Proteomics, Holm Oak (Quercus ilex L.) and Other Recalcitrant and Orphan Forest Tree Species: How do They See Each Other?

 , , , , , ,
, , , , , ,
Abstract
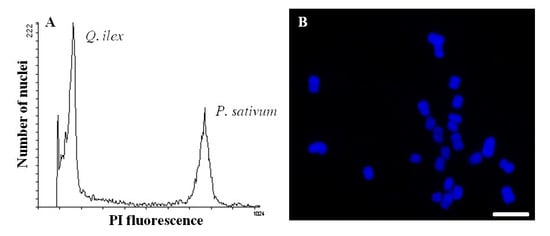
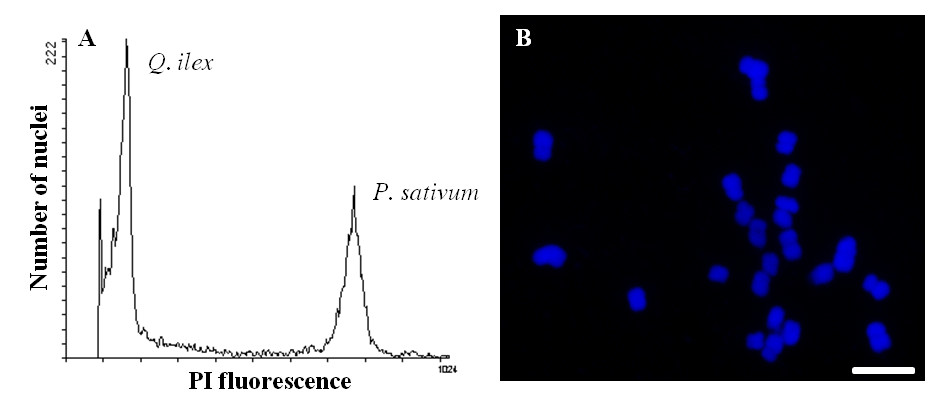
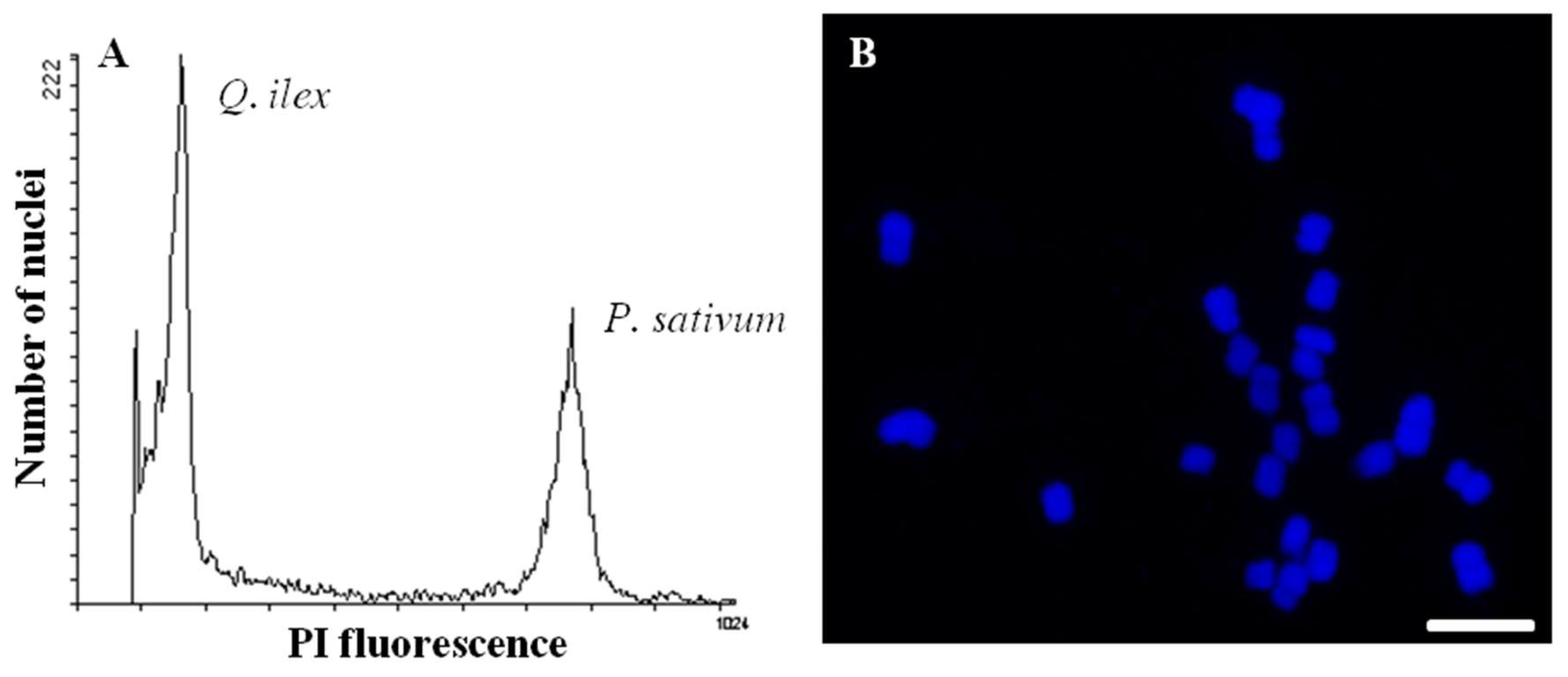
1. Introduction
2. How Quercus ilex Is Seen by Proteomics
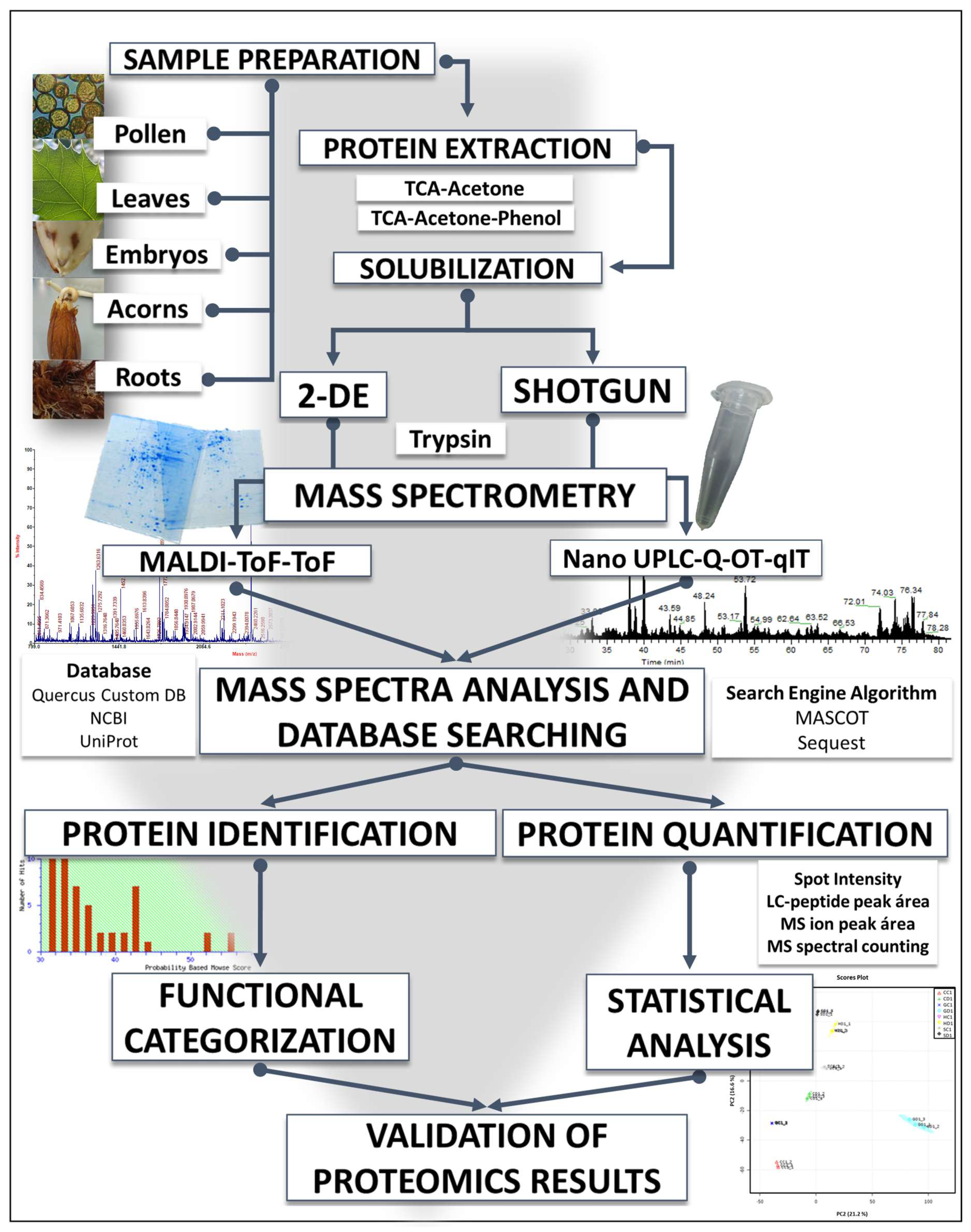
2.1. ‘Only a Small Percentage of the Total Protein Is Extracted and Solubilized, So We Deal with the Extractome Rather Than with the Real Proteome’
2.2. The Plant Proteome is Highly Variable and Therefore Requires Careful Experimental Design
2.3. Only a Small Fraction of the Present Protein Species Is Visualized and Identified by Any Given Approach
2.4. Gene Product Identification? Or Just Hits or Matches to Orthologs?
2.5. Methods and Protocols Must Be Validated and Optimized for Each Experimental System
2.6. 2-DE and Shotgun Platforms Are Complementary
2.7. How Proteomics Sees Quercus ilex
2.8. Characterizing Biodiversity
2.9. Adaptation to Biotic and Abiotic Stresses
2.10. Development: Seed Maturation and Germination
3. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olea, L.; San Miguel-Ayanz, A. The Spanish dehesa. A traditional Mediterranean silvopastoral system linking production and nature conservation. Grassl. Sci. Eur. 2006, 11, 3–13. [Google Scholar]
- De Rigo, D.; Gaudullo, G. Quercus ilex in Europe: Distribution, habitat, usage and threats. In European Atlas of Forest Tree Species; San-Miguel-Ayanz, J., Ed.; European Union: Luxembourg, 2016; pp. 130–131. [Google Scholar]
- Surová, D.; Ravera, F.; Guiomar, N.; Martínez Sastre, R.; Pinto-Correia, T. Contributions of Iberian Silvo-Pastoral Landscapes to the Well-Being of Contemporary Society. Rangel. Ecol. Manag. 2017, 71, 560–570. [Google Scholar] [CrossRef]
- Plieninger, T. Constructed and degraded? Origin and Development of the Spanish Dehesa Landscape, with a a Case Study on Two Municipalities. Die Erde 2007, 138, 25–46. [Google Scholar]
- Guzmán Álvarez, J.R. The image of a tamed landscape: Dehesa through History in Spain. Cult. Hist. Digit. J. 2016, 5, e003. [Google Scholar] [CrossRef]
- Lloret, F.; Siscart, D.; Dalmases, C. Canopy recovery after drought dieback in holm-oak Mediterranean forests of Catalonia (NE Spain). Glob. Chang. Biol. 2004, 10, 2092–2099. [Google Scholar] [CrossRef]
- Natalini, F.; Alejano, R.; Vázquez-Piqué, J.; Cañellas, I.; Gea-Izquierdo, G. The role of climate change in the widespread mortality of holm oak in open woodlands of Southwestern Spain. Dendrochronologia 2016, 38, 51–60. [Google Scholar] [CrossRef]
- Jorrín-Novo, J.V.; Navarro-Cerrillo, R.M. Variabilidad y respuesta a distintos estreses en poblaciones de encina (Quercus ilex L.) en Andalucía mediante una aproximación proteómica. Rev. Ecosistemas 2014, 23, 99–107. [Google Scholar]
- Jorge, I.; Navarro-Cerrillo, R.M.; Lenz, C.; Ariza, D.; Porras, C.; Jorrín-Novo, J.V. The holm oak leaf proteome: Analytical and biological variability in the protein expression level assessed by 2-DE and protein identification tandem mass spectrometry de novo sequencing and sequence similarity searching. Proteomics 2005, 5, 222–234. [Google Scholar] [CrossRef]
- Valledor, L.; Castillejo, M.Á.; Lenz, C.; Rodríguez, R.; Cañal, M.J. Proteomic analysis of Pinus radiata needles: 2-DE map and protein identification by LC/MS/MS and substitution-tolerant database searching. J. Proteome Res. 2008, 7, 2616–2631. [Google Scholar] [CrossRef]
- De Francisco, L.; Romero-Rodríguez, M.C.; Navarro-Cerrillo, R.M.; Miniño, V.; Perdomo, O.; Jorrín-Novo, J.V. Characterization of the orthodox Pinus occidentalis seed and pollen proteomes by using complementary gel-based and gel-free approaches. J. Proteom. 2016, 143, 382–389. [Google Scholar] [CrossRef]
- Ariza, D.; Navarro-Cerrillo, R.M.; del Campo, A. Influencia de la fecha de plantación al establecimiento de Pinus halapensis aplicación de la proteómica a estudios de Ecofisiología en campo. Soc. Española Cienc. 2008, 28, 111–117. [Google Scholar]
