High Risk of Hepatocellular Carcinoma Development in Fibrotic Liver: Role of the Hippo-YAP/TAZ Signaling Pathway

 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
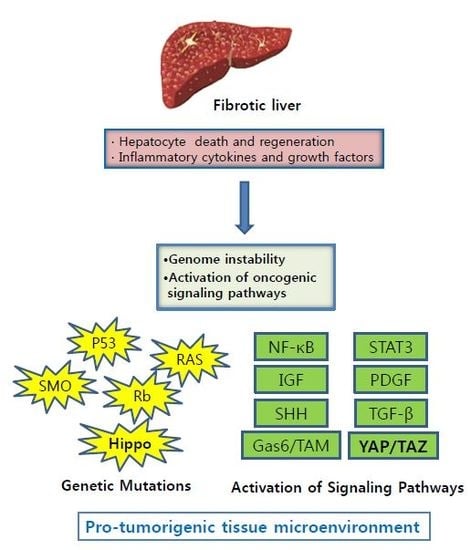
1. Introduction
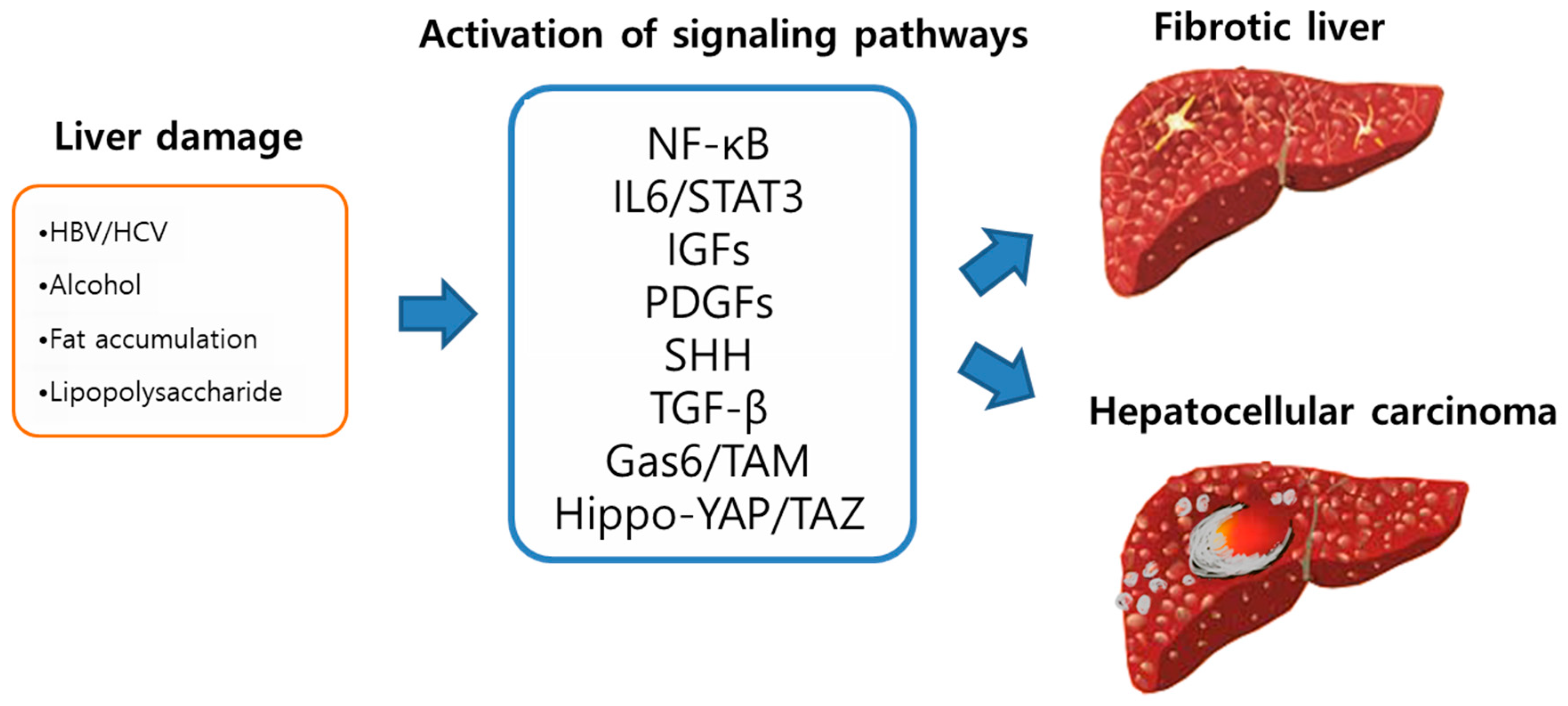
2. Liver Fibrosis
3. Genetic Instability in Fibrotic Liver
4. Increased Secretion of Growth Factors and Cytokines
4.1. Nuclear Factor Kappa B (NF-κB)
4.2. IL6/STAT3 Signaling
4.3. Insulin-Like Growth Factors (IGFs)
4.4. Platelet-Derived Growth Factor (PDGF)
4.5. Sonic Hedgehog (SHH)
4.6. Tumor Growth Factor Beta 1 (TGF-β1)
5. Gas6/TAM Pathway in Liver Fibrosis and Cancer
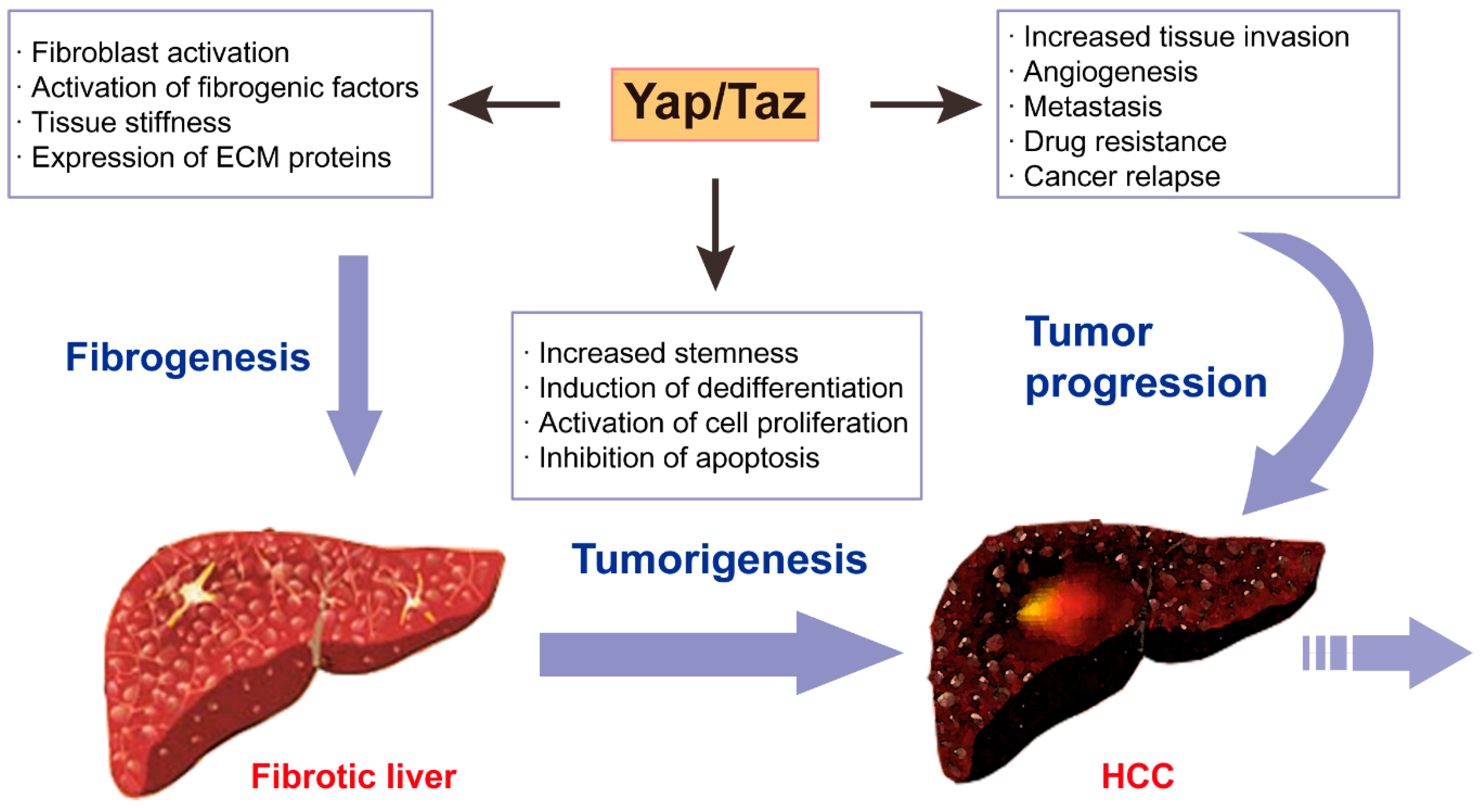
6. Hippo-YAP/TAZ Signaling in Liver Fibrosis and Cancer
6.1. Hepatic Fibrosis
6.2. Liver Cancer
6.3. YAP/TAZ Linking Hepatic Fibrosis and Cancer
7. Conclusions
Funding
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Muralidharan, P.; Raj, J.P. Update in global trends and aetiology of hepatocellular carcinoma. Contemp. Oncol. (Pozn. Pol.) 2018, 22, 141–150. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.I.; Moon, H.; Kim, D.Y.; Cho, K.J.; Ju, H.L.; Kim, D.Y.; Ahn, S.H.; Han, K.H.; Ro, S.W. Development of a transgenic mouse model of hepatocellular carcinoma with a liver fibrosis background. BMC Gastroenterol. 2016, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.I.; Moon, H.; Ju, H.L.; Cho, K.J.; Kim, D.Y.; Han, K.H.; Eun, J.W.; Nam, S.W.; Ribback, S.; Dombrowski, F.; et al. Hepatic expression of Sonic Hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. J. Hepatol. 2016, 64, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Gressner, A.M. Hepatic fibrogenesis: The puzzle of interacting cells, fibrogenic cytokines, regulatory loops, and extracellular matrix molecules. Zeitschrift fur Gastroenterologie 1992, 30 (Suppl. 1), 5–16. [Google Scholar]
- Herbst, H.; Schuppan, D.; Milani, S. [Fibrogenesis and fibrolysis in the liver]. Verhandlungen der Deutschen Gesellschaft fur Pathologie 1995, 79, 15–27. [Google Scholar] [PubMed]
- Han, K.H.; Yoon, K.T. New diagnostic method for liver fibrosis and cirrhosis. Intervirology 2008, 51 (Suppl. 1), 11–16. [Google Scholar] [CrossRef]
- Elsharkawy, A.M.; Oakley, F.; Mann, D.A. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis Int. J. Program. Cell Death 2005, 10, 927–939. [Google Scholar] [CrossRef]
- Baiocchini, A.; Montaldo, C.; Conigliaro, A.; Grimaldi, A.; Correani, V.; Mura, F.; Ciccosanti, F.; Rotiroti, N.; Brenna, A.; Montalbano, M.; et al. Extracellular Matrix Molecular Remodeling in Human Liver Fibrosis Evolution. PLoS ONE 2016, 11, e0151736. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Benyon, R.C.; Arthur, M.J. Mechanisms of hepatic fibrosis. J. Pediatr. Gastroenterol. Nutr. 1998, 27, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Burt, A.D.C.L. Cellular and molecular aspects of hepatic fibrosis. J. Pathol. 1993, 170, 105–114. [Google Scholar] [CrossRef]
- Elpek, G.O. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.; Dobbs, B.R.; Rogers, G.W. Lipoproteins and the liver sieve: The role of the fenestrated sinusoidal endothelium in lipoprotein metabolism, atherosclerosis, and cirrhosis. Hepatology 1995, 21, 863–874. [Google Scholar]
- Schwabe, R.F.; Maher, J.J. Lipids in liver disease: Looking beyond steatosis. Gastroenterology 2012, 142, 8–11. [Google Scholar] [CrossRef]
- Senoo, H.; Mezaki, Y.; Fujiwara, M. The stellate cell system (vitamin A-storing cell system). Anat. Sci. Int. 2017, 92, 387–455. [Google Scholar] [CrossRef]
- Senoo, H.; Sato, M.; Imai, K. Hepatic stellate cells—From the viewpoint of retinoid handling and function of the extracellular matrix. Kaibogaku Zasshi J. Anat. 1997, 72, 79–94. [Google Scholar]
- Wight, T.N.; Potter-Perigo, S. The extracellular matrix: An active or passive player in fibrosis? Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G950–G955. [Google Scholar] [CrossRef]
- Ellis, R.E.; Yuan, J.Y.; Horvitz, H.R. Mechanisms and functions of cell death. Annu. Rev. Cell Biol. 1991, 7, 663–698. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.C.; Zhang, Q.B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324. [Google Scholar] [CrossRef] [PubMed]
