alpha vbeta 3-induced, NF-kappa B-dependent survival factor for endothelial cells. J. Biol. Chem. 2000, 275, 20959–20962. [Google Scholar] [CrossRef]
- Clancy, P.; Koblar, S.A.; Golledge, J. Angiotensin receptor 1 blockade reduces secretion of inflammation associated cytokines from cultured human carotid atheroma and vascular cells in association with reduced extracellular signal regulated kinase expression and activation. Atherosclerosis 2014, 236, 108–115. [Google Scholar] [CrossRef]
- Chen, S.; Grover, M.; Sibai, T.; Black, J.; Rianon, N.; Rajagopal, A.; Munivez, E.; Bertin, T.; Dawson, B.; Chen, Y.; et al. Losartan increases bone mass and accelerates chondrocyte hypertrophy in developing skeleton. Mol. Genet. Metab. 2015, 115, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Min, J.K.; Kim, Y.M.; Kim, Y.M.; Kim, E.C.; Gho, Y.S.; Kang, I.J.; Lee, S.Y.; Kong, Y.Y.; Kwon, Y.G. Vascular endothelial growth factor up-regulates expression of receptor activator of NF-kappa B (RANK) in endothelial cells. Concomitant increase of angiogenic responses to RANK ligand. J. Biol. Chem. 2003, 278, 39548–39557. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Carmeliet, P. The Link Between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi-Sakamoto, M.; Isogai, E.; Holen, I. Osteoprotegerin induces cytoskeletal reorganization and activates FAK, Src, and ERK signaling in endothelial cells. Eur. J. Haematol. 2010, 85, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Veeriah, V.; Paone, R.; Chatterjee, S.; Teti, A.; Capulli, M. Osteoblasts Regulate Angiogenesis in Response to Mechanical Unloading. Calcif. Tissue Int. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zhu, Y.; Qiu, S.; Xu, J.; Chai, Y. Exosomes secreted by endothelial progenitor cells accelerate bone regeneration during distraction osteogenesis by stimulating angiogenesis. Stem Cell Res. 2019, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.J.; Scatena, M.; Kirk, E.A.; Rattazzi, M.; Varon, R.M.; Averill, M.; Schwartz, S.M.; Giachelli, C.M.; Rosenfeld, M.E. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE-/- mice. Arter. Thromb. Vasc. Biol. 2006, 26, 2117–2124. [Google Scholar] [CrossRef]
- Moran, C.S.; McCann, M.; Karan, M.; Norman, P.; Ketheesan, N.; Golledge, J. Association of osteoprotegerin with human abdominal aortic aneurysm progression. Circulation 2005, 111, 3119–3125. [Google Scholar] [CrossRef]
- Quercioli, A.; Mach, F.; Bertolotto, M.; Lenglet, S.; Vuilleumier, N.; Galan, K.; Pagano, S.; Braunersreuther, V.; Pelli, G.; Pistoia, V.; et al. Receptor activator of NF- kappaB ligand (RANKL) increases the release of neutrophil products associated with coronary vulnerability. Thromb. Haemost. 2012, 107, 124–139. [Google Scholar]
- Kobayashi, Y. The regulatory role of nitric oxide in proinflammatory cytokine expression during the induction and resolution of inflammation. J. Leukoc. Biol. 2010, 88, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Zauli, G.; Corallini, F.; Bossi, F.; Fischetti, F.; Durigutto, P.; Celeghini, C.; Tedesco, F.; Secchiero, P. Osteoprotegerin increases leukocyte adhesion to endothelial cells both in vitro and in vivo. Blood 2007, 110, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Mangan, S.H.; Van Campenhout, A.; Rush, C.; Golledge, J. Osteoprotegerin upregulates endothelial cell adhesion molecule response to tumor necrosis factor-alpha associated with induction of angiopoietin-2. Cardiovasc. Res. 2007, 76, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Peiro, C.; Lorenzo, O.; Carraro, R.; Sanchez-Ferrer, C.F. IL-1beta Inhibition in Cardiovascular Complications Associated to Diabetes Mellitus. Front. Pharmacol. 2017, 8, 363. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.B. A review of sarilumab for the treatment of rheumatoid arthritis. Immunotherapy 2018, 10, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.T.; Geerts, D.; Roseman, K.; Renaud, A.; Connelly, L. Osteoprotegerin mediates tumor-promoting effects of Interleukin-1beta in breast cancer cells. Mol. Cancer 2017, 16, 27. [Google Scholar] [CrossRef]