- Loewe, V.; Navarro-Cerrillo, R.M.; Sánchez-Lucas, R.; Ruiz Gómez, F.J.; Jorrín-Novo, J.V. Variability studies of allochthonous stone pine (Pinus pinea L.) plantations in Chile through nut protein profiling. J. Proteom. 2018, 175, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Romero-Rodríguez, M.C.; Abril, N.; Sánchez-Lucas, R.; Jorrín-Novo, J.V. Multiplex staining of 2-DE gels for an initial phosphoproteome analysis of germinating seeds and early grown seedlings from a non-orthodox specie: Quercus ilex L. subsp. ballota [Desf.] Samp. Front. Plant Sci. 2015, 6, 620. [Google Scholar] [CrossRef] [PubMed]
- Jorge, I.; Navarro-Cerrillo, R.M.; Lenz, C.; Ariza, D.; Jorrín-Novo, J.V. Variation in the holm oak leaf proteome at different plant developmental stages, between provenances and in response to drought stress. Proteomics 2006, 6, 207–214. [Google Scholar] [CrossRef]
- Valero-Galván, J.; Valledor, L.; Navarro-Cerrillo, R.M.; Pelegrín, E.G.; Jorrín-Novo, J.V. Studies of variability in Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.) through acorn protein profile analysis. J. Proteom. 2011, 74, 1244–1255. [Google Scholar]
- Sghaier-Hammami, B.; Valero-Galván, J.; Romero-Rodríguez, M.C.; Navarro-Cerrillo, R.M.; Abdelly, C.; Jorrín-Novo, J.V. Physiological and proteomics analyses of Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.) responses to Phytophthora cinnamomi. Plant Physiol. Biochem. 2013, 71, 191–202. [Google Scholar] [PubMed]
- Sghaier-Hammami, B.; Redondo-López, I.; Valero-Galván, J.; Jorrín-Novo, J.V. Protein profile of cotyledon, tegument, and embryonic axis of mature acorns from a non-orthodox plant species: Quercus ilex. Planta 2016, 243, 369–396. [Google Scholar] [CrossRef]
- Romero-Rodríguez, M.C.; Jorrín-Novo, J.V.; Castillejo, M.Á. Towards characterizing seed germination and seedling establishment in the non- orthodox forest tree species Quercus ilex through complementary gel and gel-free proteomic approaches. J. Proteom. 2018. [Google Scholar] [CrossRef]
- Echevarría-Zomeño, S.; Ariza, D.; Jorge, I.; Lenz, C.; Del Campo, A.; Jorrín-Novo, J.V.; Navarro-Cerrillo, R.M. Changes in the protein profile of Quercus ilex leaves in response to drought stress and recovery. J. Plant Physiol. 2009, 166, 233–245. [Google Scholar] [CrossRef]
- Valero-Galván, J.; González-Fernández, R.; Navarro-Cerrillo, R.M.; Gil-Pelegrín, E.; Jorrín-Novo, J.V. Physiological and proteomic analyses of drought stress response in Holm oak provenances. J. Proteome Res. 2013, 12, 5110–5123. [Google Scholar] [CrossRef]
- Simova-Stoilova, L.P.; Romero-Rodríguez, M.C.; Sánchez-Lucas, R.; Navarro-Cerrillo, R.M.; Medina-Aunon, J.A.; Jorrín-Novo, J.V. 2-DE proteomics analysis of drought treated seedlings of Quercus ilex supports a root active strategy for metabolic adaptation in response to water shortage. Front. Plant Sci. 2015, 6, 627. [Google Scholar] [CrossRef] [PubMed]
- Simova-Stoilova, L.P.; López-Hidalgo, C.; Sánchez-Lucas, R.; Valero-Galván, J.; Romero-Rodríguez, M.C.; Jorrín-Novo, J.V. Holm oak proteomic response to water limitation at seedling establishment stage reveals specific changes in different plant parts as well as interaction between roots and cotyledons. Plant Sci. 2018, 276, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Abril, N.; Gion, J.M.; Kerner, R.; Müller-Starck, G.; Navarro-Cerrillo, R.M.; Plomion, C.; Renaut, J.; Valledor, L.; Jorrín-Novo, J.V. Proteomics research on forest trees, the most recalcitrant and orphan 2plant species. Phytochemistry 2011, 72, 1219–1242. [Google Scholar] [CrossRef] [PubMed]
- Jorrín-Novo, J.V.; Maldonado-Alconada, A.M.; Castillejo, M.Á. Plant proteome analysis: A 2006 update. Proteomics 2007, 7, 2947–2962. [Google Scholar] [CrossRef] [PubMed]
- Jorrín-Novo, J.V.; Maldonado-Alconada, A.M.; Echevarría-Zomeño, S.; Valledor, L.; Castillejo, M.Á.; Curto, M.; Valero-Galván, J.; Sghaier-Hammami, B.; Donoso, G.; Redondo-López, I. Plant proteomics update (2007–2008): Second-generation proteomic techniques, an appropriate experimental design, and data analysis to fulfill MIAPE standards, increase plant proteome coverage and expand biological knowledge. J. Proteom. 2009, 72, 285–314. [Google Scholar] [CrossRef]
- Jorrín-Novo, J.V.; Komatsu, S.; Weckwerth, W.; Wienkoop, S. Plant Proteomics—Methods and Protocols, 2nd ed.; Humana Press: Totowa, NJ, USA, 2014; ISBN 9781627036306. [Google Scholar]
