Curbing Lipids: Impacts ON Cancer and Viral Infection



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Cancers and Infection Related Cancers
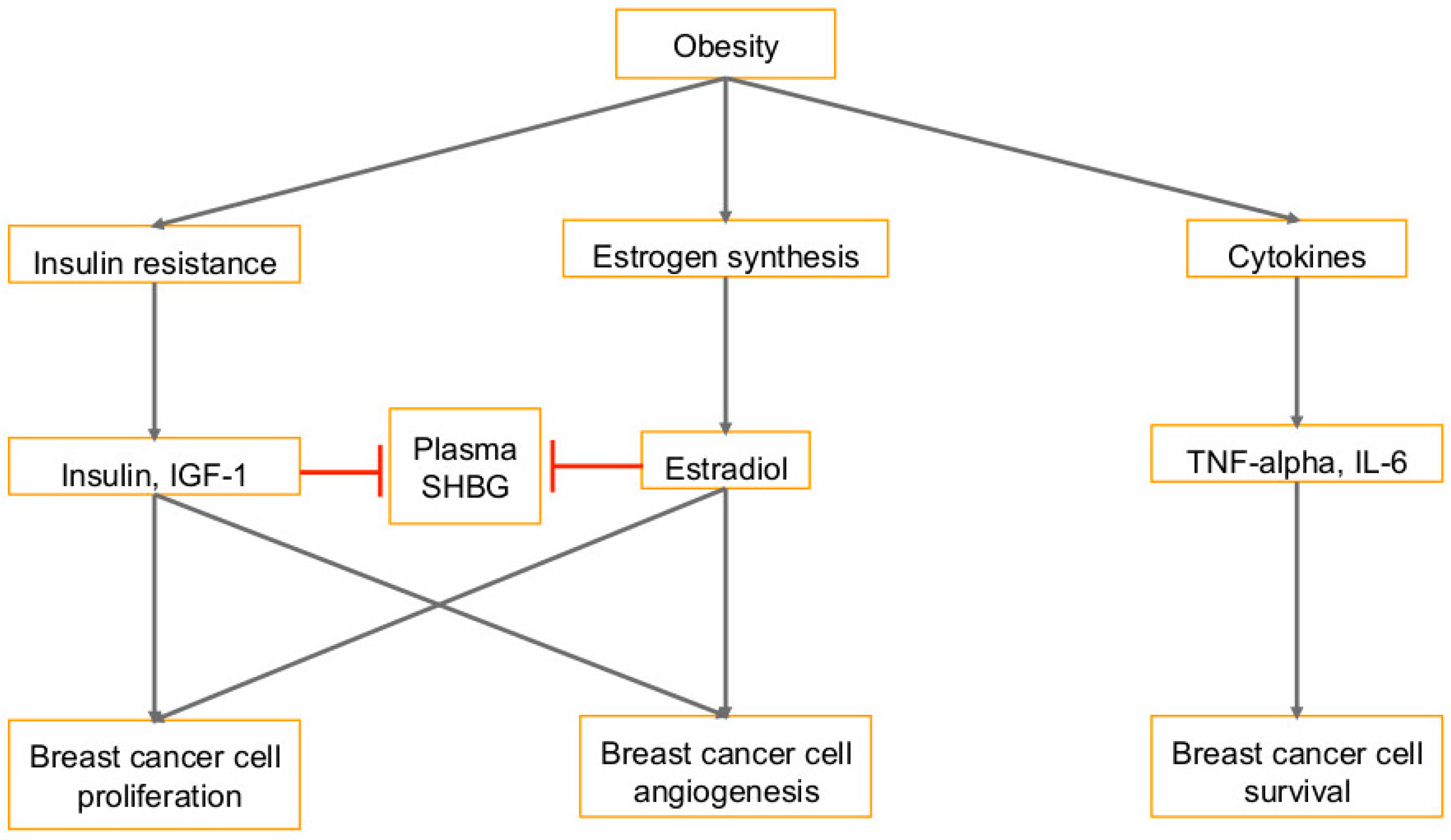
1.2. Diet and Obesity in Cancer
2. Lipid Synthesis in Human Cancers and Viral Infection Linked Cancers
2.1. Regulation of Lipids in Membrane Microdomains
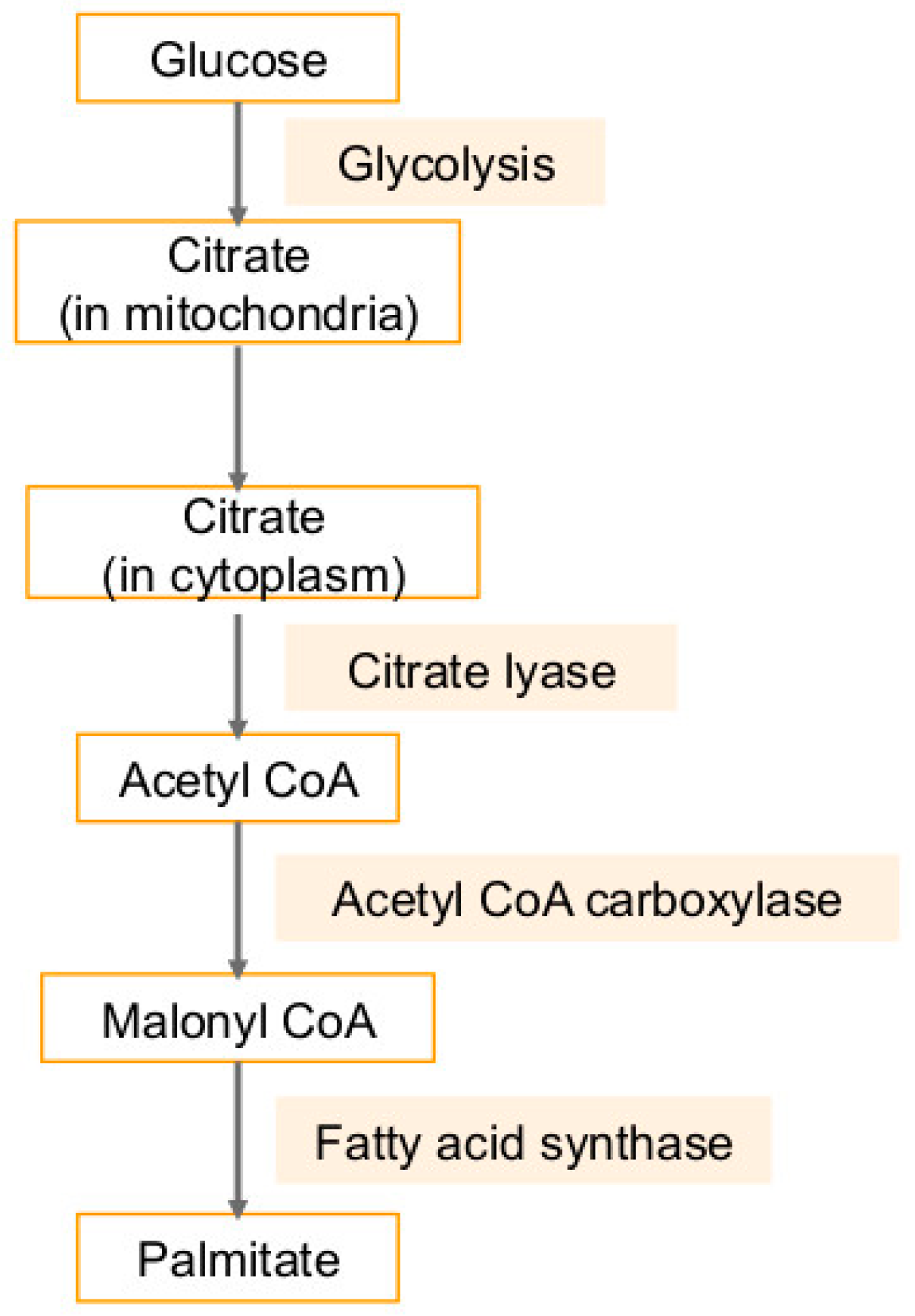
2.2. Association of Lipid Pathways with Glycolysis, Fatty Acid Synthesis, and Glutaminolysis
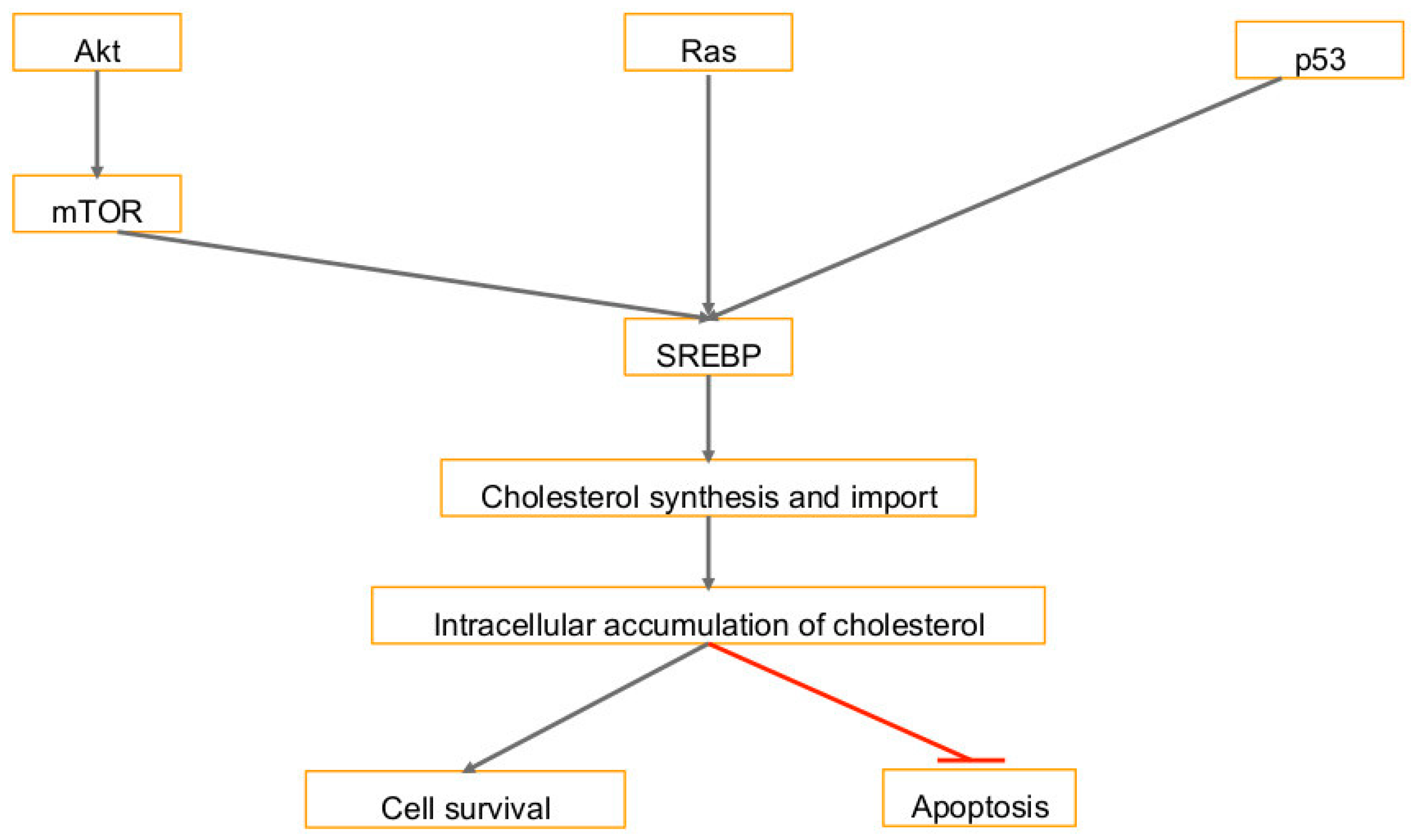
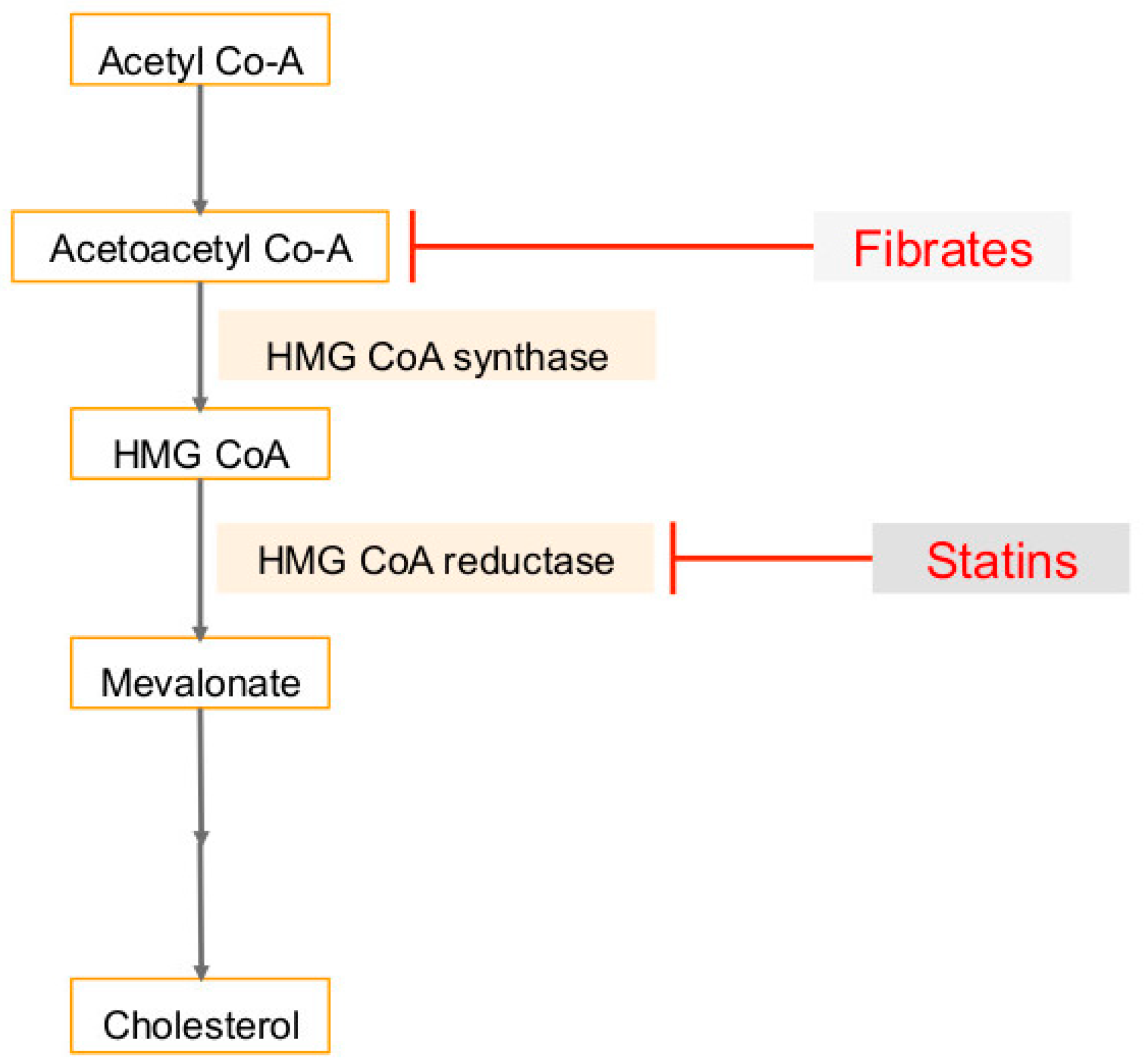
3. Cholesterol Synthesis in Human Cancers and Viral Infection Linked Cancers
4. Fatty Acid Synthase (FASN) in Cancers and Viral Infection-Associated Cancers
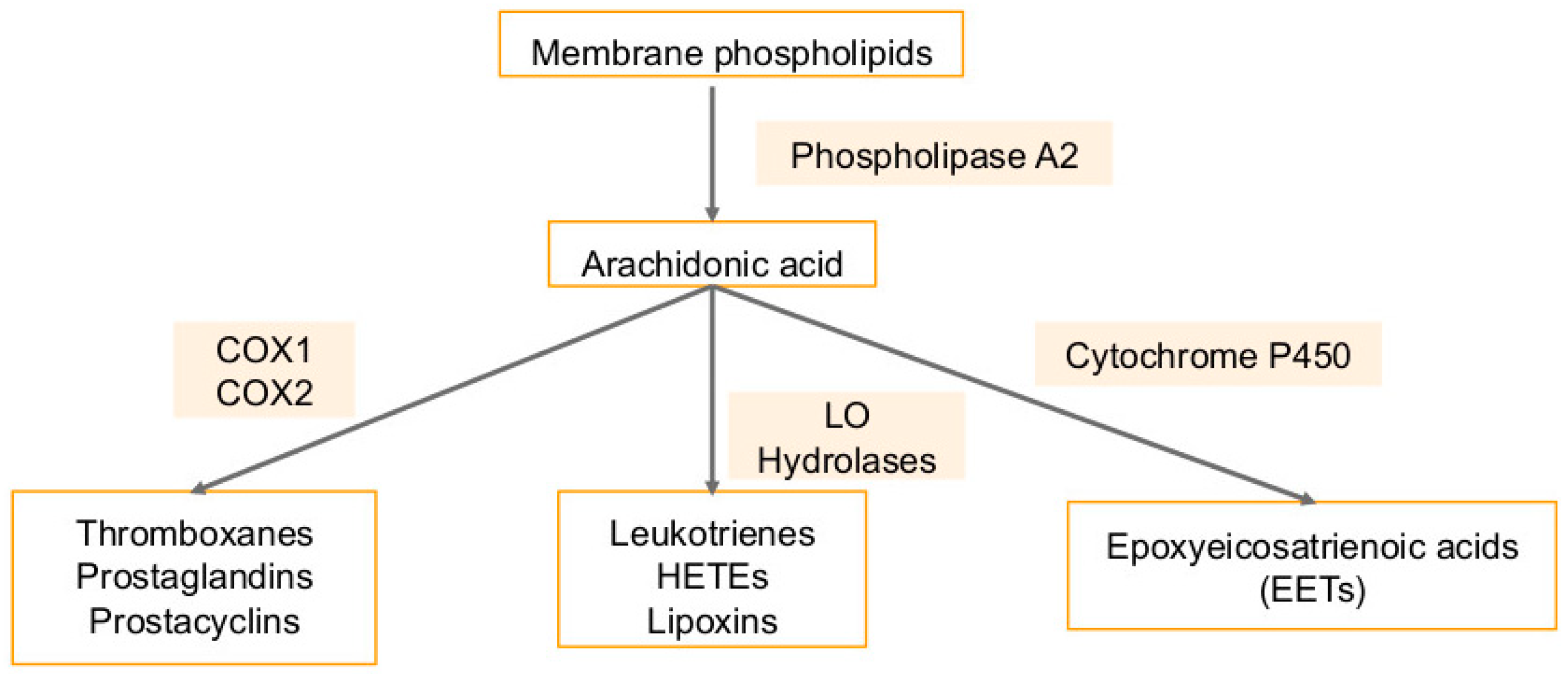
5. Arachidonic Acid Pathway Metabolites in Cancers and Viral Infection-Associated Cancers
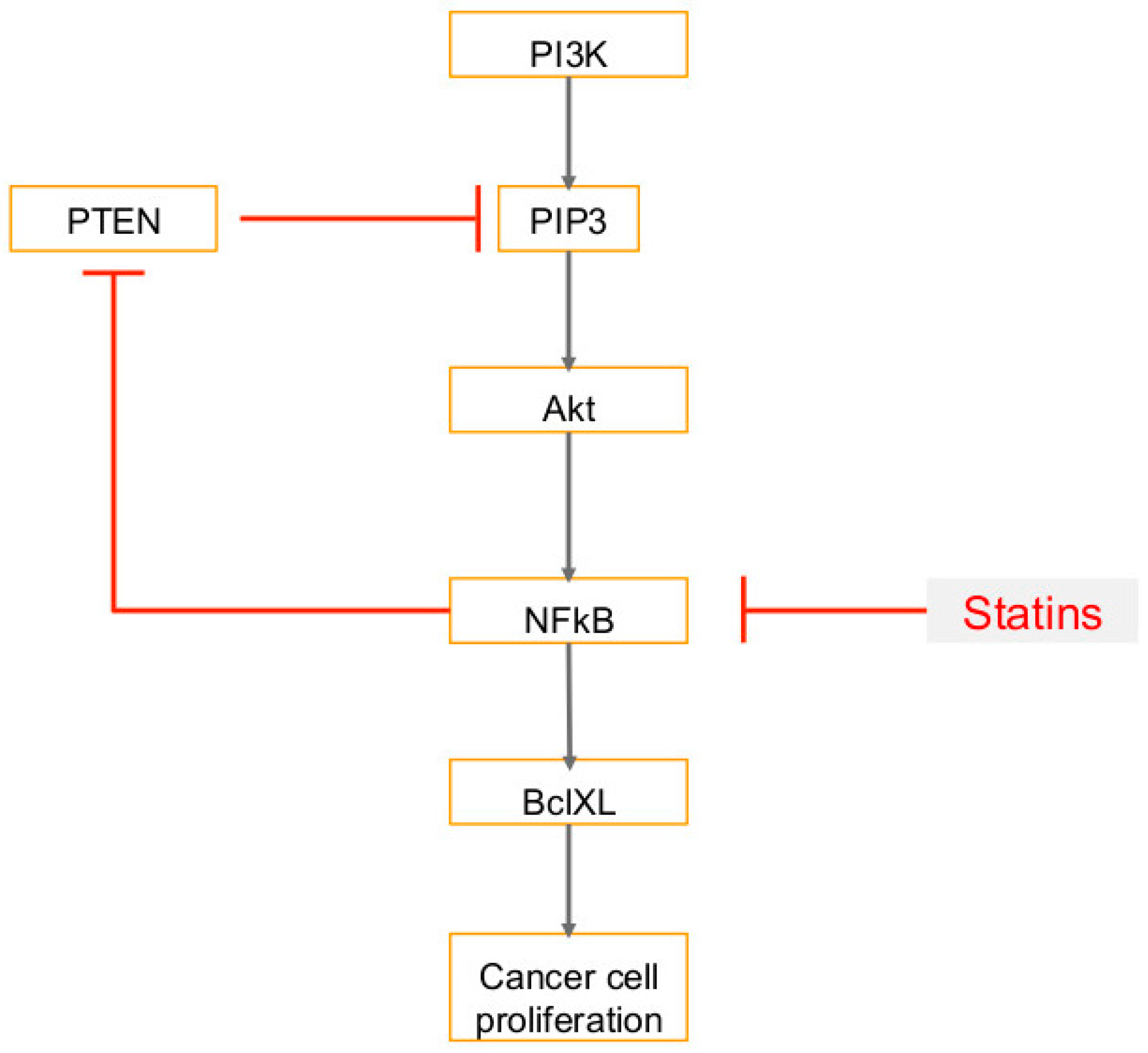
6. Use of Statins in Cancers, Viral Infections and Associated Cancers
7. Use of Fibrates (PPARα Agonist) in Cancers, Viral Infections and Associated Cancers
8. Other Lipid Metabolites and Pathways in Cancer and Viral Infections
9. Summary
Funding
Conflicts of Interest
Abbreviations
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| MCPyV | Merkel cell polyomavirus—a polyoma virus |
| MCC | Merkel cell carcinoma |
| EBV | Epstein–Barr virus |
| HPV | human papilloma virus |
| HTLV | human T lymphotropic virus |
| cART | combination of anti-retroviral therapy |
| IGF-1 | insulin-like growth factor 1 |
| SHBG | sex-hormone binding globulin |
| BARD1 | BRCA1-associated RING domain protein 1 |
| TNFα | tumor necrosis factor-alpha |
| MAPK | mitogen activated protein kinase |
| HMEC | human mammary epithelial cells |
| OPG | osteoprotegerin |
| SREBF2 | Sterol regulatory element-binding protein transcription factor 2 |
| LXR | Liver x receptors |
| PPARs | Peroxisome proliferator—activated receptors |
| HCC | hepatocellular carcinoma |
| LKB1 | liver kinase B1 |
| HMG-CoA synthase | 3-hydroxy-3-methylglutaryl-coenzyme A synthase |
| MCM | minichromosome maintenance |
| PTEN | phosphatase and tensin homolog |
| LTB4 | leukotriene B4 |
| MMPs | matrix metalloproteinases |
| PLD | Phospholipase D |
| ACLY | ATP citrate lyase |
| VEGF | vascular endothelial growth factor |
| FGF2 | fibroblast growth factor 2 |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| EETs | epoxyeicosatrienoic acid |
| STAR | steroidogenic acute regulatory |
| FASN | fatty acid synthase |
| ABCA1 | ATP-binding cassette transporter |
| ACSL1 | Acyl-CoA Synthetase Long Chain Family Member 1 |
| AGPAT1 | 1-Acylglycerol-3-Phosphate O-Acyltransferase 1 |
| SCD | Stearoyl-CoA desaturase (Δ-9-desaturase) |
| RAC | Rectal adenocarcinoma |
| PDAC | Pancreatic ductal adenocarcinoma |
| MUFAs | mono-unsaturated fatty acids |
| SCFA | Short-chain fatty acids |
| PI3K | phosphatidylinositol 3-kinase |
| HIFs | hypoxia inducible factors |
| ESCRT | endosomal sorting complexes required for transport |
| ELOVL6 | fatty acid elongase |
| CPT | Carnitine palmitoyltransferase. |
References
- Zali, H.; Rezaei-Tavirani, M.; Azodi, M. Gastric cancer: Prevention, risk factors and treatment. Gastroenterol. Hepatol. Bed Bench 2011, 4, 175–185. [Google Scholar] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin. Chest Med. 2011, 32, 605–644. [Google Scholar] [CrossRef] [PubMed]
- Ahern, T.P.; Pedersen, L.; Tarp, M.; Cronin-Fenton, D.P.; Garne, J.P.; Silliman, R.A.; Sorensen, H.T.; Lash, T.L. Statin prescriptions and breast cancer recurrence risk: A Danish nationwide prospective cohort study. J. Natl. Cancer Inst. 2011, 103, 1461–1468. [Google Scholar] [CrossRef] [PubMed]
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.C.; Jiang, J.Y.; Goggins, W.B.; Liang, M.; Fang, Y.; Fung, F.D.; Leung, C.; Wang, H.H.; Wong, G.L.; Wong, V.W.; et al. International incidence and mortality trends of liver cancer: A global profile. Sci. Rep. 2017, 7, 45846. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.B. Viruses and human cancer. Yale J. Biol. Med. 2006, 79, 115–122. [Google Scholar]
- Piccaluga, P.P.; Weber, A.; Ambrosio, M.R.; Ahmed, Y.; Leoncini, L. Epstein-Barr Virus-Induced Metabolic Rearrangements in Human B-Cell Lymphomas. Front. Microbiol. 2018, 9, 1233. [Google Scholar] [CrossRef]
- Stanfield, B.A.; Luftig, M.A. Recent advances in understanding Epstein-Barr virus. F1000Res 2017, 6, 386. [Google Scholar] [CrossRef]
- Cohen, J.I. Epstein-barr virus vaccines. Clin. Transl. Immunol. 2015, 4, e32. [Google Scholar] [CrossRef]