- Maziere, C.; Salle, V.; Gomila, C.; Maziere, J.C. Oxidized low density lipoprotein increases RANKL level in human vascular cells. Involvement of oxidative stress. Biochem. Biophys. Res. Commun 2013, 440, 295–299. [Google Scholar] [CrossRef]
- Holvoet, P.; Jenny, N.S.; Schreiner, P.J.; Tracy, R.P.; Jacobs, D.R.; Multi-Ethnic Study of, A. The relationship between oxidized LDL and other cardiovascular risk factors and subclinical CVD in different ethnic groups: The Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis 2007, 194, 245–252. [Google Scholar] [CrossRef]
- Shimamura, M.; Nakagami, H.; Osako, M.K.; Kurinami, H.; Koriyama, H.; Zhengda, P.; Tomioka, H.; Tenma, A.; Wakayama, K.; Morishita, R. OPG/RANKL/RANK axis is a critical inflammatory signaling system in ischemic brain in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 8191–8196. [Google Scholar] [CrossRef]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef]
- Domouzoglou, E.M.; Naka, K.K.; Vlahos, A.P.; Papafaklis, M.I.; Michalis, L.K.; Tsatsoulis, A.; Maratos-Flier, E. Fibroblast growth factors in cardiovascular disease: The emerging role of FGF21. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1029–H1038. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Wang, S.; Cao, X.; Liu, X.; Fu, K.; Hao, P.; Liu, J. Fibroblast growth factor 21 attenuates calcification of vascular smooth muscle cells in vitro. J. Pharm. Pharm. 2017, 69, 1802–1816. [Google Scholar] [CrossRef] [PubMed]
- Katsimpardi, L.; Litterman, N.K.; Schein, P.A.; Miller, C.M.; Loffredo, F.S.; Wojtkiewicz, G.R.; Chen, J.W.; Lee, R.T.; Wagers, A.J.; Rubin, L.L. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 2014, 344, 630–634. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Growth and differentiation factor 11 (GDF11): Functions in the regulation of erythropoiesis and cardiac regeneration. Pharm. Ther. 2015, 156, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, L.; Zhou, C.; Zhang, S.; Jing, J.; Xie, L.; Sun, N.; Duan, X.; Jing, W.; Liang, X.; et al. GDF11 decreases bone mass by stimulating osteoclastogenesis and inhibiting osteoblast differentiation. Nat. Commun. 2016, 7, 12794. [Google Scholar] [CrossRef] [PubMed]
- Zimmers, T.A.; Jiang, Y.; Wang, M.; Liang, T.W.; Rupert, J.E.; Au, E.D.; Marino, F.E.; Couch, M.E.; Koniaris, L.G. Exogenous GDF11 induces cardiac and skeletal muscle dysfunction and wasting. Basic Res. Cardiol. 2017, 112, 48. [Google Scholar] [CrossRef] [PubMed]
- Stangl, K.; Stangl, V. The ubiquitin-proteasome pathway and endothelial (dys)function. Cardiovasc. Res. 2010, 85, 281–290. [Google Scholar] [CrossRef]
- Laina, A.; Stellos, K.; Stamatelopoulos, K. Vascular ageing: Underlying mechanisms and clinical implications. Exp. Gerontol. 2018, 109, 16–30. [Google Scholar] [CrossRef]
- Depre, C.; Wang, Q.; Yan, L.; Hedhli, N.; Peter, P.; Chen, L.; Hong, C.; Hittinger, L.; Ghaleh, B.; Sadoshima, J.; et al. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation 2006, 114, 1821–1828. [Google Scholar] [CrossRef]
- Ueland, T.; Yndestad, A.; Oie, E.; Florholmen, G.; Halvorsen, B.; Froland, S.S.; Simonsen, S.; Christensen, G.; Gullestad, L.; Aukrust, P. Dysregulated osteoprotegerin/RANK ligand/RANK axis in clinical and experimental heart failure. Circulation 2005, 111, 2461–2468. [Google Scholar] [CrossRef]