- Jorrín-Novo, J.V.; Pascual, J.; Sánchez-Lucas, R.; Romero-Rodríguez, M.C.; Rodríguez-Ortega, M.J.; Lenz, C.; Valledor, L. Fourteen years of plant proteomics reflected in Proteomics: Moving from model species and 2DE-based approaches to orphan species and gel-free platforms. Proteomics 2015, 15, 1089–1112. [Google Scholar] [CrossRef] [PubMed]
- Jorrín-Novo, J.V.; Valledor, L. Translational proteomics special issue. J. Proteom. 2013, 93, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Lucas, R.; Mehta, A.; Valledor, L.; Cabello-Hurtado, F.; Romero-Rodríguez, M.C.; Simova-Stoilova, L.P.; Demir, S.; Rodríguez de Francisco, L.; Maldonado-Alconada, A.M.; Jorrín-Prieto, A.L.; et al. A year (2014–2015) of plants in Proteomics journal. Progress in wet and dry methodologies, moving from protein catalogs, and the view of classic plant biochemists. Proteomics 2016, 16, 866–876. [Google Scholar] [CrossRef]
- Komatsu, S.; Jorrín-Novo, J.V. Food and Crop Proteomics. J. Proteom. 2016, 143, 1–2. [Google Scholar] [CrossRef]
- Jorrín-Novo, J.V.; Komatsu, S.; Sánchez-Lucas, R.; de Francisco, L.E.R. Gel electrophoresis-based plant proteomics: Past, present, and future. Happy 10th anniversary Journal of Proteomics! J. Proteom. 2018. [Google Scholar] [CrossRef]
- Jorrín-Novo, J.V. Scientific standards and MIAPEs in plant proteomics research and publications. Front. Plant Sci. 2015, 6, 473. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Lei, Y.; Zhang, S. Differences in resistance to nitrogen and phosphorus deficiencies explain male-biased populations of poplar in nutrient-deficient habitats. J. Proteom. 2018, 178, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Holliday, J.A.; Aitken, S.N.; Cooke, J.E.K.; Fady, B.; Gonzalez-Martinez, S.C.; Heuertz, M.; Jaramillo-Correa, J.P.; Lexer, C.; Staton, M.; Whetten, R.W.; et al. Advances in ecological genomics in forest trees and applications to genetic resources conservation and breeding. Mol. Eco. 2017, 26, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Valledor, L.; Jorrín-Novo, J.V. Back to the basics: Maximizing the information obtained by quantitative two dimensional gel electrophoresis analyses by an appropriate experimental design and statistical analyses. J. Proteom. 2011, 74, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Alconada, A.M.; Echevarría-Zomeño, S.; Jean-Baptiste, S.; Hernández de la Torre, M.; Jorrín-Novo, J.V. Evaluation of three different protocols of protein extraction for Arabidopsis thaliana leaf proteome analysis by two-dimensional electrophoresis. J. Proteom. 2008, 71, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Valero-Galván, J.; Valledor, L.; González-Fernández, R.; Navarro-Cerrillo, R.M.; Jorrín-Novo, J.V. Proteomic analysis of Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.) pollen. J. Proteom. 2012, 75, 2736–2744. [Google Scholar]
- López-Hidalgo, C.; Guerrero-Sanchez, V.M.; Gómez-Gálvez, I.; Sánchez-Lucas, R.; Castillejo, M.Á.; Maldonado-Alconada, A.M.; Valledor, L.; Jorrín-Novo, J.V. A multi-omics analysis pipeline for the metabolic pathway reconstruction in the orphan species Quercus ilex. Front. Plant Sci. 2018, 9, 935. [Google Scholar] [CrossRef]
- Guerrero-Sanchez, V.M.; Maldonado-Alconada, A.M.; Amil-Ruiz, F.; Jorrín-Novo, J.V. Holm Oak (Quercus ilex) transcriptome. De novo sequencing and assembly analysis. Front. Mol. Biosci. 2017, 4, 70. [Google Scholar] [CrossRef]
- Romero-Rodríguez, M.C.; Pascual, J.; Valledor, L.; Jorrín-Novo, J.V. Improving the quality of protein identification in non-model species. Characterization of Quercus ilex seed and Pinus radiata needle proteomes by using SEQUEST and custom databases. J. Proteom. 2014, 105, 85–91. [Google Scholar] [CrossRef]
- Kao, S.H.; Wong, H.K.; Chiang, C.Y.; Chen, H.M. Evaluating the compatibility of three colorimetric protein assays for two-dimensional electrophoresis experiments. Proteomics 2008, 8, 2178–2184. [Google Scholar] [CrossRef]