- Wu, T.T.; Qian, J.; Ang, J.; Sun, R. Vaccine prospect of Kaposi sarcoma-associated herpesvirus. Curr. Opin. Virol. 2012, 2, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Lin, Z.; Jiang, W.; Flemington, E.K.; Qin, Z. Lipids, lipid metabolism and Kaposi’s sarcoma-associated herpesvirus pathogenesis. Virol. Sin. 2017, 32, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.H.; Hearps, A.C.; Martin, G.E.; Williams, K.C.; Crowe, S.M. The importance of monocytes and macrophages in HIV pathogenesis, treatment, and cure. AIDS 2014, 28, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—An update. Curr. Opin. Virol. 2013, 3, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Mosam, A.; Shaik, F.; Uldrick, T.S.; Esterhuizen, T.; Friedland, G.H.; Scadden, D.T.; Aboobaker, J.; Coovadia, H.M. A randomized controlled trial of highly active antiretroviral therapy versus highly active antiretroviral therapy and chemotherapy in therapy-naive patients with HIV-associated Kaposi sarcoma in South Africa. J. Acquir. Immune Defic. Syndr. 2012, 60, 150–157. [Google Scholar] [CrossRef]
- Letang, E.; Almeida, J.M.; Miro, J.M.; Ayala, E.; White, I.E.; Carrilho, C.; Bastos, R.; Nhampossa, T.; Menendez, C.; Campbell, T.B.; et al. Predictors of immune reconstitution inflammatory syndrome-associated with kaposi sarcoma in mozambique: A prospective study. J. Acquir. Immune Defic. Syndr. 2010, 53, 589–597. [Google Scholar] [CrossRef]
- Borok, M.; Fiorillo, S.; Gudza, I.; Putnam, B.; Ndemera, B.; White, I.E.; Gwanzura, L.; Schooley, R.T.; Campbell, T.B. Evaluation of plasma human herpesvirus 8 DNA as a marker of clinical outcomes during antiretroviral therapy for AIDS-related Kaposi sarcoma in Zimbabwe. Clin. Infect. Dis. 2010, 51, 342–349. [Google Scholar] [CrossRef]
- MacLachlan, J.H.; Cowie, B.C. Hepatitis B virus epidemiology. Cold Spring Harb. Perspect. Med. 2015, 5, a021410. [Google Scholar] [CrossRef]
- Lorizate, M.; Krausslich, H.G. Role of lipids in virus replication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004820. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; McPherson, M.; Gail Darlington, L. Obesity and Cancer: Existing and New Hypotheses for a Causal Connection. EBioMedicine 2018, 30, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Llaverias, G.; Danilo, C.; Mercier, I.; Daumer, K.; Capozza, F.; Williams, T.M.; Sotgia, F.; Lisanti, M.P.; Frank, P.G. Role of cholesterol in the development and progression of breast cancer. Am. J. Pathol. 2011, 178, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, A.M.; Sukumar, S. Molecular links between obesity and breast cancer. Endocr. Relat. Cancer 2006, 13, 279–292. [Google Scholar] [CrossRef]
- Bianchini, F.; Kaaks, R.; Vainio, H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002, 3, 565–574. [Google Scholar] [CrossRef]
- Bagga, D.; Wang, L.; Farias-Eisner, R.; Glaspy, J.A.; Reddy, S.T. Differential effects of prostaglandin derived from omega-6 and omega-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc. Natl. Acad. Sci. USA 2003, 100, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Polyunsaturated fatty acids and inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2006, 75, 197–202. [Google Scholar] [CrossRef]
- Calder, P.C. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006, 83, 1505S–1519S. [Google Scholar] [CrossRef]
- Goswami, S.; Sharma-Walia, N. Osteoprotegerin secreted by inflammatory and invasive breast cancer cells induces aneuploidy, cell proliferation and angiogenesis. BMC Cancer 2015, 15, 935. [Google Scholar] [CrossRef]
- Goswami, S.; Sharma-Walia, N. Crosstalk between osteoprotegerin (OPG), fatty acid synthase (FASN) and, cycloxygenase-2 (COX-2) in breast cancer: Implications in carcinogenesis. Oncotarget 2016, 7, 58953–58974. [Google Scholar] [CrossRef]
- Goswami, S.; Sharma-Walia, N. Osteoprotegerin rich tumor microenvironment: Implications in breast cancer. Oncotarget 2016, 7, 42777–42791. [Google Scholar] [CrossRef] [PubMed]
- Suburu, J.; Shi, L.; Wu, J.; Wang, S.; Samuel, M.; Thomas, M.J.; Kock, N.D.; Yang, G.; Kridel, S.; Chen, Y.Q. Fatty acid synthase is required for mammary gland development and milk production during lactation. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1132–E1143. [Google Scholar] [CrossRef] [PubMed]
- Dal Maso, L.; Zucchetto, A.; Talamini, R.; Serraino, D.; Stocco, C.F.; Vercelli, M.; Falcini, F.; Franceschi, S. Prospective Analysis of Case-control studies on Environmental, f.; health study, g. Effect of obesity and other lifestyle factors on mortality in women with breast cancer. Int. J. Cancer 2008, 123, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, J.; Zhou, Y.; Qiao, L. Obesity and gastric cancer. Front. Biosci. 2012, 17, 2383–2390. [Google Scholar] [CrossRef]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Model. Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Zalba, S.; Ten Hagen, T.L. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat. Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Delmas, P.; Coste, B.; Gamper, N.; Shapiro, M.S. Phosphoinositide lipid second messengers: New paradigms for calcium channel modulation. Neuron 2005, 47, 179–182. [Google Scholar] [CrossRef]