- Fowell, A.J.; Collins, J.E.; Duncombe, D.R.; Pickering, J.A.; Rosenberg, W.M.; Benyon, R.C. Silencing tissue inhibitors of metalloproteinases (TIMPs) with short interfering RNA reveals a role for TIMP-1 in hepatic stellate cell proliferation. Biochem. Biophys. Res. Commun. 2011, 407, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Kisseleva, T. Reversibility of liver fibrosis. Clin. Res. Hepatol. Gastroenterol. 2015, 39 (Suppl. 1), S60–S63. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Deng, X.; Liang, J. Modulation of hepatic stellate cells and reversibility of hepatic fibrosis. Exp. Cell Res. 2017, 352, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Liver regeneration after partial hepatectomy: Critical analysis of mechanistic dilemmas. Am. J. Pathol. 2010, 176, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Swiderska-Syn, M.; Jewell, M.L.; Premont, R.T.; Diehl, A.M. Liver regeneration requires Yap1-TGFbeta-dependent epithelial-mesenchymal transition in hepatocytes. J. Hepatol. 2018, 69, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B. Understanding the origins of human cancer. Science 2015, 350, 1175–1177. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, M.; Enjoji, M.; Iguchi, H.; Yokota, M.; Iwamoto, H.; Nakamuta, M.; Sakai, H.; Nawata, H. Telomerase activity is repressed during differentiation along the hepatocytic and biliary epithelial lineages: Verification on immortal cell lines from the same origin. Cell Biochem. Funct. 2001, 19, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Low, K.C.; Tergaonkar, V. Telomerase: Central regulator of all of the hallmarks of cancer. Trends Biochem. Sci. 2013, 38, 426–434. [Google Scholar] [CrossRef]
- Shay, J.W.; Zou, Y.; Hiyama, E.; Wright, W.E. Telomerase and cancer. Hum. Mol. Genet. 2001, 10, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, A.; Manns, M.P.; Rudolph, K.L. Telomeres and telomerase: A dual role in hepatocarcinogenesis. Hepatology 2004, 40, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.; Moch, A.; Saab, S. Relationship between Telomere Maintenance and Liver Disease. Gut Liver 2018, 13, 11–15. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Kawai, Y.; Nuka, E. Abundance of DNA adducts of 4-oxo-2-alkenals, lipid peroxidation-derived highly reactive genotoxins. J. Clin. Biochem. Nutr. 2018, 62, 3–10. [Google Scholar] [CrossRef]
- Morry, J.; Ngamcherdtrakul, W.; Yantasee, W. Oxidative stress in cancer and fibrosis: Opportunity for therapeutic intervention with antioxidant compounds, enzymes, and nanoparticles. Redox Biol. 2017, 11, 240–253. [Google Scholar] [CrossRef]
- Yeo, C.Q.X.; Alexander, I.; Lin, Z.; Lim, S.; Aning, O.A.; Kumar, R.; Sangthongpitag, K.; Pendharkar, V.; Ho, V.H.B.; Cheok, C.F. p53 Maintains Genomic Stability by Preventing Interference between Transcription and Replication. Cell Rep. 2016, 15, 132–146. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Tan, X.; Zeng, G.; Misse, A.; Singh, S.; Kim, Y.; Klaunig, J.E.; Monga, S.P. Conditional beta-catenin loss in mice promotes chemical hepatocarcinogenesis: Role of oxidative stress and platelet-derived growth factor receptor alpha/phosphoinositide 3-kinase signaling. Hepatology 2010, 52, 954–965. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Schrodl, W.; Buchler, R.; Wendler, S.; Reinhold, P.; Muckova, P.; Reindl, J.; Rhode, H. Acute phase proteins as promising biomarkers: Perspectives and limitations for human and veterinary medicine. Proteomics Clin. Appl. 2016, 10, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Lubbers, R.; van Essen, M.F.; van Kooten, C.; Trouw, L.A. Production of complement components by cells of the immune system. Clin. Exp. Immunol. 2017, 188, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Minute, L.; Ajona, D.; Corrales, L.; Melero, I.; Pio, R. Innate immune mediators in cancer: Between defense and resistance. Immunol. Rev. 2016, 274, 290–306. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. The liver as a lymphoid organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Baird, A.W.; O’Farrelly, C. Microanatomy of the liver immune system. Semin. Immunopathol. 2009, 31, 333–343. [Google Scholar] [CrossRef] [PubMed]