- Jorrín-Novo, J.V.; Ardilla, H.; Castillejo, M.Á.; Curto, M.; Echevarría-Zomeño, S.; Hernández de la Torre, M.; Gómez-Gálvez, F.; González-Fernández, R. From 2003 to 2011: Proteomics investigation at the agroforestry and plant biochemistry and proteomics research group (University of Cordoba, Spain). In 3rd International Symposium on Frontiers in Agriculture Proteome Research: Contribution of Proteomics Technology in Agricultural Sciences. Frontiers; Mock, H.P., Wang, Z.Y., Komatsu, S., Eds.; Frontiers in Agriculture Proteome Research. Contribution of Proteomics Technology in Agricultural Sciences; NARO Institute of Crop Science: Ibaraki, Japan, 2011; pp. 130–137. [Google Scholar]
- Yeoh, H.H.; Wee, Y.C. Leaf protein contents and nitrogen-to-protein conversion factors for 90 plant species. Food Chem. 1994, 49, 245–250. [Google Scholar] [CrossRef]
- Valero-Galván, J.; González-Fernández, R.; Valledor, L.; Navarro-Cerrillo, R.M.; Jorrín-Novo, J.V. Proteotyping of Holm oak (Quercus ilex subsp. ballota) provenances through proteomic analysis of acorn flour. In Plant Proteomics—Methods and Protocols; Humana Press: Totowa, NJ, USA, 2014; pp. 709–724. ISBN 9780470988879. [Google Scholar]
- Romero-Rodríguez, M.C.; Maldonado-Alconada, A.M.; Valledor, L.; Jorrín-Novo, J.V. Back to Osborne. Sequential protein extraction and LC-MS analysis for the characterization of the Holm oak seed proteome. In Plant Proteomics—Methods and Protocols; Humana Press: Totowa, NJ, USA, 2007; pp. 379–390. ISBN 9780470988879. [Google Scholar]
- Valero-Galván, J.; Jorrín-Novo, J.V.; Gómez Cabrera, A.; Ariza, D.; García-Olmo, J.; Navarro-Cerrillo, R.M. Population variability based on the morphometry and chemical composition of the acorn in Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.). Eur. J. Res. 2012, 131, 893–904. [Google Scholar]
- Valledor, L.; Romero-Rodríguez, M.C.; Jorrín-Novo, J.V. Standardization of data processing and statistical analysis in comparative plant proteomics experiment. In Plant Proteomics—Methods and Protocols; Humana Press: Totowa, NJ, USA, 2014; pp. 51–60. ISBN 9780470988879. [Google Scholar]
- Maitra, S.; Yan, J. Principle component analysis and partial least squares: Two dimension reduction techniques for regression. Appl. Multivar. Statist. Models 2008, 79, 79–90. [Google Scholar]
- Plomion, C.; Aury, J.M.; Amselem, J.; Alaeitabar, T.; Barbe, V.; Belser, C.; Bergès, H.; Bodénès, C.; Boudet, N.; Boury, C.; et al. Decoding the oak genome: Public release of sequence data, assembly, annotation and publication strategies. Mol. Ecol. Resour. 2016, 16, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Sork, V.L.; Fitz-Gibbon, S.T.; Puiu, D.; Crepeau, M.; Gugger, P.F.; Sherman, R.; Stevens, K.; Langley, C.H.; Pellegrini, M.; Salzberg, S.L. First draft assembly and annotation of the genome of a california endemic oak Quercus lobata Nee (Fagaceae). G3 (Bethesda) 2016, 6, 3485–3495. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.M.; Usié, A.; Barbosa, P.; Barros, P.M.; Capote, T.; Chaves, I.; Simöes, F.; Abreu, I.; Carrasquinho, I.; Faro, C.; et al. The draft genome sequence of cork oak. Sci. Data 2018, 5, 180069. [Google Scholar] [CrossRef] [PubMed]
- Kremer, A.; Casasoli, M.; Barreneche, T.; Bódenès, C.; Sisco, P.; Kubisiak, T.; Scalfi, M.; Leonardi, S.; Bakker, E.; Buiteveld, J.; et al. Genome Mapping and Molecular Breeding in Plants; Chittaranjan, K., Ed.; Springer: Heidelberg/Berlin, Germany, 2007; Volume 7, pp. 165–187. [Google Scholar]
- Lesur, I.; Le Provost, G.; Bento, P.; Da Silva, C.; Leplé, J.C.; Murat, F.; Ueno, S.; Bartholomé, J.; Lalanne, C.; Ehrenmann, F.; et al. The oak gene expression atlas: Insights into Fagaceae genome evolution and the discovery of genes regulated during bud dormancy release. BMC Genom. 2015, 16. [Google Scholar] [CrossRef]
- Rey, M.D.; Guerrero-Sánchez, V.M.; Sánchez-Lucas, R.; López-Hidalgo, C.; Maldonado-Alconada, A.M.; Jorrín-Novo, J.V. The Use of -Omics Technologies to Progress in the Quercus Ilex Biology; XIV RBMP: Salamanca, Spain, 2018; p. 20. [Google Scholar]
- Zoldos, V.; Papes, D.; Brown, S.; Panaud, O.; Siljak-Yakovlev, S. Protocol for flow cytometric assay Genome size and base composition of seven Quercus species: Inter- and intra-population variation. Genome 1998, 41, 162–168. [Google Scholar] [CrossRef]
- Chen, S.C.; Cannon, C.H.; Kua, C.S.; Liu, J.J.; Galbraith, D.W. Genome size variation in the Fagaceae and its implications for trees. Tree Genet. Genomes 2014, 10, 977–988. [Google Scholar] [CrossRef]