- Di Giuseppe, R.; Biemann, R.; Wirth, J.; Menzel, J.; Isermann, B.; Stangl, G.I.; Fritsche, A.; Boeing, H.; Schulze, M.B.; Weikert, C. Plasma osteoprotegerin, its correlates, and risk of heart failure: A prospective cohort study. Eur. J. Epidemiol. 2017, 32, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Saliques, S.; Teyssier, J.R.; Vergely, C.; Lorgis, L.; Lorin, J.; Donzel, A.; Sicard, P.; Berchoud, J.; Ragot, S.; Touzery, C.; et al. Smoking and FOS expression from blood leukocyte transcripts in patients with coronary artery disease. Atherosclerosis 2011, 219, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C. Molecular pathways: Osteoclast-dependent and osteoclast-independent roles of the RANKL/RANK/OPG pathway in tumorigenesis and metastasis. Clin. Cancer Res. 2012, 18, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.J.; Jung, S.H.; Lee, H.C.; Han, N.K.; Bae, I.H.; Lee, M.; Han, Y.H.; Kang, Y.S.; Lee, S.J.; Park, H.J.; et al. Identification of novel therapeutic targets in the secretome of ionizing radiationinduced senescent tumor cells. Oncol. Rep. 2016, 35, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, P.; Mechtcheriakova, D.; Meshcheryakova, A.; Foger-Samwald, U.; Ellinger, I. Immunology of Osteoporosis: A Mini-Review. Gerontology 2016, 62, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Bartoli-Leonard, F.; Wilkinson, F.L.; Langford-Smith, A.W.W.; Alexander, M.Y.; Weston, R. The Interplay of SIRT1 and Wnt Signaling in Vascular Calcification. Front. Cardiovasc. Med. 2018, 5, 183. [Google Scholar] [CrossRef] [PubMed]
- Davaine, J.M.; Quillard, T.; Brion, R.; Laperine, O.; Guyomarch, B.; Merlini, T.; Chatelais, M.; Guilbaud, F.; Brennan, M.A.; Charrier, C.; et al. Osteoprotegerin, pericytes and bone-like vascular calcification are associated with carotid plaque stability. PLoS ONE 2014, 9, e107642. [Google Scholar] [CrossRef] [PubMed]
- Navarro, R.; Compte, M.; Alvarez-Vallina, L.; Sanz, L. Immune Regulation by Pericytes: Modulating Innate and Adaptive Immunity. Front. Immunol. 2016, 7, 480. [Google Scholar] [CrossRef]
- Hung, C.F.; Mittelsteadt, K.L.; Brauer, R.; McKinney, B.L.; Hallstrand, T.S.; Parks, W.C.; Chen, P.; Schnapp, L.M.; Liles, W.C.; Duffield, J.S.; et al. Lung pericyte-like cells are functional interstitial immune sentinel cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L556–L567. [Google Scholar] [CrossRef]
- Benslimane-Ahmim, Z.; Heymann, D.; Dizier, B.; Lokajczyk, A.; Brion, R.; Laurendeau, I.; Bieche, I.; Smadja, D.M.; Galy-Fauroux, I.; Colliec-Jouault, S.; et al. Osteoprotegerin, a new actor in vasculogenesis, stimulates endothelial colony-forming cells properties. J. Thromb. Haemost. JTH 2011, 9, 834–843. [Google Scholar] [CrossRef]
- Abu El-Asrar, A.M.; Struyf, S.; Mohammad, G.; Gouwy, M.; Rytinx, P.; Siddiquei, M.M.; Hernandez, C.; Alam, K.; Mousa, A.; De Hertogh, G.; et al. Osteoprotegerin Is a New Regulator of Inflammation and Angiogenesis in Proliferative Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3189–3201. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.R.; Baily, J.E.; Chen, W.C.W.; Dar, A.; Gonzalez, Z.N.; Jensen, A.R.; Petrigliano, F.A.; Deb, A.; Henderson, N.C. Skeletal and cardiac muscle pericytes: Functions and therapeutic potential. Pharm. 2017, 171, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Okada, M.; Proto, J.D.; Gao, X.; Sekiya, N.; Beckman, S.A.; Corselli, M.; Crisan, M.; Saparov, A.; Tobita, K.; et al. Human pericytes for ischemic heart repair. Stem Cells 2013, 31, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular calcification: An update on mechanisms and challenges in treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Schneeweis, L.A.; Willard, D.; Milla, M.E. Functional dissection of osteoprotegerin and its interaction with receptor activator of NF-kappaB ligand. J. Biol. Chem. 2005, 280, 41155–41164. [Google Scholar] [CrossRef] [PubMed]