- O’Farrelly, C.; Crispe, I.N. Prometheus through the looking glass: Reflections on the hepatic immune system. Immunol. Today 1999, 20, 394–398. [Google Scholar] [CrossRef]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.C.; Logan, C.Y.; Fish, M.; Anbarchian, T.; Aguisanda, F.; Alvarez-Varela, A.; Wu, P.; Jin, Y.; Zhu, J.; Li, B.; et al. Inflammatory Cytokine TNFalpha Promotes the Long-Term Expansion of Primary Hepatocytes in 3D Culture. Cell 2018, 175, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- Grunebaum, E.; Avitzur, Y. Liver-associated immune abnormalities. Autoimmun. Rev. 2018, 18, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Mazza, E.; Nava, A.; Hahnloser, D.; Jochum, W.; Bajka, M. The mechanical response of human liver and its relation to histology: An in vivo study. Med. Image Anal. 2007, 11, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, L. Liver regeneration microenvironment of hepatocellular carcinoma for prevention and therapy. Oncotarget 2017, 8, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Fu, J.; Xu, A.; Yu, L.; Zhu, J.; Dai, R.; Su, B.; Luo, T.; Li, N.; Qin, W.; et al. Gankyrin drives malignant transformation of chronic liver damage-mediated fibrosis via the Rac1/JNK pathway. Cell Death Dis. 2015, 6, e1751. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, C.M.; Viscardi, G.; Papaccio, F.; Esposito, G.; Martini, G.; Ciardiello, D.; Martinelli, E.; Ciardiello, F.; Morgillo, F. Implication of the Hedgehog pathway in hepatocellular carcinoma. World J. Gastroenterol. 2017, 23, 4330–4340. [Google Scholar] [CrossRef] [PubMed]
- Lemberger, U.J.; Fuchs, C.D.; Karer, M.; Haas, S.; Stojakovic, T.; Schofer, C.; Marschall, H.U.; Wrba, F.; Taketo, M.M.; Egger, G.; et al. Hepatocyte specific expression of an oncogenic variant of beta-catenin results in cholestatic liver disease. Oncotarget 2016, 7, 86985–86998. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. BioMed Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef]
- Kodama, Y.; Kisseleva, T.; Iwaisako, K.; Miura, K.; Taura, K.; De Minicis, S.; Osterreicher, C.H.; Schnabl, B.; Seki, E.; Brenner, D.A. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology 2009, 137, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Tsai, W.L.; Shao, R.X.; Wu, G.; Peng, L.F.; Barlow, L.L.; Chung, W.J.; Zhang, L.; Zhao, H.; Jang, J.Y.; et al. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 2010, 138, 2509–2518. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ogata, H.; Kamio, M.; Joo, A.; Shiraishi, H.; Tokunaga, Y.; Sata, M.; Nagai, H.; Yoshimura, A. SOCS1 is a suppressor of liver fibrosis and hepatitis-induced carcinogenesis. J. Exp. Med. 2004, 199, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Saile, B.; Matthes, N.; El Armouche, H.; Neubauer, K.; Ramadori, G. The bcl, NFkappaB and p53/p21WAF1 systems are involved in spontaneous apoptosis and in the anti-apoptotic effect of TGF-beta or TNF-alpha on activated hepatic stellate cells. Eur. J. Cell Biol. 2001, 80, 554–561. [Google Scholar] [CrossRef]
- Kuhnel, F.; Zender, L.; Paul, Y.; Tietze, M.K.; Trautwein, C.; Manns, M.; Kubicka, S. NFkappaB mediates apoptosis through transcriptional activation of Fas (CD95) in adenoviral hepatitis. J. Biol. Chem. 2000, 275, 6421–6427. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- West, A.P.; Koblansky, A.A.; Ghosh, S. Recognition and signaling by toll-like receptors. Annu. Rev. Cell Dev. Biol. 2006, 22, 409–437. [Google Scholar] [CrossRef]