- Rey, M.D.; Moore, G.; Martin, A.C. Identification and comparison of individual chromosomes of three Hordeum chilense accessions, Hordeum vulgare and Triticum aestivum by FISH. Genome 2018, 61, 387–396. [Google Scholar] [CrossRef]
- Guerrero-Sanchez, V.M.; Maldonado-Alconada, A.M.; Amil-Ruiz, F.; Verardi, A.; Jorrín-Novo, J.V.; Rey, M.D. Ion torrent and Illumina, two complementary RNA-Seq platforms fo constructing the holm oak (Quercus ilex) transcriptome. PLoS ONE 2019, 14, e0210356. [Google Scholar] [CrossRef] [PubMed]
- Neale, D.B.; McGuire, P.E.; Wheeler, N.C.; Stevens, K.A.; Crepeau, M.W.; Cardeno, C.; Zimin, A.V.; Puiu, D.; Pertea, G.M.; Sezen, U.U.; et al. The Douglas-Fir Genome Sequence Reveals Specialization of the Photosynthetic Apparatus in Pinaceae. G3 (Bethesda) 2017, 3157–3167. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gálvez, I.M.; Castillejo, M.A.; Márquez, C.; Jorrín-Novo, J.V. Unravelling Mechanisms of Tolerance in Holm Oak through a Physiological and Molecular Approach; INPPO: Padova, Italy, 2018. [Google Scholar]
- Curto, M.; Valledor, L.; Navarrete, C.; Gutiérrez, D.; Sychrova, H.; Ramos, J.; Jorrín-Novo, J.V. 2-DE based proteomic analysis of Saccharomyces cerevisiae wild and K+ transport-affected mutant (trk1, 2) strains at the growth exponential and stationary phases. J. Proteom. 2010, 73, 2316–2335. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Alconada, A.M.; Echevarría-Zomeño, S.; Lindermayr, C.; Redondo-López, I.; Durner, J.; Jorrín-Novo, J.V. Proteomic analysis of Arabidopsis protein S-nitrosylation in response to inoculation with Pseudomonas syringae. Acta Physiol. Plant. 2011, 33, 1493–1514. [Google Scholar] [CrossRef]
- González-Fernández, R.; Aloria, K.; Arizmendi, J.M.; Jorrín-Novo, J.V. Application of label-free shotgun nUPLC-MSEand 2-DE approaches in the study of Botrytis cinerea mycelium. J. Proteome Res. 2013, 12, 3042–3056. [Google Scholar] [CrossRef] [PubMed]
- Lenz, C.; Seymour, S.; Shilov, I.; Jorrín-Novo, J.V. Protein Pilot: Accommodating genetic diversity in mass spectrometry-based plant proteome research. In II “Plant Proteomics in Europe” Meeting (Working Group 2): Will Plant Proteomics Research Help in Facing Food, Health and Environmental Concerns? Changes Induced by the Pepper Mild Mottle Tobamovirus on the Chloroplast Proteome of Nicotiana Benthamiana; University of Córdoba (UCO): Córdoba, Spain, 2008. [Google Scholar]
- Romero-Rodríguez, M.C.; Valledor, L.; Lenz, C.; Hurlaub, H.; Jorrín-Novo, J.V. Proteomics workflows and protocols for the study of orphan species. In HUPO 2014. The Proteome Quest to Understand Biology and Disease. Non-Human and Food Proteomics Section; HUPO: Madrid, Spain, 2014. [Google Scholar]
- Tong, Z.; Wang, D.; Sun, Y.; Yang, Q.; Meng, X.R.; Wang, L.M.; Feng, W.Q.; Li, L.; Wurtele, E.S.; Wang, X.C. Comparative Proteomics of Rubber Latex Revealed Multiple Protein Species of REF/SRPP Family Respond Diversely to Ethylene Stimulation among Different Rubber Tree Clones. Int. J. Mol. Sci. 2017, 18, 958. [Google Scholar] [CrossRef] [PubMed]
- Lenz, C.; Jorrín-Novo, J.V.; Urlaub, H. Quercus ilex: Protein identification strategies for an orphan tree species. In Proceedings of the 61st ASMS Conference on Mass Spectrometry and Allied Topics, Baltimore, MD, USA, 6–13 June 2013. [Google Scholar]
- Valero-Galván, J.; Sghaier-Hammami, B.; Navarro-Cerrillo, R.M.; Jorrín-Novo, J.V. Natural variability and responses to stresses in andalusia Holm oak (Quercus ilex subsp. ballota) populations. In Oak: Ecology, Types and Management; Chuteira, C.A., Grão, A.B., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2012; pp. 193–206. ISBN 978-1-61942-492-0. [Google Scholar]
- Zhang, X.L.; Zhang, J.; Guo, Y.H.; Sun, P.; Jia, H.X.; Fan, W.; Lu, M.Z.; Hu, J.J. Comparative proteomic analysis of mature pollen in triploid and diploid Populus deltoids. Int. J. Mol. Sci. 2016, 17, 1475. [Google Scholar] [CrossRef]
- Fernández i Marti, A.; Romero-Rodríguez, C.; Navarro-Cerrillo, R.; Abril, N.; Jorrín-Novo, J.V.; Dodd, R. Population genetic diversity of Quercus ilex subsp. ballota (Desf.) Samp. reveals divergence in recent and evolutionary migration rates in the Spanish dehesas. Forests 2018, 9, 337. [Google Scholar] [CrossRef]