- Moolenaar, W.H. Lysophosphatidic acid, a multifunctional phospholipid messenger. J. Biol. Chem. 1995, 270, 12949–12952. [Google Scholar] [CrossRef]
- Wang, J.; Lv, X.W.; Shi, J.P.; Hu, X.S. Mechanisms involved in ceramide-induced cell cycle arrest in human hepatocarcinoma cells. World J. Gastroenterol. 2007, 13, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Guenther, G.G.; Peralta, E.R.; Rosales, K.R.; Wong, S.Y.; Siskind, L.J.; Edinger, A.L. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 17402–17407. [Google Scholar] [CrossRef] [PubMed]
- Watters, R.J.; Wang, H.G.; Sung, S.S.; Loughran, T.P.; Liu, X. Targeting sphingosine-1-phosphate receptors in cancer. Anticancer Agents Med. Chem. 2011, 11, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Long, J.S.; Fujiwara, Y.; Edwards, J.; Tannahill, C.L.; Tigyi, G.; Pyne, S.; Pyne, N.J. Sphingosine 1-phosphate receptor 4 uses HER2 (ERBB2) to regulate extracellular signal regulated kinase-1/2 in MDA-MB-453 breast cancer cells. J. Biol. Chem. 2010, 285, 35957–35966. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.; Long, J.S.; Orange, C.; Tannahill, C.L.; Mallon, E.; McGlynn, L.M.; Pyne, S.; Pyne, N.J.; Edwards, J. High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am. J. Pathol. 2010, 177, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 2008, 9, 162–176. [Google Scholar] [CrossRef]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef]
- Robbins, S.M.; Quintrell, N.A.; Bishop, J.M. Myristoylation and differential palmitoylation of the HCK protein-tyrosine kinases govern their attachment to membranes and association with caveolae. Mol. Cell. Biol. 1995, 15, 3507–3515. [Google Scholar] [CrossRef]
- Resh, M.D. Fatty acylation of proteins: New insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Et Biophys. Acta 1999, 1451, 1–16. [Google Scholar] [CrossRef]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Et Biophys. Acta 2016, 1864, 1372–1401. [Google Scholar] [CrossRef]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E. Structural Basis for Regulation of Phosphoinositide Kinases and Their Involvement in Human Disease. Mol. Cell 2018, 71, 653–673. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, Y.; Ren, J. Autophagic Regulation of Lipid Homeostasis in Cardiometabolic Syndrome. Front. Cardiovasc. Med. 2018, 5, 38. [Google Scholar] [CrossRef]
- Fernandez, L.P.; Ramos-Ruiz, R.; Herranz, J.; Martin-Hernandez, R.; Vargas, T.; Mendiola, M.; Guerra, L.; Reglero, G.; Feliu, J.; Ramirez de Molina, A. The transcriptional and mutational landscapes of lipid metabolism-related genes in colon cancer. Oncotarget 2018, 9, 5919–5930. [Google Scholar] [CrossRef] [PubMed]
- Fernandes Messias, M.C.; Mecatti, G.C.; Figueiredo Angolini, C.F.; Eberlin, M.N.; Credidio, L.; Real Martinez, C.A.; Rodrigues Coy, C.S.; de Oliveira Carvalho, P. Plasma Lipidomic Signature of Rectal Adenocarcinoma Reveals Potential Biomarkers. Front. Oncol. 2017, 7, 325. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers (Basel) 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef]
- Popescu, C.I.; Riva, L.; Vlaicu, O.; Farhat, R.; Rouille, Y.; Dubuisson, J. Hepatitis C virus life cycle and lipid metabolism. Biology (Basel) 2014, 3, 892–921. [Google Scholar] [CrossRef]
- Lo, A.K.; Lung, R.W.; Dawson, C.W.; Young, L.S.; Ko, C.W.; Yeung, W.W.; Kang, W.; To, K.F.; Lo, K.W. Activation of SREBP1-mediated lipogenesis by the Epstein-Barr Virus-encoded LMP1 promotes cell proliferation and progression of nasopharyngeal carcinoma. J. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gorres, K.L.; Daigle, D.; Mohanram, S.; Miller, G. Activation and repression of Epstein-Barr Virus and Kaposi’s sarcoma-associated herpesvirus lytic cycles by short- and medium-chain fatty acids. J. Virol. 2014, 88, 8028–8044. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr.; Menaker, N.F.; Raab-Traub, N. Epstein-Barr virus LMP1 modulates lipid raft microdomains and the vimentin cytoskeleton for signal transduction and transformation. J. Virol. 2013, 87, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.D.; Gadelha, S.R.; Marin, L.J.; Brito-Melo, G.E.; Martins, C.P.; Fonseca, F.G.; Barbosa-Stancioli, E.F. Are lipid disorders involved in the predominance of human T-lymphotropic virus-1 infections in women? Rev. Soc. Bras. Med. Trop. 2015, 48, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, H. Cellular Factors Involved in HTLV-1 Entry and Pathogenicit. Front. Microbiol. 2012, 3, 222. [Google Scholar] [CrossRef] [PubMed]