- Aoyama, T.; Inokuchi, S.; Brenner, D.A.; Seki, E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology 2010, 52, 1390–1400. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Pascale, R.M.; Feo, F. Dissection of signal transduction pathways as a tool for the development of targeted therapies of hepatocellular carcinoma. Rev. Recent Clin. Trials 2007, 2, 217–236. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-kappaB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Brenner, D.A. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: Role of IKK, JNK, and ROS pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G583–G589. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, G.; Rastogi, A.; Trehanpati, N.; Sen, B.; Khosla, R.; Sarin, S.K. From cirrhosis to hepatocellular carcinoma: New molecular insights on inflammation and cellular senescence. Liver Cancer 2013, 2, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Kaisho, T.; Takeda, K.; Tsujimura, T.; Kawai, T.; Nomura, F.; Terada, N.; Akira, S. IkappaB kinase alpha is essential for mature B cell development and function. J. Exp. Med. 2001, 193, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Tai, D.I.; Tsai, S.L.; Chang, Y.H.; Huang, S.N.; Chen, T.C.; Chang, K.S.; Liaw, Y.F. Constitutive activation of nuclear factor kappaB in hepatocellular carcinoma. Cancer 2000, 89, 2274–2281. [Google Scholar] [CrossRef]
- He, G.; Karin, M. NF-kappaB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, M.J.; Chow, D.C.; Brevnova, E.E.; Garcia, K.C. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science 2003, 300, 2101–2104. [Google Scholar] [CrossRef]
- Ward, L.D.; Howlett, G.J.; Discolo, G.; Yasukawa, K.; Hammacher, A.; Moritz, R.L.; Simpson, R.J. High affinity interleukin-6 receptor is a hexameric complex consisting of two molecules each of interleukin-6, interleukin-6 receptor, and gp-130. J. Biol. Chem. 1994, 269, 23286–23289. [Google Scholar] [PubMed]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Svinka, J.; Mikulits, W.; Eferl, R. STAT3 in hepatocellular carcinoma: New perspectives. Hepatic Oncol. 2014, 1, 107–120. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef]
- Takahashi, Y. The Role of Growth Hormone and Insulin-Like Growth Factor-I in the Liver. Int. J. Mol. Sci. 2017, 18, 1447. [Google Scholar] [CrossRef]
- Adamek, A.; Kasprzak, A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef]
- Nishizawa, H.; Takahashi, M.; Fukuoka, H.; Iguchi, G.; Kitazawa, R.; Takahashi, Y. GH-independent IGF-I action is essential to prevent the development of nonalcoholic steatohepatitis in a GH-deficient rat model. Biochem. Biophys. Res. Commun. 2012, 423, 295–300. [Google Scholar] [CrossRef]
- Nishizawa, H.; Iguchi, G.; Fukuoka, H.; Takahashi, M.; Suda, K.; Bando, H.; Matsumoto, R.; Yoshida, K.; Odake, Y.; Ogawa, W.; et al. IGF-I induces senescence of hepatic stellate cells and limits fibrosis in a p53-dependent manner. Sci. Rep. 2016, 6, 34605. [Google Scholar] [CrossRef]
- Sanz, S.; Pucilowska, J.B.; Liu, S.; Rodriguez-Ortigosa, C.M.; Lund, P.K.; Brenner, D.A.; Fuller, C.R.; Simmons, J.G.; Pardo, A.; Martinez-Chantar, M.L.; et al. Expression of insulin-like growth factor I by activated hepatic stellate cells reduces fibrogenesis and enhances regeneration after liver injury. Gut 2005, 54, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Ling, X. IGF-1 promotes the growth and metastasis of hepatocellular carcinoma via the inhibition of proteasome-mediated cathepsin B degradation. World J. Gastroenterol. 2015, 21, 10137–10149. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Friedman, S.L.; Goossens, N.; Hoshida, Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J. Hepatol. 2018, 68, 526–549. [Google Scholar] [CrossRef] [PubMed]
- Abboud, H.E.; Grandaliano, G.; Pinzani, M.; Knauss, T.; Pierce, G.F.; Jaffer, F. Actions of platelet-derived growth factor isoforms in mesangial cells. J. Cell. Physiol. 1994, 158, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Pinzani, M.; Milani, S.; Grappone, C.; Weber, F.L., Jr.; Gentilini, P.; Abboud, H.E. Expression of platelet-derived growth factor in a model of acute liver injury. Hepatology 1994, 19, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.; Yamasaki, G.; Johnson, R.J.; Friedman, S.L. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J. Clin. Investig. 1994, 94, 1563–1569. [Google Scholar] [CrossRef]
- Friedman, S.L.; Wei, S.; Blaner, W.S. Retinol release by activated rat hepatic lipocytes: Regulation by Kupffer cell-conditioned medium and PDGF. Am. J. Physiol. 1993, 264, G947–G952. [Google Scholar] [CrossRef]
