Antioxidant Properties of Fucoidan Alleviate Acceleration and Exacerbation of Hippocampal Neuronal Death Following Transient Global Cerebral Ischemia in High-Fat Diet-Induced Obese Gerbils

 ,
,  and
and
Abstract
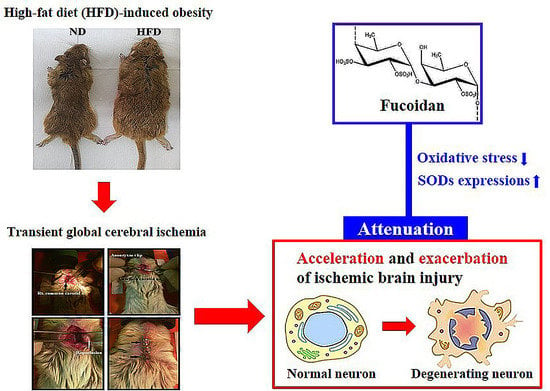
1. Introduction
2. Results
2.1. Physiological Characteristics
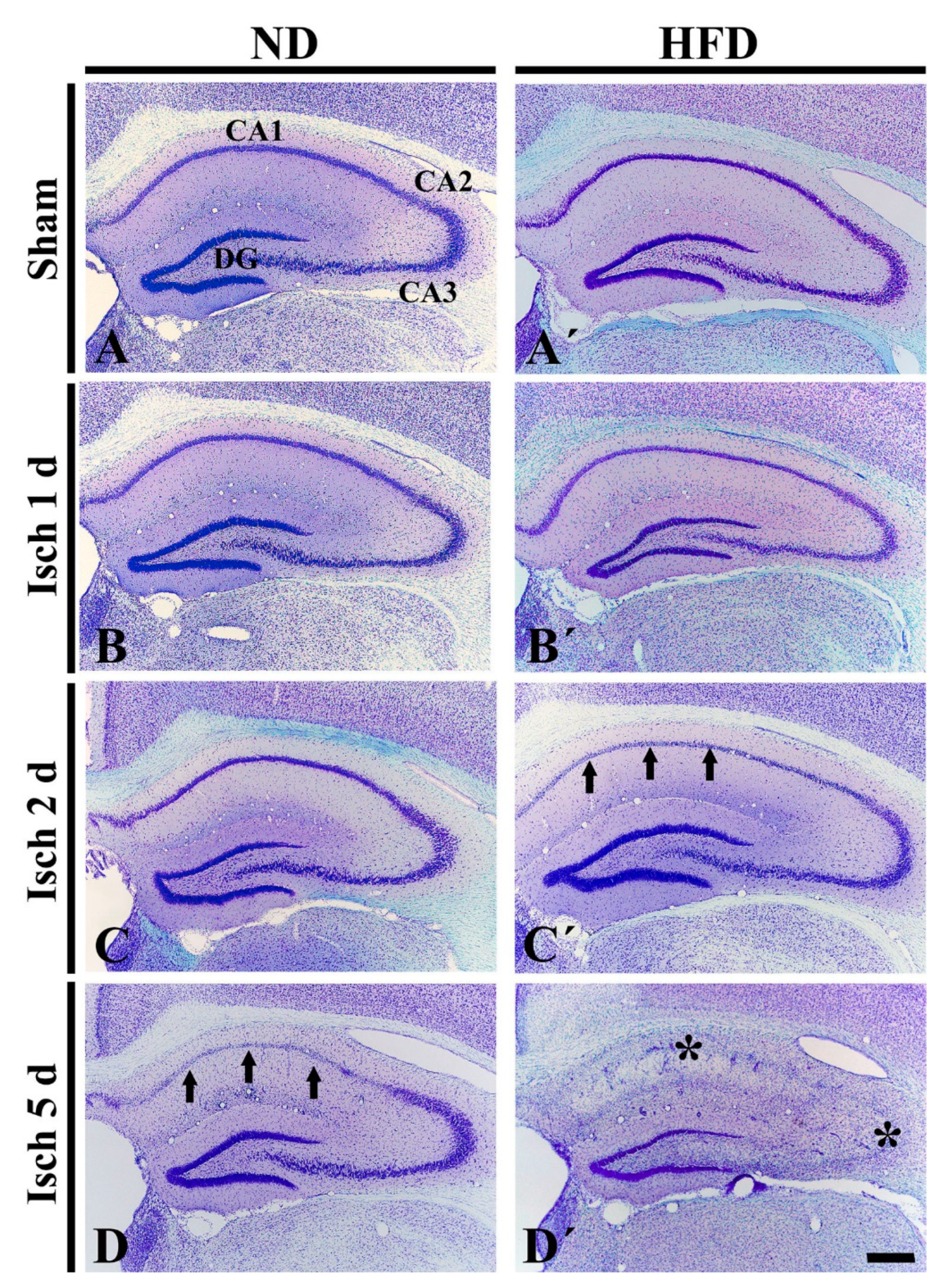
2.2. Neuronal Death in HFD-Ischemia Group
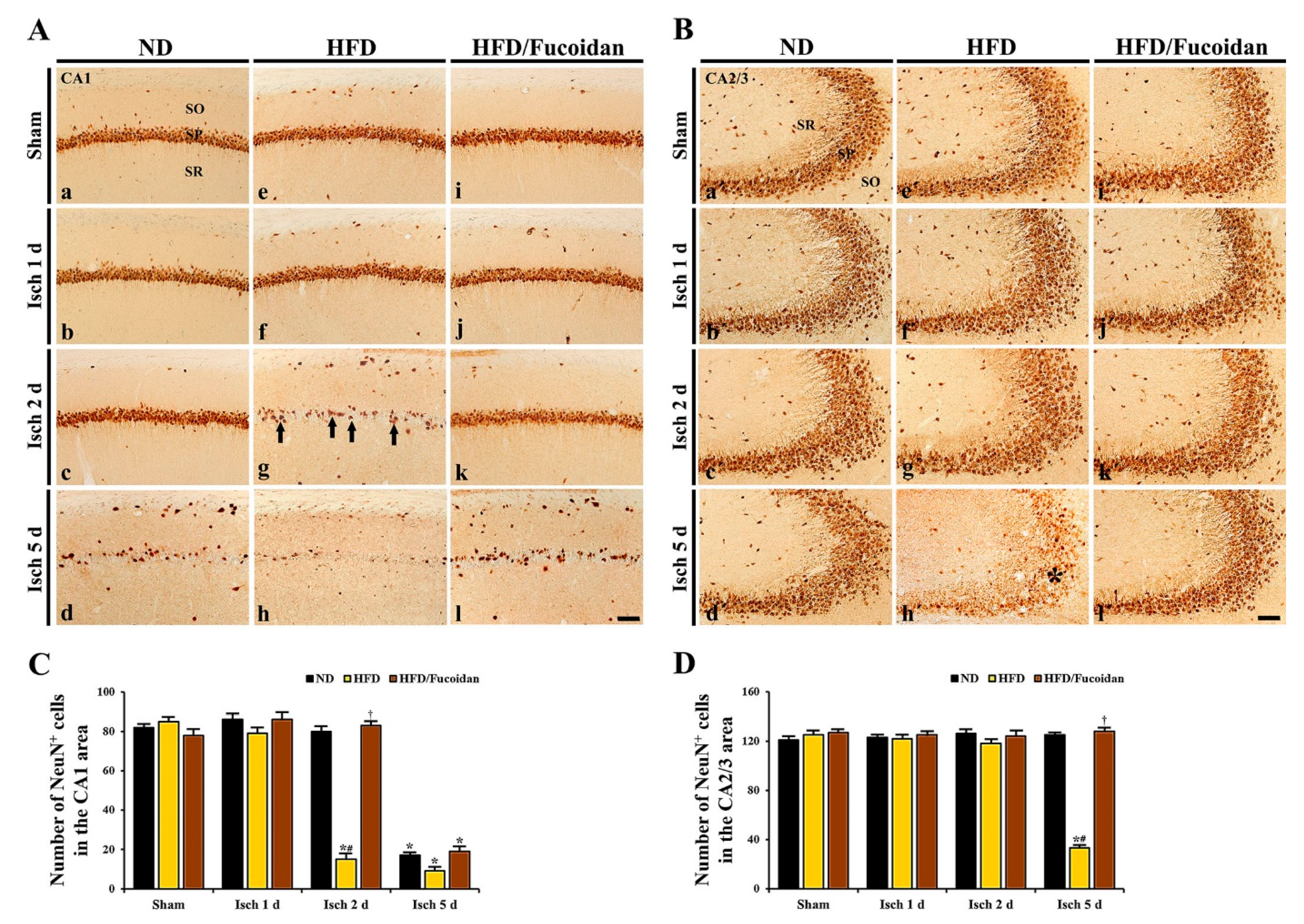