- Currer, R.; Van Duyne, R.; Jaworski, E.; Guendel, I.; Sampey, G.; Das, R.; Narayanan, A.; Kashanchi, F. HTLV tax: A fascinating multifunctional co-regulator of viral and cellular pathways. Front. Microbiol. 2012, 3, 406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, L.; Cai, S.H.; Cheng, H. Identification of TBK1 and IKKepsilon, the non-canonical IκB kinases, as crucial pro-survival factors in HTLV-1-transformed T lymphocytes. Leukemia Res. 2016, 46, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Takahashi, Y.; Liu, X.; Loughran, T.P.; Sun, S.C.; Wang, H.G.; Cheng, H. HTLV-1 Tax deregulates autophagy by recruiting autophagic molecules into lipid raft microdomains. Oncogene 2015, 34, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ren, T.; Guan, H.; Jiang, Y.; Cheng, H. HTLV-1 Tax is a critical lipid raft modulator that hijacks IκB kinases to the microdomains for persistent activation of NF-κB. J. Biol. Chem. 2009, 284, 6208–6217. [Google Scholar] [CrossRef] [PubMed]
- Tso, F.Y.; Kossenkov, A.V.; Lidenge, S.J.; Ngalamika, O.; Ngowi, J.R.; Mwaiselage, J.; Wickramasinghe, J.; Kwon, E.H.; West, J.T.; Lieberman, P.M.; et al. RNA-Seq of Kaposi’s sarcoma reveals alterations in glucose and lipid metabolism. PLoS Pathog. 2018, 14, e1006844. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.L.; Pulliam, T.H.; Dimaio, T.A.; Thalhofer, A.B.; Delgado, T.; Lagunoff, M. Glycolysis, Glutaminolysis, and Fatty Acid Synthesis Are Required for Distinct Stages of Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Veettil, M.V.; Kumar, B.; Ansari, M.A.; Dutta, D.; Iqbal, J.; Gjyshi, O.; Bottero, V.; Chandran, B. ESCRT-0 Component Hrs Promotes Macropinocytosis of Kaposi’s Sarcoma-Associated Herpesvirus in Human Dermal Microvascular Endothelial Cells. J. Virol. 2016, 90, 3860–3872. [Google Scholar] [CrossRef] [PubMed]
- Raghu, H.; Sharma-Walia, N.; Veettil, M.V.; Sadagopan, S.; Caballero, A.; Sivakumar, R.; Varga, L.; Bottero, V.; Chandran, B. Lipid rafts of primary endothelial cells are essential for Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8-induced phosphatidylinositol 3-kinase and RhoA-GTPases critical for microtubule dynamics and nuclear delivery of viral DNA but dispensable for binding and entry. J. Virol. 2007, 81, 7941–7959. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, N.; Li, W.; Zhu, F.; Wang, Y.; Yuan, Y. Mono-ubiquitylated ORF45 Mediates Association of KSHV Particles with Internal Lipid Rafts for Viral Assembly and Egress. PLoS Pathog. 2015, 11, e1005332. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.P.; Jacobs, S.R.; Freemerman, A.J.; Makowski, L.; Rathmell, J.C.; Dittmer, D.P.; Damania, B. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA. 2012, 109, 11818–11823. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.P.; Damania, B. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front. Immunol. 2012, 3, 401. [Google Scholar] [CrossRef] [PubMed]
- Angius, F.; Uda, S.; Piras, E.; Spolitu, S.; Ingianni, A.; Batetta, B.; Pompei, R. Neutral lipid alterations in human herpesvirus 8-infected HUVEC cells and their possible involvement in neo-angiogenesis. BMC Microbiol. 2015, 15, 74. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Lang, F.; Pei, Y.; Jha, H.C.; Robertson, E.S. Metabolic reprogramming of Kaposi’s sarcoma associated herpes virus infected B-cells in hypoxia. PLoS Pathog. 2018, 14, e1007062. [Google Scholar] [CrossRef]
- Huang, C.; Freter, C. Lipid metabolism, apoptosis and cancer therapy. Int. J. Mol. Sci. 2015, 16, 924–949. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Park, M.J.; Ye, S.K.; Kim, C.W.; Kim, Y.N. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am. J. Pathol. 2006, 168, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.A.; Ito, Y.; Morimoto, J.; Otsuki, Y. Lovastatin inhibits tumor growth and lung metastasis in mouse mammary carcinoma model: A p53-independent mitochondrial-mediated apoptotic mechanism. Carcinogenesis 2004, 25, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Ye, J. Reliance of host cholesterol metabolic pathways for the life cycle of hepatitis C virus. PLoS Pathog. 2007, 3, e108. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Longnecker, R. Cholesterol is critical for Epstein-Barr virus latent membrane protein 2A trafficking and protein stability. Virology 2007, 360, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Pandyra, A.A.; Mullen, P.J.; Goard, C.A.; Ericson, E.; Sharma, P.; Kalkat, M.; Yu, R.; Pong, J.T.; Brown, K.R.; Hart, T.; et al. Genome-wide RNAi analysis reveals that simultaneous inhibition of specific mevalonate pathway genes potentiates tumor cell death. Oncotarget 2015, 6, 26909–26921. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.E.; El-Sohemy, A.; Archer, M.C. Mevalonate promotes the growth of tumors derived from human cancer cells in vivo and stimulates proliferation in vitro with enhanced cyclin-dependent kinase-2 activity. J. Biol. Chem. 2004, 279, 33079–33084. [Google Scholar] [CrossRef] [PubMed]
- Serquina, A.K.P.; Kambach, D.M.; Sarker, O.; Ziegelbauer, J.M. Viral MicroRNAs Repress the Cholesterol Pathway, and 25-Hydroxycholesterol Inhibits Infection. mBio 2017, 8. [Google Scholar] [CrossRef]