- Geilfus, C.M.; Carpentier, S.C.; Zavisic, A.; Polle, A. Changes in the fine root proteome of Fagus sylvatica L. trees associated with P-deficiency and amelioration of P-deficiency. J. Proteom. 2017, 169, 33–40. [Google Scholar] [CrossRef]
- Fan, F.H.; Ding, G.J.; Wen, X.P. Proteomic analyses provide new insights into the responses of Pinus massoniana seedlings to phosphorus deficiency. Proteomics 2015, 16, 504–515. [Google Scholar] [CrossRef]
- Yu, J.J.; Jin, X.; Sun, X.M.; Gao, T.X.; Chen, X.M.; She, Y.M.; Jiang, T.B.; Chen, S.X.; Dai, S.J. Hydrogen peroxide response in leaves of Poplar (Populus simonii × Populus nigra) revealed from physiological and proteomic analyses. Int. J. Mol. Sci. 2017, 18, 2085. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.X.; Peng, M.; Zhang, X.L.; Lei, P.; Ji, X.M.; Chow, W.; Meng, F.J.; Sun, G.Y. Comparative mitochondrial proteomic, physiological, biochemical and ultrastructural profiling reveal factors underpinning salt tolerance in tetraploid black locust (Robinia pseudoacacia L.). Bmc Genom. 2017, 18, 648. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.J.; Dong, Y.P.; Zhao, Z.L.; Li, Y.; Fan, G.Q. Dissecting the proteome dynamics of the salt stress induced changes in the leaf of diploid and autotetraploid Paulownia fortunei. PLoS ONE 2017, 12, e0181937. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, L.L.; Zhou, K.K.; Li, Y.M.; Zhao, Z. Changes in protein profile of Platycladus orientalis (L.) roots and leaves in response to drought stress. Tree Genet. Genomes 2017, 4, 76. [Google Scholar] [CrossRef]
- Taibi, K.; del Campo, A.D.; Vilagrosa, A.; Belles, J.M.; Lopez-Gresa, M.P.; Pla, D.; Calvete, J.J.; Lopez-Nicolas, J.M.; Mulet, J.M. Drought tolerance in Pinus halepensis seed sources as identified by distinctive physiological and molecular markers. Front. Plant Sci. 2017, 8, 1202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, H.G.; Pang, Q.Y. Physiological evaluation of the responses of Larix olgensis families to drought stress and proteomic analysis of the superior family. Genet. Mol. Res. 2015, 14, 15577–15586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Feng, L.H.; Jiang, H.; Zhang, Y.B.; Zhang, S. Different proteome profiles between male and female Populus cathayana exposed to UVB radiation. Front. Plant Sci. 2018, 8, 320. [Google Scholar]
- Pascual, J.; Canal, M.J.; Escandon, M.; Meijon, M.; Weckwerth, W.; Valledor, L. Integrated physiological, proteomic, and metabolomic analysis of ultra violet (UV) stress responses and adaptation mechanisms in Pinus radiata. Mol. Cell. Proteom. 2018, 16, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Alegre, S.; Nagler, M.; Escandon, M.; Annacondia, M.L.; Weckwerth, W.; Valledor, L.; Canal, M.J. The variations in the nuclear proteome reveal new transcription factors and mechanisms involved in UK stress response in Pinus radiata. J. Proteom. 2016, 143, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Q.; Li, X.; Yang, S.H.; Zhou, Y.L.; Dong, C.; Ren, J.; Sun, X.D.; Yang, Y.P. Comparative physiological and proteomic analysis reveals the leaf response to cadmium-induced stress in Poplar (Populus yunnanensis). PLoS ONE 2015, 10, e0137396. [Google Scholar] [CrossRef]
- Cao, X.B.; Fan, G.Q.; Dong, Y.P.; Zhao, Z.L.; Deng, M.J.; Wang, Z.; Liu, W.S. Proteome profiling of Paulownia seedlings infected with phytoplasma. Front. Plant Sci. 2017, 8, 342. [Google Scholar] [CrossRef] [PubMed]
- Castillejo, M.Á.; Maldonado-Alconada, A.M.; Ogueta, S.; Jorrín-Novo, J.V. Proteomic analysis of responses to drought stress in sunflower (Helianthus annuus) leaves by 2DE gel electrophoresis and mass spectrometry. Open Proteom. J. 2008, 1, 59–71. [Google Scholar] [CrossRef]
- Sghaier-Hammami, B.; Valledor, L.; Redondo-López, I.; Weckwerth, W.; Jorrín-Novo, J.V. 2-DE-based and LC-label-free proteomics studies of seed development in holm oak (Quercus ilex). In Proceedings of the HUPO 2011 10th World Congress, Geneva, Switzerland, 3–7 September 2011; p. 277. [Google Scholar]