- Ying, H.Z.; Chen, Q.; Zhang, W.Y.; Zhang, H.H.; Ma, Y.; Zhang, S.Z.; Fang, J.; Yu, C.H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review). Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef]
- Li, X.; Eriksson, U. Novel PDGF family members: PDGF-C and PDGF-D. Cytokine Growth Factor Rev. 2003, 14, 91–98. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef]
- Alvarez, R.H.; Kantarjian, H.M.; Cortes, J.E. Biology of platelet-derived growth factor and its involvement in disease. Mayo Clin. Proc. 2006, 81, 1241–1257. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.S.; Hughes, S.D.; Gilbertson, D.G.; Palmer, T.E.; Holdren, M.S.; Haran, A.C.; Odell, M.M.; Bauer, R.L.; Ren, H.P.; Haugen, H.S.; et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 3389–3394. [Google Scholar] [CrossRef] [PubMed]
- Maass, T.; Thieringer, F.R.; Mann, A.; Longerich, T.; Schirmacher, P.; Strand, D.; Hansen, T.; Galle, P.R.; Teufel, A.; Kanzler, S. Liver specific overexpression of platelet-derived growth factor-B accelerates liver cancer development in chemically induced liver carcinogenesis. Int. J. Cancer 2011, 128, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Chen, Y.; Wurmbach, E.; Roayaie, S.; Fiel, M.I.; Schwartz, M.; Thung, S.N.; Khitrov, G.; Zhang, W.; Villanueva, A.; et al. A molecular signature to discriminate dysplastic nodules from early hepatocellular carcinoma in HCV cirrhosis. Gastroenterology 2006, 131, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Stock, P.; Monga, D.; Tan, X.; Micsenyi, A.; Loizos, N.; Monga, S.P. Platelet-derived growth factor receptor-alpha: A novel therapeutic target in human hepatocellular cancer. Mol. Cancer Ther. 2007, 6, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.E.; Scott, M.P. Communicating with Hedgehogs. Nat. Rev. Mol. Cell Biol. 2005, 6, 306–317. [Google Scholar] [CrossRef]
- Omenetti, A.; Choi, S.; Michelotti, G.; Diehl, A.M. Hedgehog signaling in the liver. J. Hepatol. 2011, 54, 366–373. [Google Scholar] [CrossRef]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef]
- Omenetti, A.; Yang, L.; Li, Y.X.; McCall, S.J.; Jung, Y.; Sicklick, J.K.; Huang, J.; Choi, S.; Suzuki, A.; Diehl, A.M. Hedgehog-mediated mesenchymal-epithelial interactions modulate hepatic response to bile duct ligation. Lab. Investig. J. Tech. Methods Pathol. 2007, 87, 499–514. [Google Scholar] [CrossRef]
- Syn, W.K.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereira, T.A.; Zdanowicz, M.; Malladi, P.; et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011, 53, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Philips, G.M.; Chan, I.S.; Swiderska, M.; Schroder, V.T.; Guy, C.; Karaca, G.F.; Moylan, C.; Venkatraman, T.; Feuerlein, S.; Syn, W.K.; et al. Hedgehog signaling antagonist promotes regression of both liver fibrosis and hepatocellular carcinoma in a murine model of primary liver cancer. PLoS ONE 2011, 6, e23943. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Choi, S.S.; Michelotti, G.A.; Chan, I.S.; Swiderska-Syn, M.; Karaca, G.F.; Xie, G.; Moylan, C.A.; Garibaldi, F.; Premont, R.; et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 2012, 143, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Omenetti, A.; Syn, W.K.; Diehl, A.M. The role of Hedgehog signaling in fibrogenic liver repair. Int. J. Biochem. Cell Biol. 2011, 43, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Li, Y.X.; Jayaraman, A.; Kannangai, R.; Qi, Y.; Vivekanandan, P.; Ludlow, J.W.; Owzar, K.; Chen, W.; Torbenson, M.S.; et al. Dysregulation of the Hedgehog pathway in human hepatocarcinogenesis. Carcinogenesis 2006, 27, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Eichenmuller, M.; Gruner, I.; Hagl, B.; Haberle, B.; Muller-Hocker, J.; von Schweinitz, D.; Kappler, R. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology 2009, 49, 482–490. [Google Scholar] [CrossRef]
- Patil, M.A.; Zhang, J.; Ho, C.; Cheung, S.T.; Fan, S.T.; Chen, X. Hedgehog signaling in human hepatocellular carcinoma. Cancer Biol. Ther. 2006, 5, 111–117. [Google Scholar] [CrossRef]
- Pasca di Magliano, M.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Schuster, N.; Krieglstein, K. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res. 2002, 307, 1–14. [Google Scholar] [CrossRef]