- Alvisi, G.; Madan, V.; Bartenschlager, R. Hepatitis C virus and host cell lipids: an intimate connection. RNA Biol. 2011, 8, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Katano, H.; Pesnicak, L.; Cohen, J.I. Simvastatin induces apoptosis of Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines and delays development of EBV lymphomas. Proc. Natl. Acad. Sci. USA 2004, 101, 4960–4965. [Google Scholar] [CrossRef] [PubMed]
- Berquin, I.M.; Edwards, I.J.; Kridel, S.J.; Chen, Y.Q. Polyunsaturated fatty acid metabolism in prostate cancer. Cancer Metast. Rev. 2011, 30, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.Y.; Kim, W.Y. Stearoyl co-A desaturase 1 as a ccRCC therapeutic target: Death by stress. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3111–3113. [Google Scholar] [CrossRef] [PubMed]
- Fenner, A. Kidney cancer: Stearoyl-CoA desaturase: A novel therapeutic target for RCC. Nat. Rev. Urol. 2013, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2368–2380. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Tomita, N.; Kawakita, Y.; Ito, Y.; Ono, K.; Nii, N.; Miyazaki, T.; Yonemori, K.; Tawada, M.; Sumi, H.; et al. Discovery of Novel and Potent Stearoyl Coenzyme A Desaturase 1 (SCD1) Inhibitors as Anticancer Agents. Bioorg. Med. Chem. 2017, 25, 3768–3779. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, S.; Sumi, H.; Satoh, Y.; Yamamoto, Y.; Kitazawa, S.; Honda, K.; Araki, H.; Kakoi, K.; Imamura, K.; Sasaki, M.; et al. In vitro and in vivo antitumor activities of T-3764518, a novel and orally available small molecule stearoyl-CoA desaturase 1 inhibitor. Eur. J. Pharm. 2017, 807, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Goswami, S.; Sharma-Walia, N. Implications of a peroxisome proliferator-activated receptor alpha (PPARalpha) ligand clofibrate in breast cancer. Oncotarget 2016, 7, 15577–15599. [Google Scholar] [CrossRef]
- Kuhajda, F.P. Fatty acid synthase and cancer: New application of an old pathway. Cancer Res. 2006, 66, 5977–5980. [Google Scholar] [CrossRef]
- Kuhajda, F.P. Fatty-acid synthase and human cancer: New perspectives on its role in tumor biology. Nutrition 2000, 16, 202–208. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Zhan, R.; Wang, Y.; Pai, S.K.; Hirota, S.; Hosobe, S.; Takano, Y.; Saito, K.; Furuta, E.; Iiizumi, M.; et al. Mechanism of apoptosis induced by the inhibition of fatty acid synthase in breast cancer cells. Cancer Res. 2006, 66, 5934–5940. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, Y.Y.; Rychahou, P.G.; Le, A.T.; Scott, T.L.; Flight, R.M.; Kim, J.T.; Harris, J.; Liu, J.; Wang, C.; Morris, A.J.; et al. Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer. Oncotarget 2018, 9, 24787–24800. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.X.; Huang, C.J.; Yang, Z.G. Impact of hepatitis B virus infection on hepatic metabolic signaling pathway. World J. Gastroenterol. 2016, 22, 8161–8167. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Webster-Cyriaque, J.; Tomlinson, C.C.; Yohe, M.; Kenney, S. Fatty acid synthase expression is induced by the Epstein-Barr virus immediate-early protein BRLF1 and is required for lytic viral gene expression. J. Virol. 2004, 78, 4197–4206. [Google Scholar] [CrossRef] [PubMed]
- Sychev, Z.E.; Hu, A.; DiMaio, T.A.; Gitter, A.; Camp, N.D.; Noble, W.S.; Wolf-Yadlin, A.; Lagunoff, M. Integrated systems biology analysis of KSHV latent infection reveals viral induction and reliance on peroxisome mediated lipid metabolism. PLoS Pathog. 2017, 13, e1006256. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Qiu, J.; Li, Q.; Shi, Z. Prostaglandin E2 Signaling: Alternative Target for Glioblastoma? Trends Cancer 2017, 3, 75–78. [Google Scholar] [CrossRef]
- Larsson, K.; Jakobsson, P.J. Inhibition of microsomal prostaglandin E synthase-1 as targeted therapy in cancer treatment. Prostaglandins Other Lipid Mediat. 2015, 120, 161–165. [Google Scholar] [CrossRef]
- Harris, R.E. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology 2009, 17, 55–67. [Google Scholar] [CrossRef]
- Nuvoli, B.; Galati, R. Cyclooxygenase-2, epidermal growth factor receptor, and aromatase signaling in inflammation and mesothelioma. Mol. Cancer 2013, 12, 844–852. [Google Scholar] [CrossRef]