- Moothoo-Padayachie, A.; Macdonald, A.; Varghese, B.; Pammenter, N.W.; Govender, P. Uncovering the basis of viability loss in desiccation sensitive Trichilia dregeana seeds using differential quantitative protein expression profiling by iTRAQ. J. Plant Physiol. 2018, 221, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, D.; Shen, H.L.; Li, Y.H.; Nie, Y.Z. Proteome analysis of dormancy-released seeds of Fraxinus mandshurica Rupr. in response to re-dehydration under different conditions. Int. J. Mol. Sci. 2015, 16, 4713–4730. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.P.; Yang, L.; Shen, H.L. Proteomic analysis of immature Fraxinus mandshurica cotyledon tissues during somatic embryogenesis: Effects of explant browning on somatic embryogenesis. Int. J. Mol. Sci. 2015, 16, 13692–13713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, W.Q.; Liu, S.J.; Moller, I.M.; Song, S.Q. Proteome analysis of Poplar seed vigor. PLoS ONE 2015, 10, e0132509. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, E.; Kalemba, E.M.; Pukacka, S. Age-related changes in protein metabolism of beech (Fagus sylvatica L.) seeds during alleviation of dormancy and in the early stage of germination. Plant Physiol. Biochem. 2015, 94, 114–121. [Google Scholar] [CrossRef]
- Pawlowski, T.A.; Staszak, A.M. Analysis of the embryo proteome of sycamore (Acer pseudoplatanus L.) seeds reveals a distinct class of proteins regulating dormancy release. J. Plant Physiol. 2016, 195, 9–22. [Google Scholar]
- Jing, D.L.; Zhang, J.W.; Xia, Y.; Kong, L.S.; Ou Yang, F.Q.; Zhang, S.G.; Zhang, H.G.; Wang, J.H. Proteomic analysis of stress-related proteins and metabolic pathways in Picea asperata somatic embryos during partial desiccation. Plant Biotechnol. J. 2017, 15, 27–38. [Google Scholar] [CrossRef]
- Loijon, F.; Melzer, M.; Zhou, Q.; Srivastava, V.; Bulone, V. Proteomic analysis of plasmodesmata from populus cell suspension cultures in relation with callose biosynthesis. Front. Plant Sci. 2018, 9, 1681. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Dominguez, P.G.; Kumar, M.; Bygdell, J.; Miroshnichenko, S.; Sundberg, B.; Wingsle, G.; Niittyla, T. Cellulose synthase stoichiometry in aspen differs from Arabidopsis and Norway spruce. Plant Physiol. 2018, 177, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Guzicka, M.; Pawlowski, T.A.; Staszak, A.; Rozkowski, R.; Chmura, D.J. Molecular and structural changes in vegetative buds of Norway spruce during dormancy in natural weather conditions. Tree Physiol. 2018, 38, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.F.; Zhang, S.G.; Wang, J.H. Transcriptomic and proteomic analyses of embryogenic tissues in Picea balfouriana treated with 6-benzylaminopurine. Physiol. Plant. 2015, 154, 95–113. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.; Valledor, L.; Hancock, R.D.; Renaut, J.; Pascual, J.; Soares, A.M.V.M.; Pinto, G. Integrated proteomics and metabolomics to unlock global and clonal responses of Eucalyptus globulus recovery from water deficit. Metabolomics 2016, 12, 141. [Google Scholar] [CrossRef]
- Zheng, W.; Komatsu, S.; Zhu, W.; Zhang, L.; Li, X.M.; Cui, L.; Tian, J.K. Response and Defense Mechanisms of Taxus chinensis leaves Under UVA radiation are revealed using comparative proteomics and metabolomics analyses. Plant Cell Physiol. 2016, 57, 1839–1853. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, W.; Wang, G.; Cao, F.; Cai, J.F.; Wang, H. Comparative Proteomic and Physiological analysis reveals the variation mechanisms of leaf coloration and carbon fixation in a Xantha Mutant of Ginkgo biloba L. Int. J. Mol. Sci. 2016, 17, 1794. [Google Scholar] [CrossRef] [PubMed]
- McKenna, O.E.; Posselt, G.; Briza, P.; Lackner, P.; Schmitt, A.O.; Gadermaier, G.; Wessler, S.; Ferreira, F. Multi-approach analysis for the identification of proteases within birch pollen. Int. J. Mol. Sci. 2017, 7, 1433. [Google Scholar] [CrossRef]
- Mousavi, F.; Majd, A.; Shahali, Y.; Ghahremaninejad, F.; Shoormasti, R.S.; Pourpak, Z. Immunoproteomics of tree of heaven (Ailanthus atltissima) pollen allergens. J. Proteom. 2017, 154, 94–101. [Google Scholar] [CrossRef]