- Hellerbrand, C.; Stefanovic, B.; Giordano, F.; Burchardt, E.R.; Brenner, D.A. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J. Hepatol. 1999, 30, 77–87. [Google Scholar] [CrossRef]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.G.; Thomas, B.; Koff, J. TGF-beta: Master regulator of inflammation and fibrosis. Respirology 2018, 23, 1096–1097. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M.; Baek, J.Y.; Koszarek, A.; Kanngurn, S.; Knoblaugh, S.E.; Grady, W.M. Transforming growth factor-beta signaling promotes hepatocarcinogenesis induced by p53 loss. Hepatology 2012, 55, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Sugimachi, K.; Goto, H.; Wang, J.; Morikawa, T.; Miyachi, Y.; Takano, Y.; Hikasa, H.; Itoh, T.; Suzuki, S.O.; et al. Dysregulated YAP1/TAZ and TGF-beta signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice. Proc. Natl. Acad. Sci. USA 2016, 113, E71–E80. [Google Scholar] [CrossRef]
- Yang, L.; Inokuchi, S.; Roh, Y.S.; Song, J.; Loomba, R.; Park, E.J.; Seki, E. Transforming growth factor-beta signaling in hepatocytes promotes hepatic fibrosis and carcinogenesis in mice with hepatocyte-specific deletion of TAK1. Gastroenterology 2013, 144, 1042–1054. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hikiba, Y.; Hirata, Y.; Font-Burgada, J.; Sakamoto, K.; Hayakawa, Y.; Taniguchi, K.; Umemura, A.; Kinoshita, H.; Sakitani, K.; et al. Loss of liver E-cadherin induces sclerosing cholangitis and promotes carcinogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Ju, H.L.; Chung, S.I.; Cho, K.J.; Eun, J.W.; Nam, S.W.; Han, K.H.; Calvisi, D.F.; Ro, S.W. Transforming Growth Factor-beta Promotes Liver Tumorigenesis in Mice via Up-regulation of Snail. Gastroenterology 2017, 153, 1378–1391. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Eun, J.W.; Lee, K.; Kim, H.S.; Yang, H.D.; Kim, S.Y.; Lee, E.K.; Kim, T.; Kang, K.; Kim, S.; et al. Barrier to autointegration factor 1, procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3, and splicing factor 3b subunit 4 as early-stage cancer decision markers and drivers of hepatocellular carcinoma. Hepatology 2018, 67, 1360–1377. [Google Scholar] [CrossRef] [PubMed]
- Bellido-Martin, L.; de Frutos, P.G. Vitamin K-dependent actions of Gas6. Vitam. Horm. 2008, 78, 185–209. [Google Scholar] [CrossRef] [PubMed]
- Holstein, E.; Binder, M.; Mikulits, W. Dynamics of Axl Receptor Shedding in Hepatocellular Carcinoma and Its Implication for Theranostics. Int. J. Mol. Sci. 2018, 19, 4111. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.K.; Wilhelm, A.; Antoniades, C.G. TAM receptor tyrosine kinase function and the immunopathology of liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G899–G905. [Google Scholar] [CrossRef] [PubMed]
- Qi, N.; Liu, P.; Zhang, Y.; Wu, H.; Chen, Y.; Han, D. Development of a spontaneous liver disease resembling autoimmune hepatitis in mice lacking tyro3, axl and mer receptor tyrosine kinases. PLoS ONE 2013, 8, e66604. [Google Scholar] [CrossRef] [PubMed]
- Lafdil, F.; Chobert, M.N.; Couchie, D.; Brouillet, A.; Zafrani, E.S.; Mavier, P.; Laperche, Y. Induction of Gas6 protein in CCl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology 2006, 44, 228–239. [Google Scholar] [CrossRef]
- Bellan, M.; Pogliani, G.; Marconi, C.; Minisini, R.; Franzosi, L.; Alciato, F.; Magri, A.; Avanzi, G.C.; Pirisi, M.; Sainaghi, P.P. Gas6 as a putative noninvasive biomarker of hepatic fibrosis. Biomark. Med. 2016, 10, 1241–1249. [Google Scholar] [CrossRef]
- Barcena, C.; Stefanovic, M.; Tutusaus, A.; Joannas, L.; Menendez, A.; Garcia-Ruiz, C.; Sancho-Bru, P.; Mari, M.; Caballeria, J.; Rothlin, C.V.; et al. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J. Hepatol. 2015, 63, 670–678. [Google Scholar] [CrossRef]