- Fleming, I. Vascular cytochrome p450 enzymes: Physiology and pathophysiology. Trends Cardiovasc. Med. 2008, 18, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Rand, A.A.; Barnych, B.; Morisseau, C.; Cajka, T.; Lee, K.S.S.; Panigrahy, D.; Hammock, B.D. Cyclooxygenase-derived proangiogenic metabolites of epoxyeicosatrienoic acids. Proc. Natl. Acad. Sci. USA 2017, 114, 4370–4375. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R. Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Res. 2007, 9, 210. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, D.; Ang, R.; Waxman, J. COX inhibitors and breast cancer. Br. J. Cancer 2006, 94, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.G.; Chandran, B.; Sharma-Walia, N. Cyclooxygenase-2-prostaglandin E2-eicosanoid receptor inflammatory axis: A key player in Kaposi’s sarcoma-associated herpes virus associated malignancies. Transl. Res. 2013, 162, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Sharma-Walia, N.; Chandran, K.; Patel, K.; Veettil, M.V.; Marginean, A. The Kaposi’s sarcoma-associated herpesvirus (KSHV)-induced 5-lipoxygenase-leukotriene B4 cascade plays key roles in KSHV latency, monocyte recruitment, and lipogenesis. J. Virol. 2014, 88, 2131–2156. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, H.; Ma, A.H.; Yu, W.; Zimmermann, M.; Yang, J.; Hwang, S.H.; Zhu, D.; Lin, T.Y.; Malfatti, M.; et al. COX-2/sEH Dual Inhibitor PTUPB Potentiates the Antitumor Efficacy of Cisplatin. Mol. Cancer 2018, 17, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Goulitquer, S.; Croyal, M.; Lalande, J.; Royer, A.L.; Guitton, Y.; Arzur, D.; Durand, S.; Le Jossic-Corcos, C.; Bouchereau, A.; Potin, P.; et al. Consequences of blunting the mevalonate pathway in cancer identified by a pluri-omics approach. Cell Death Dis. 2018, 9, 745. [Google Scholar] [CrossRef]
- Hajar, R. Statins: Past and present. Heart Views Off. J. Gulf Heart Assoc. 2011, 12, 121–127. [Google Scholar] [CrossRef]
- Lee, J.S.; Roberts, A.; Juarez, D.; Vo, T.T.; Bhatt, S.; Herzog, L.O.; Mallya, S.; Bellin, R.J.; Agarwal, S.K.; Salem, A.H.; et al. Statins enhance efficacy of venetoclax in blood cancers. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Walther, U.; Emmrich, K.; Ramer, R.; Mittag, N.; Hinz, B. Lovastatin lactone elicits human lung cancer cell apoptosis via a COX-2/PPARgamma-dependent pathway. Oncotarget 2016, 7, 10345–10362. [Google Scholar] [CrossRef] [PubMed]
- Brewer, T.M.; Masuda, H.; Liu, D.D.; Shen, Y.; Liu, P.; Iwamoto, T.; Kai, K.; Barnett, C.M.; Woodward, W.A.; Reuben, J.M.; et al. Statin use in primary inflammatory breast cancer: A cohort study. Br. J. Cancer 2013, 109, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Takeda, T.; Kino, T.; Obata, N.; Itoh, T.; Imano, M.; Mashimo, K.; Fujiwara, D.; Sakaguchi, K.; Satou, T.; et al. Statins improve survival by inhibiting spontaneous metastasis and tumor growth in a mouse melanoma model. Am. J. Cancer Res. 2015, 5, 3186–3197. [Google Scholar] [PubMed]
- Karlic, H.; Thaler, R.; Gerner, C.; Grunt, T.; Proestling, K.; Haider, F.; Varga, F. Inhibition of the mevalonate pathway affects epigenetic regulation in cancer cells. Cancer Genet. 2015, 208, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Liao, W.C.; Lin, H.J.; Hsu, Y.M.; Lin, C.L.; Chen, Y.A.; Feng, C.L.; Chen, C.J.; Kao, M.C.; Lai, C.H.; et al. Statins Attenuate Helicobacter pylori CagA Translocation and Reduce Incidence of Gastric Cancer: In Vitro and Population-Based Case-Control Studies. PLoS ONE 2016, 11, e0146432. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Zhu, G.Q.; Wang, Y.; Zheng, J.N.; Ruan, L.Y.; Cheng, Z.; Hu, B.; Fu, S.W.; Zheng, M.H. Systematic review with network meta-analysis: Statins and risk of hepatocellular carcinoma. Oncotarget 2016, 7, 21753–21762. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhury, N.; Mandal, C.C.; Ghosh-Choudhury, N.; Ghosh Choudhury, G. Simvastatin induces derepression of PTEN expression via NFkappaB to inhibit breast cancer cell growth. Cell Signal. 2010, 22, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, L.; Gong, Z.; Shen, L.; Kao, C.; Hock, J.M.; Sun, L.; Li, X. Lovastatin enhances adenovirus-mediated TRAIL induced apoptosis by depleting cholesterol of lipid rafts and affecting CAR and death receptor expression of prostate cancer cells. Oncotarget 2015, 6, 3055–3070. [Google Scholar] [CrossRef]
- Ahern, T.P.; Lash, T.L.; Damkier, P.; Christiansen, P.M.; Cronin-Fenton, D.P. Statins and breast cancer prognosis: Evidence and opportunities. Lancet Oncol. 2014, 15, e461–e468. [Google Scholar] [CrossRef]
- Lee, J.; Hong, E.M.; Jang, J.A.; Park, S.W.; Koh, D.H.; Choi, M.H.; Jang, H.J.; Kae, S.H. Simvastatin Induces Apoptosis and Suppresses Insulin-Like Growth Factor 1 Receptor in Bile Duct Cancer Cells. Gut Liver 2016, 10, 310–317. [Google Scholar] [CrossRef]
- Xie, F.; Liu, J.; Li, C.; Zhao, Y. Simvastatin blocks TGF-beta1-induced epithelial-mesenchymal transition in human prostate cancer cells. Oncol. Lett. 2016, 11, 3377–3383. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Seah, S.; Loh, X.; Chan, C.W.; Hartman, M.; Goh, B.C.; Lee, S.C. Simvastatin-induced breast cancer cell death and deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by metabolic products of the mevalonate pathway. Oncotarget 2016, 7, 2532–2544. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cao, F.; Li, J.; Wang, Z.; Ren, Y.; Liang, Z.; Liu, P. Simvastatin exerts anti-hepatitis B virus activity by inhibiting expression of minichromosome maintenance protein 7 in HepG2.2.15 cells. Mol. Med. Rep. 2016, 14, 5334–5342. [Google Scholar] [CrossRef] [PubMed]