- Panizo, S.; Cardus, A.; Encinas, M.; Parisi, E.; Valcheva, P.; Lopez-Ongil, S.; Coll, B.; Fernandez, E.; Valdivielso, J.M. RANKL increases vascular smooth muscle cell calcification through a RANK-BMP4-dependent pathway. Circ. Res. 2009, 104, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sanchez, C.; Posadas-Romero, C.; Posadas-Sanchez, R.; Carreon-Torres, E.; Rodriguez-Perez, J.M.; Juarez-Rojas, J.G.; Martinez-Sanchez, C.; Fragoso, J.M.; Gonzalez-Pacheco, H.; Vargas-Alarcon, G.; et al. Low concentrations of phospholipids and plasma HDL cholesterol subclasses in asymptomatic subjects with high coronary calcium scores. Atherosclerosis 2015, 238, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Shioi, A.; Katagi, M.; Okuno, Y.; Mori, K.; Jono, S.; Koyama, H.; Nishizawa, Y. Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: Roles of tumor necrosis factor-alpha and oncostatin M derived from macrophages. Circ. Res. 2002, 91, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Morisawa, T.; Nakagomi, A.; Kohashi, K.; Kosugi, M.; Kusama, Y.; Atarashi, H.; Shimizu, W. Osteoprotegerin is Associated with Endothelial Function and Predicts Early Carotid Atherosclerosis in Patients With Coronary Artery Disease. Int. Heart J. 2015, 56, 605–612. [Google Scholar] [CrossRef]
- Panh, L.; Ruidavets, J.B.; Rousseau, H.; Petermann, A.; Bongard, V.; Berard, E.; Taraszkiewicz, D.; Lairez, O.; Galinier, M.; Carrie, D.; et al. Association between serum alkaline phosphatase and coronary artery calcification in a sample of primary cardiovascular prevention patients. Atherosclerosis 2017, 260, 81–86. [Google Scholar] [CrossRef]
- Schnabel, R.B.; Larson, M.G.; Yamamoto, J.F.; Kathiresan, S.; Rong, J.; Levy, D.; Keaney, J.F., Jr.; Wang, T.J.; Vasan, R.S.; Benjamin, E.J. Relation of multiple inflammatory biomarkers to incident atrial fibrillation. Am. J. Cardiol. 2009, 104, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Willeit, K.; Pechlaner, R.; Willeit, P.; Skroblin, P.; Paulweber, B.; Schernthaner, C.; Toell, T.; Egger, G.; Weger, S.; Oberhollenzer, M.; et al. Association Between Vascular Cell Adhesion Molecule 1 and Atrial Fibrillation. Jama Cardiol 2017, 2, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.F.; Appelman-Dijkstra, N.M.; van der Burg, S.H.; Kroep, J.R. The anti-tumor effect of RANKL inhibition in malignant solid tumors—A systematic review. Cancer Treat. Rev. 2018, 62, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kobayashi, Y.; Uehara, S.; Suzuki, T.; Koide, M.; Yamashita, T.; Nakamura, M.; Takahashi, N.; Kato, H.; Udagawa, N.; et al. A Jak1/2 inhibitor, baricitinib, inhibits osteoclastogenesis by suppressing RANKL expression in osteoblasts in vitro. PLoS ONE 2017, 12, e0181126. [Google Scholar] [CrossRef] [PubMed]
- St Hilaire, C.; Ziegler, S.G.; Markello, T.C.; Brusco, A.; Groden, C.; Gill, F.; Carlson-Donohoe, H.; Lederman, R.J.; Chen, M.Y.; Yang, D.; et al. NT5E mutations and arterial calcifications. N. Engl. J. Med. 2011, 364, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Ghalamfarsa, G.; Kazemi, M.H.; Raoofi Mohseni, S.; Masjedi, A.; Hojjat-Farsangi, M.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. CD73 as a potential opportunity for cancer immunotherapy. Expert Opin. Ther. Targets 2019, 23, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magid, A.F. Inhibitors of CD73 May Provide a Treatment for Cancer and Autoimmune Diseases. ACS Med. Chem. Lett. 2017, 8, 781–782. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rochette, L.; Meloux, A.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. The Role of Osteoprotegerin and Its Ligands in Vascular Function. Int. J. Mol. Sci. 2019, 20, 705. https://doi.org/10.3390/ijms20030705
Rochette L, Meloux A, Rigal E, Zeller M, Cottin Y, Vergely C. The Role of Osteoprotegerin and Its Ligands in Vascular Function. International Journal of Molecular Sciences. 2019; 20(3):705. https://doi.org/10.3390/ijms20030705
Chicago/Turabian StyleRochette, Luc, Alexandre Meloux, Eve Rigal, Marianne Zeller, Yves Cottin, and Catherine Vergely. 2019. "The Role of Osteoprotegerin and Its Ligands in Vascular Function" International Journal of Molecular Sciences 20, no. 3: 705. https://doi.org/10.3390/ijms20030705
APA StyleRochette, L., Meloux, A., Rigal, E., Zeller, M., Cottin, Y., & Vergely, C. (2019). The Role of Osteoprotegerin and Its Ligands in Vascular Function. International Journal of Molecular Sciences, 20(3), 705. https://doi.org/10.3390/ijms20030705