- Wu, G.; Ma, Z.; Hu, W.; Wang, D.; Gong, B.; Fan, C.; Jiang, S.; Li, T.; Gao, J.; Yang, Y. Molecular insights of Gas6/TAM in cancer development and therapy. Cell Death Dis. 2017, 8, e2700. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Ma, Z.; Cheng, Y.; Hu, W.; Deng, C.; Jiang, S.; Li, T.; Chen, F.; Yang, Y. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol. Cancer 2018, 17, 20. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zheng, Y.; Dong, J.; Klusza, S.; Deng, W.M.; Pan, D. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev. Cell 2010, 18, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bai, H.; David, K.K.; Dong, J.; Zheng, Y.; Cai, J.; Giovannini, M.; Liu, P.; Anders, R.A.; Pan, D. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev. Cell 2010, 19, 27–38. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef]
- Goulev, Y.; Fauny, J.D.; Gonzalez-Marti, B.; Flagiello, D.; Silber, J.; Zider, A. SCALLOPED interacts with YORKIE, the nuclear effector of the hippo tumor-suppressor pathway in Drosophila. Curr. Biol. 2008, 18, 435–441. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Yu, F.X.; Gong, R.; Brown, J.H.; Guan, K.L. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev. 2012, 26, 2138–2143. [Google Scholar] [CrossRef] [PubMed]
- Loforese, G.; Malinka, T.; Keogh, A.; Baier, F.; Simillion, C.; Montani, M.; Halazonetis, T.D.; Candinas, D.; Stroka, D. Impaired liver regeneration in aged mice can be rescued by silencing Hippo core kinases MST1 and MST2. EMBO Mol. Med. 2017, 9, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; He, Z.; Kong, L.L.; Chen, Q.; Yuan, Q.; Zhang, S.; Ye, J.; Liu, H.; Sun, X.; Geng, J.; et al. Pharmacological targeting of kinases MST1 and MST2 augments tissue repair and regeneration. Sci. Transl. Med. 2016, 8, 352ra108. [Google Scholar] [CrossRef] [PubMed]
- Konishi, T.; Schuster, R.M.; Lentsch, A.B. Proliferation of hepatic stellate cells, mediated by YAP and TAZ, contributes to liver repair and regeneration after liver ischemia-reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G471–G482. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, N.; Hata, S.; Itoh, T.; Tanaka, M.; Nishio, M.; Itoh, M.; Ogawa, Y.; Terai, S.; Sakaida, I.; Suzuki, A.; et al. YAP determines the cell fate of injured mouse hepatocytes in vivo. Nat. Commun. 2017, 8, 16017. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef]
- Caliari, S.R.; Perepelyuk, M.; Cosgrove, B.D.; Tsai, S.J.; Lee, G.Y.; Mauck, R.L.; Wells, R.G.; Burdick, J.A. Stiffening hydrogels for investigating the dynamics of hepatic stellate cell mechanotransduction during myofibroblast activation. Sci. Rep. 2016, 6, 21387. [Google Scholar] [CrossRef]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investing. 2018, 128, 45–53. [Google Scholar] [CrossRef]
- Zhang, K.; Chang, Y.; Shi, Z.; Han, X.; Han, Y.; Yao, Q.; Hu, Z.; Cui, H.; Zheng, L.; Han, T.; et al. omega-3 PUFAs ameliorate liver fibrosis and inhibit hepatic stellate cells proliferation and activation by promoting YAP/TAZ degradation. Sci. Rep. 2016, 6, 30029. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, Z.; Caviglia, J.M.; Corey, K.E.; Herfel, T.M.; Cai, B.; Masia, R.; Chung, R.T.; Lefkowitch, J.H.; Schwabe, R.F.; et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016, 24, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Moya, I.M.; Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Li, Y.; Liu, Q.; Chen, Q.; Chen, L.; Zhou, D. The Hippo Signaling Pathway in Regenerative Medicine. Methods Mol. Biol. 2019, 1893, 353–370. [Google Scholar] [CrossRef] [PubMed]
- Urtasun, R.; Latasa, M.U.; Demartis, M.I.; Balzani, S.; Goni, S.; Garcia-Irigoyen, O.; Elizalde, M.; Azcona, M.; Pascale, R.M.; Feo, F.; et al. Connective tissue growth factor autocriny in human hepatocellular carcinoma: Oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology 2011, 54, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Juric, V.; Chen, C.C.; Lau, L.F. Fas-mediated apoptosis is regulated by the extracellular matrix protein CCN1 (CYR61) in vitro and in vivo. Mol. Cell. Biol. 2009, 29, 3266–3279. [Google Scholar] [CrossRef]
- Huo, X.; Zhang, Q.; Liu, A.M.; Tang, C.; Gong, Y.; Bian, J.; Luk, J.M.; Xu, Z.; Chen, J. Overexpression of Yes-associated protein confers doxorubicin resistance in hepatocellullar carcinoma. Oncol. Rep. 2013, 29, 840–846. [Google Scholar] [CrossRef]