- Tsan, Y.T.; Lee, C.H.; Wang, J.D.; Chen, P.C. Statins and the risk of hepatocellular carcinoma in patients with hepatitis B virus infection. J. Clin. Oncol. 2012, 30, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, E.; Armendariz, A.; Kim, K.; Heo, S.J.; Boffelli, D.; Tantisira, K.; Rotter, J.I.; Krauss, R.M.; Medina, M.W. Statin-induced changes in gene expression in EBV-transformed and native B-cells. Hum. Mol. Genet. 2014, 23, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. HMG CoA reductase inhibitors (statins) to treat Epstein-Barr virus-driven lymphoma. Br. J. Cancer 2005, 92, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.F.; Schaefer, E.A.; Maloof, N.; Skaff, A.; Berical, A.; Belon, C.A.; Heck, J.A.; Lin, W.; Frick, D.N.; Allen, T.M.; et al. Ceestatin, a novel small molecule inhibitor of hepatitis C virus replication, inhibits 3-hydroxy-3-methylglutaryl-coenzyme A synthase. J. Infect. Dis. 2011, 204, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.C.; Liu, Y.; Ye, Y.Y.; Hao, F.B.; Wang, K.; Gong, J.P. Meta-analysis of studies using statins as a reducer for primary liver cancer risk. Sci. Rep. 2016, 6, 26256. [Google Scholar] [CrossRef] [PubMed]
- Lalloyer, F.; Staels, B. Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 894–899. [Google Scholar] [CrossRef]
- Okopien, B.; Buldak, L.; Boldys, A. Fibrates in the management of atherogenic dyslipidemia. Expert Rev. Cardiovasc. Ther. 2017, 15, 913–921. [Google Scholar] [CrossRef]
- Lian, X.; Wang, G.; Zhou, H.; Zheng, Z.; Fu, Y.; Cai, L. Anticancer Properties of Fenofibrate: A Repurposing Use. J. Cancer 2018, 9, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Kaipainen, A.; Huang, S.; Butterfield, C.E.; Barnes, C.M.; Fannon, M.; Laforme, A.M.; Chaponis, D.M.; Folkman, J.; Kieran, M.W. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 985–990. [Google Scholar] [CrossRef]
- Moosmann, B.; Behl, C. Selenoprotein synthesis and side-effects of statins. Lancet 2004, 363, 892–894. [Google Scholar] [CrossRef]
- Miziorko, H.M. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch. Biochem. Biophys. 2011, 505, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Groux-Degroote, S.; Rodriguez-Walker, M.; Dewald, J.H.; Daniotti, J.L.; Delannoy, P. Gangliosides in Cancer Cell Signaling. Prog. Mol. Biol. Transl. Sci. 2018, 156, 197–227. [Google Scholar] [CrossRef] [PubMed]
- Ramenskaia, G.V.; Melnik, E.V.; Petukhov, A.E. Phospholipase D: Its role in metabolism processes and disease development. Biomed. Khim. 2018, 64, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.A.; Thomas, P.G.; Lindsley, C.W. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat. Rev. Drug Discov. 2017, 16, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Utter, M.; Chakraborty, S.; Goren, L.; Feuser, L.; Zhu, Y.S.; Foster, D.A. Elevated phospholipase D activity in androgen-insensitive prostate cancer cells promotes both survival and metastatic phenotypes. Cancer Lett. 2018, 423, 28–35. [Google Scholar] [CrossRef]
- Noble, A.R.; Maitland, N.J.; Berney, D.M.; Rumsby, M.G. Phospholipase D inhibitors reduce human prostate cancer cell proliferation and colony formation. Br. J. Cancer 2018, 118, 189–199. [Google Scholar] [CrossRef]
- Rabachini, T.; Boccardo, E.; Andrade, R.; Perez, K.R.; Nonogaki, S.; Cuccovia, I.M.; Villa, L.L. HPV-16 E7 expression up-regulates phospholipase D activity and promotes rapamycin resistance in a pRB-dependent manner. BMC Cancer 2018, 18, 485. [Google Scholar] [CrossRef]
- Wang, D.; Yin, L.; Wei, J.; Yang, Z.; Jiang, G. ATP citrate lyase is increased in human breast cancer, depletion of which promotes apoptosis. Tumour. Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Khwairakpam, A.D.; Shyamananda, M.S.; Sailo, B.L.; Rathnakaram, S.R.; Padmavathi, G.; Kotoky, J.; Kunnumakkara, A.B. ATP citrate lyase (ACLY): A promising target for cancer prevention and treatment. Curr. Drug Targets 2015, 16, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Torres, A.; Henry, R.A.; Trefely, S.; Wallace, M.; Lee, J.V.; Carrer, A.; Sengupta, A.; Campbell, S.L.; Kuo, Y.M.; et al. ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep. 2016, 17, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Lucenay, K.S.; Doostan, I.; Karakas, C.; Bui, T.; Ding, Z.; Mills, G.B.; Hunt, K.K.; Keyomarsi, K. Cyclin E Associates with the Lipogenic Enzyme ATP-Citrate Lyase to Enable Malignant Growth of Breast Cancer Cells. Cancer Res. 2016, 76, 2406–2418. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.V.; Berry, C.T.; Kim, K.; Sen, P.; Kim, T.; Carrer, A.; Trefely, S.; Zhao, S.; Fernandez, S.; Barney, L.E.; et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca(2+)-NFAT signaling. Genes Dev. 2018, 32, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.F.; Wu, H.C.; Hsieh, W.C.; Tsai, H.W.; Su, I.J. Activation of ATP citrate lyase by mTOR signal induces disturbed lipid metabolism in hepatitis B virus pre-S2 mutant tumorigenesis. J. Virol. 2015, 89, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Vysochan, A.; Sengupta, A.; Weljie, A.M.; Alwine, J.C.; Yu, Y. ACSS2-mediated acetyl-CoA synthesis from acetate is necessary for human cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 2017, 114, E1528–E1535. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef]