- Bygdell, J.; Srivastava, V.; Obudulu, O.; Srivastava, M.K.; Nilsson, R.; Sundberg, B.; Trygg, J.; Mellerowicz, E.J.; Wingsle, G. Protein expression in tension wood formation monitored at high tissue resolution in Populus. J. Exp. Bot. 2017, 13, 3405–3417. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Author | Year | Plant Organ | Protein Yield (mg g−1 DW Tissue) a | Proteomic Strategy | Features c | Identified Proteins | Proteome Database e |
|---|---|---|---|---|---|---|---|
| Jorge [9] | 2005 | Leaf | Data not reported *; L | 2-DE MALDI TOF/TOF | 350 | 20 out of 100 spots | NCBI: restriction to Viridiplantae |
| Jorge [15] | 2006 | Data not reported *; L | 400 | 24 out of 100 spots | |||
| Echevarría-Zomeño [20] | 2009 | 7 *; L | 390 | 12 out of 46 spots | SwissProt, trEMBL and NCBI: restriction to Viridiplantae | ||
| Valero-Galván [16] | 2011 | Seed | 6 *; B | 240 | 16 out of 56 spots | NCBI: restriction to Viridiplantae | |
| Valero-Galván [38] | 2012 | Pollen | 15 §; B | 2-DE MALDI-TOF/TOF | 600 | 77 out of 100 spots | UniProtKB restricted to Arabidopsis; Phytozome restricted to Populus and Eucaliptus; Custom-build database from Quercus ESTs f |
| Shotgun (nLC-MS/MS) b | Data not reported | 273 | |||||
| Valero-Galván [21] | 2013 | Leaf | 10 §; B | 2-DE MALDI-TOF/TOF | 230 | 18 out of 28 spots | NCBI: restriction to Viridiplantae |
| Sghaier-Hammami [17] | 2013 | 40 §; B | 480 | 80 out of 480 spots | |||
| Simova-Stoilova [22] | 2015 | Root | 3 §; B | 360 | 79 out of 90 spots | NCBI and UniProtKB: restriction to Viridiplantae | |
| Romero-Rodríguez [14] | 2015 | Embryo | 150 §; B | 480 | 20 d out of 55 spots | NCBI, UniProtKB: restriction to Viridiplantae and Custom Quercus database f | |
| Sghaier-Hammami [18] | 2016 | Cotyledon | 2 §; B | 440 | 50 out of 153 spots | NCBI: restriction to Viridiplantae | |
| Embryo | 80 §; B | 470 | 50 out of 153 spots | ||||
| Tegument | 0,4 §; B | 420 | 40 out of 153 spots | ||||
| López-Hidalgo [39] | 2018 | Pool of tissues: acorn, embryo, cotyledon, leaf and root | 40 §; B | Shotgun (nLC-MS/MS) b | 58600 | 2830 | SwissProt: restriction to Viridiplantae/ Custom-build specie database f |
| Romero-Rodríguez [19] | 2018 | Seed | 25 §; B | 2-DE MALDI-TOF/TOF | 540 | 90 out of 103 spots | NCBI, UniProtKB/TrEMBL and UniProtKB/SwissProt restricted to Viridiplantae; Custom-build Q. ilex database f |
| Shotgun (nLC-MS/MS) b | 3113 | 1650 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rey, M.-D.; Castillejo, M.Á.; Sánchez-Lucas, R.; Guerrero-Sanchez, V.M.; López-Hidalgo, C.; Romero-Rodríguez, C.; Valero-Galván, J.; Sghaier-Hammami, B.; Simova-Stoilova, L.; Echevarría-Zomeño, S.; et al. Proteomics, Holm Oak (Quercus ilex L.) and Other Recalcitrant and Orphan Forest Tree Species: How do They See Each Other? Int. J. Mol. Sci. 2019, 20, 692. https://doi.org/10.3390/ijms20030692
Rey M-D, Castillejo MÁ, Sánchez-Lucas R, Guerrero-Sanchez VM, López-Hidalgo C, Romero-Rodríguez C, Valero-Galván J, Sghaier-Hammami B, Simova-Stoilova L, Echevarría-Zomeño S, et al. Proteomics, Holm Oak (Quercus ilex L.) and Other Recalcitrant and Orphan Forest Tree Species: How do They See Each Other? International Journal of Molecular Sciences. 2019; 20(3):692. https://doi.org/10.3390/ijms20030692
Chicago/Turabian StyleRey, María-Dolores, María Ángeles Castillejo, Rosa Sánchez-Lucas, Victor M. Guerrero-Sanchez, Cristina López-Hidalgo, Cristina Romero-Rodríguez, José Valero-Galván, Besma Sghaier-Hammami, Lyudmila Simova-Stoilova, Sira Echevarría-Zomeño, and et al. 2019. "Proteomics, Holm Oak (Quercus ilex L.) and Other Recalcitrant and Orphan Forest Tree Species: How do They See Each Other?" International Journal of Molecular Sciences 20, no. 3: 692. https://doi.org/10.3390/ijms20030692
APA StyleRey, M.-D., Castillejo, M. Á., Sánchez-Lucas, R., Guerrero-Sanchez, V. M., López-Hidalgo, C., Romero-Rodríguez, C., Valero-Galván, J., Sghaier-Hammami, B., Simova-Stoilova, L., Echevarría-Zomeño, S., Jorge, I., Gómez-Gálvez, I., Papa, M. E., Carvalho, K., Rodríguez de Francisco, L. E., Maldonado-Alconada, A. M., Valledor, L., & Jorrín-Novo, J. V. (2019). Proteomics, Holm Oak (Quercus ilex L.) and Other Recalcitrant and Orphan Forest Tree Species: How do They See Each Other? International Journal of Molecular Sciences, 20(3), 692. https://doi.org/10.3390/ijms20030692