- Kim, M.; Jho, E.H. Cross-talk between Wnt/beta-catenin and Hippo signaling pathways: A brief review. BMB Rep. 2014, 47, 540–545. [Google Scholar] [CrossRef]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef]
- Mohseni, M.; Sun, J.; Lau, A.; Curtis, S.; Goldsmith, J.; Fox, V.L.; Wei, C.; Frazier, M.; Samson, O.; Wong, K.K.; et al. A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat. Cell Biol. 2014, 16, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 2014, 7, ra42. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011, 30, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.C.; Pepe-Mooney, B.; Galli, G.G.; Dill, M.T.; Huang, H.T.; Hao, M.; Wang, Y.; Liang, H.; Calogero, R.A.; Camargo, F.D. NUAK2 is a critical YAP target in liver cancer. Nat. Commun. 2018, 9, 4834. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Chen, J.; Feng, H.; Peng, S.; Adams, U.; Bai, Y.; Huang, L.; Li, J.; Huang, J.; Meng, S.; et al. YAP/TEAD-mediated transcription controls cellular senescence. Cancer Res. 2013, 73, 3615–3624. [Google Scholar] [CrossRef]
- Han, S.X.; Bai, E.; Jin, G.H.; He, C.C.; Guo, X.J.; Wang, L.J.; Li, M.; Ying, X.; Zhu, Q. Expression and clinical significance of YAP, TAZ, and AREG in hepatocellular carcinoma. J. Immunol. Res. 2014, 2014, 261365. [Google Scholar] [CrossRef]
- Kim, G.J.; Kim, H.; Park, Y.N. Increased expression of Yes-associated protein 1 in hepatocellular carcinoma with stemness and combined hepatocellular-cholangiocarcinoma. PLoS ONE 2013, 8, e75449. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Jiang, N.; Zhou, B.; Liu, Q.; Du, C. TAZ regulates cell proliferation and epithelial-mesenchymal transition of human hepatocellular carcinoma. Cancer Sci. 2015, 106, 151–159. [Google Scholar] [CrossRef]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.; Halder, G.; Finegold, M.J.; Lee, J.S.; et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar] [CrossRef]
- Song, H.; Mak, K.K.; Topol, L.; Yun, K.; Hu, J.; Garrett, L.; Chen, Y.; Park, O.; Chang, J.; Simpson, R.M.; et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. USA 2010, 107, 1431–1436. [Google Scholar] [CrossRef]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Z.; Yao, T.J.; Lee, N.P.; Ng, I.O.; Chan, Y.T.; Zender, L.; Lowe, S.W.; Poon, R.T.; Luk, J.M. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer 2009, 115, 4576–4585. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Karin, M. Inflammation and liver tumorigenesis. Front. Med. 2013, 7, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Khan, S.K.; Liu, Y.; Xu, R.; Park, O.; He, Y.; Cha, B.; Gao, B.; Yang, Y. Hepatic Hippo signaling inhibits protumoural microenvironment to suppress hepatocellular carcinoma. Gut 2018, 67, 1692–1703. [Google Scholar] [CrossRef] [PubMed]
- Hagenbeek, T.J.; Webster, J.D.; Kljavin, N.M.; Chang, M.T.; Pham, T.; Lee, H.J.; Klijn, C.; Cai, A.G.; Totpal, K.; Ravishankar, B.; et al. The Hippo pathway effector TAZ induces TEAD-dependent liver inflammation and tumors. Sci. Signal. 2018, 11, eaaj1757. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, H.; Cho, K.; Shin, S.; Kim, D.Y.; Han, K.-H.; Ro, S.W. High Risk of Hepatocellular Carcinoma Development in Fibrotic Liver: Role of the Hippo-YAP/TAZ Signaling Pathway. Int. J. Mol. Sci. 2019, 20, 581. https://doi.org/10.3390/ijms20030581
Moon H, Cho K, Shin S, Kim DY, Han K-H, Ro SW. High Risk of Hepatocellular Carcinoma Development in Fibrotic Liver: Role of the Hippo-YAP/TAZ Signaling Pathway. International Journal of Molecular Sciences. 2019; 20(3):581. https://doi.org/10.3390/ijms20030581
Chicago/Turabian StyleMoon, Hyuk, Kyungjoo Cho, Sunyeong Shin, Do Young Kim, Kwang-Hyub Han, and Simon Weonsang Ro. 2019. "High Risk of Hepatocellular Carcinoma Development in Fibrotic Liver: Role of the Hippo-YAP/TAZ Signaling Pathway" International Journal of Molecular Sciences 20, no. 3: 581. https://doi.org/10.3390/ijms20030581
APA StyleMoon, H., Cho, K., Shin, S., Kim, D. Y., Han, K.-H., & Ro, S. W. (2019). High Risk of Hepatocellular Carcinoma Development in Fibrotic Liver: Role of the Hippo-YAP/TAZ Signaling Pathway. International Journal of Molecular Sciences, 20(3), 581. https://doi.org/10.3390/ijms20030581