- Sansone, A.; Tolika, E.; Louka, M.; Sunda, V.; Deplano, S.; Melchiorre, M.; Anagnostopoulos, D.; Chatgilialoglu, C.; Formisano, C.; Di Micco, R.; et al. Hexadecenoic Fatty Acid Isomers in Human Blood Lipids and Their Relevance for the Interpretation of Lipidomic Profiles. PLoS ONE 2016, 11, e0152378. [Google Scholar] [CrossRef]
- Wang, X.; Lin, H.; Gu, Y. Multiple roles of dihomo-gamma-linolenic acid against proliferation diseases. Lipids Health Dis. 2012, 11, 25. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Brusselmans, K.; Verhoeven, G. Increased lipogenesis in cancer cells: New players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.G.; Sharma-Walia, N.; Chandran, B. Targeting KSHV/HHV-8 latency with COX-2 selective inhibitor nimesulide: A potential chemotherapeutic modality for primary effusion lymphoma. PLoS ONE 2011, 6, e24379. [Google Scholar] [CrossRef]
- Paul, A.G.; Chandran, B.; Sharma-Walia, N. Concurrent targeting of eicosanoid receptor 1/eicosanoid receptor 4 receptors and COX-2 induces synergistic apoptosis in Kaposi’s sarcoma-associated herpesvirus and Epstein-Barr virus associated non-Hodgkin lymphoma cell lines. Transl. Res. 2013, 161, 447–468. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, J.; Gaur, N.; Khera, L.; Kaul, R.; Robertson, E.S. COX-2 induces lytic reactivation of EBV through PGE2 by modulating the EP receptor signaling pathway. Virology 2015, 484, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rader, J.S.; Sill, M.W.; Beumer, J.H.; Lankes, H.A.; Benbrook, D.M.; Garcia, F.; Trimble, C.; Tate Thigpen, J.; Lieberman, R.; Zuna, R.E.; et al. A stratified randomized double-blind phase II trial of celecoxib for treating patients with cervical intraepithelial neoplasia: The potential predictive value of VEGF serum levels: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 145, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Siddiqui, A. Hepatitis C virus stimulates the expression of cyclooxygenase-2 via oxidative stress: Role of prostaglandin E2 in RNA replication. J. Virol. 2005, 79, 9725–9734. [Google Scholar] [CrossRef] [PubMed]
- Bassiouny, A.R.; Zaky, A.; Neenaa, H.M. Synergistic effect of celecoxib on 5-fluorouracil-induced apoptosis in hepatocellular carcinoma patients. Ann. Hepatol. 2010, 9, 410–418. [Google Scholar]
- Chandrasekharan, J.A.; Huang, X.M.; Hwang, A.C.; Sharma-Walia, N. Altering the Anti-inflammatory Lipoxin Microenvironment: A New Insight into Kaposi’s Sarcoma-Associated Herpesvirus Pathogenesis. J. Virol. 2016, 90, 11020–11031. [Google Scholar] [CrossRef]
- Roy, D.; Sin, S.H.; Lucas, A.; Venkataramanan, R.; Wang, L.; Eason, A.; Chavakula, V.; Hilton, I.B.; Tamburro, K.M.; Damania, B.; et al. mTOR inhibitors block Kaposi sarcoma growth by inhibiting essential autocrine growth factors and tumor angiogenesis. Cancer Res. 2013, 73, 2235–2246. [Google Scholar] [CrossRef]
- Dittmer, D.P.; Krown, S.E. Targeted therapy for Kaposi’s sarcoma and Kaposi’s sarcoma-associated herpesvirus. Curr. Opin. Oncol. 2007, 19, 452–457. [Google Scholar] [CrossRef]
- Sin, S.H.; Roy, D.; Wang, L.; Staudt, M.R.; Fakhari, F.D.; Patel, D.D.; Henry, D.; Harrington, W.J., Jr.; Damania, B.A.; Dittmer, D.P. Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood 2007, 109, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Molinolo, A.A.; Marsh, C.; El Dinali, M.; Gangane, N.; Jennison, K.; Hewitt, S.; Patel, V.; Seiwert, T.Y.; Gutkind, J.S. mTOR as a molecular target in HPV-associated oral and cervical squamous carcinomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2558–2568. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutta, A.; Sharma-Walia, N. Curbing Lipids: Impacts ON Cancer and Viral Infection. Int. J. Mol. Sci. 2019, 20, 644. https://doi.org/10.3390/ijms20030644
Dutta A, Sharma-Walia N. Curbing Lipids: Impacts ON Cancer and Viral Infection. International Journal of Molecular Sciences. 2019; 20(3):644. https://doi.org/10.3390/ijms20030644
Chicago/Turabian StyleDutta, Anika, and Neelam Sharma-Walia. 2019. "Curbing Lipids: Impacts ON Cancer and Viral Infection" International Journal of Molecular Sciences 20, no. 3: 644. https://doi.org/10.3390/ijms20030644
APA StyleDutta, A., & Sharma-Walia, N. (2019). Curbing Lipids: Impacts ON Cancer and Viral Infection. International Journal of Molecular Sciences, 20(3), 644. https://doi.org/10.3390/ijms20030644