2.3. Neuroprotection by Fucoidan in HFD-Ischemia Group
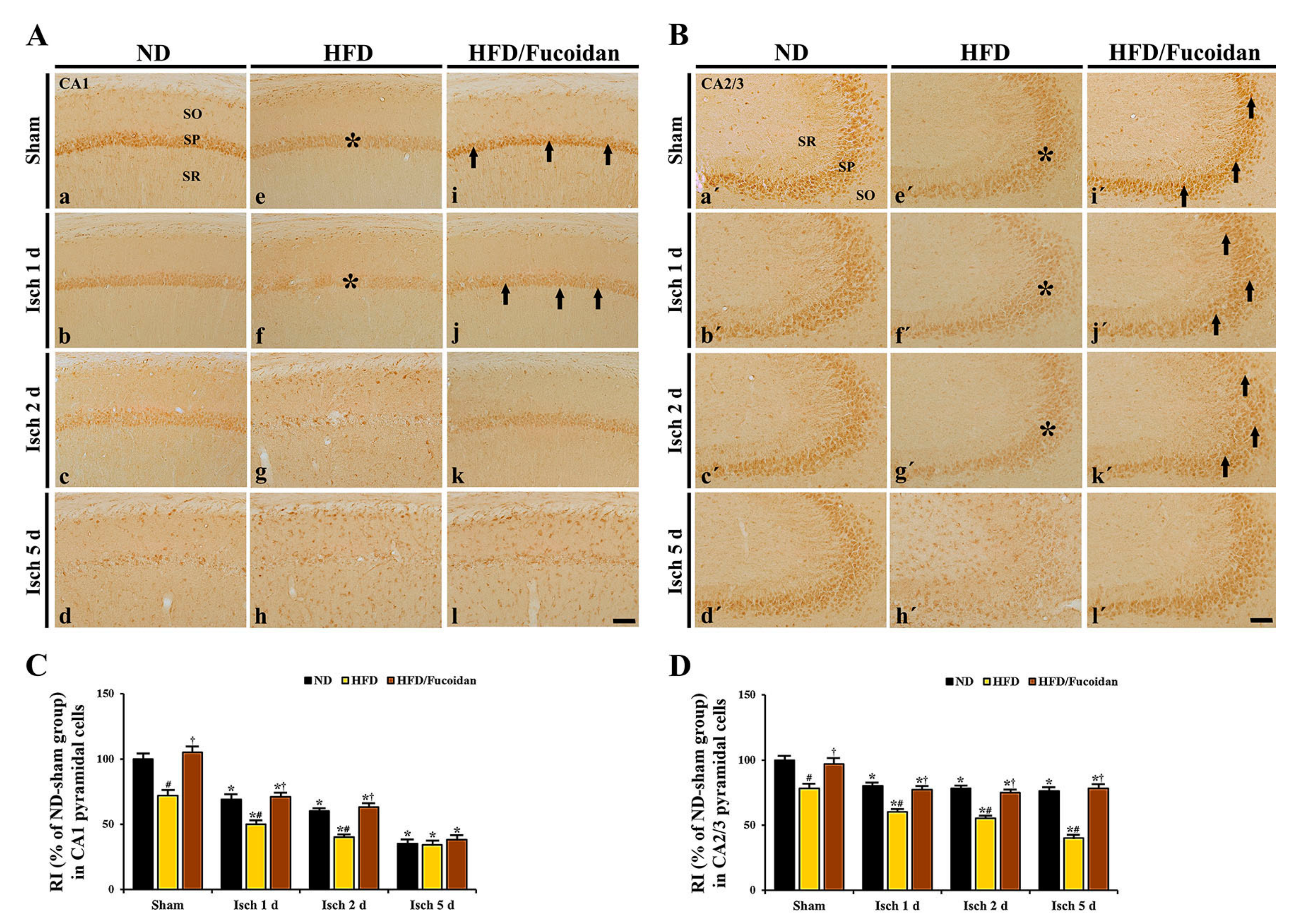
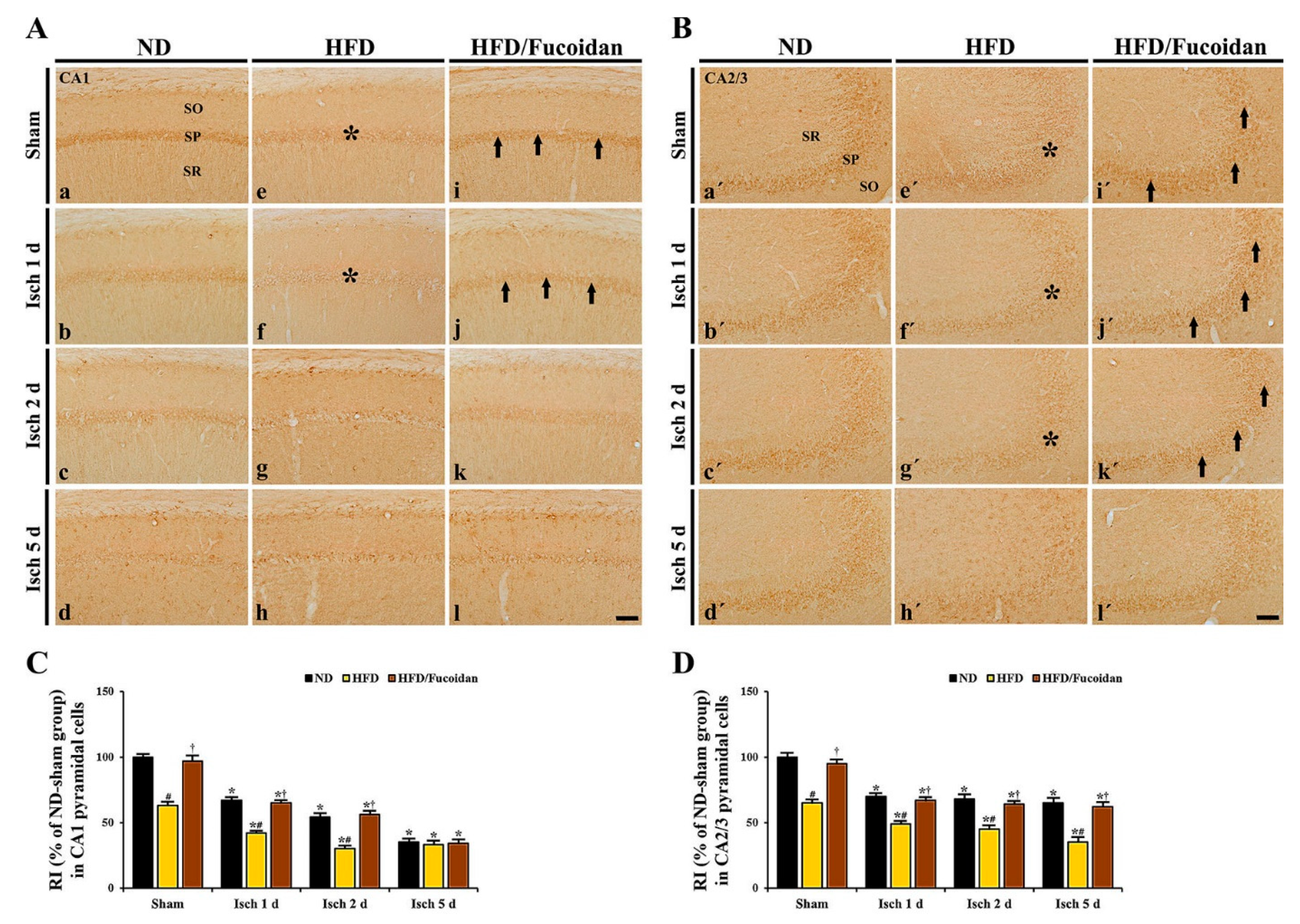
2.3.1. NeuN+ Cells
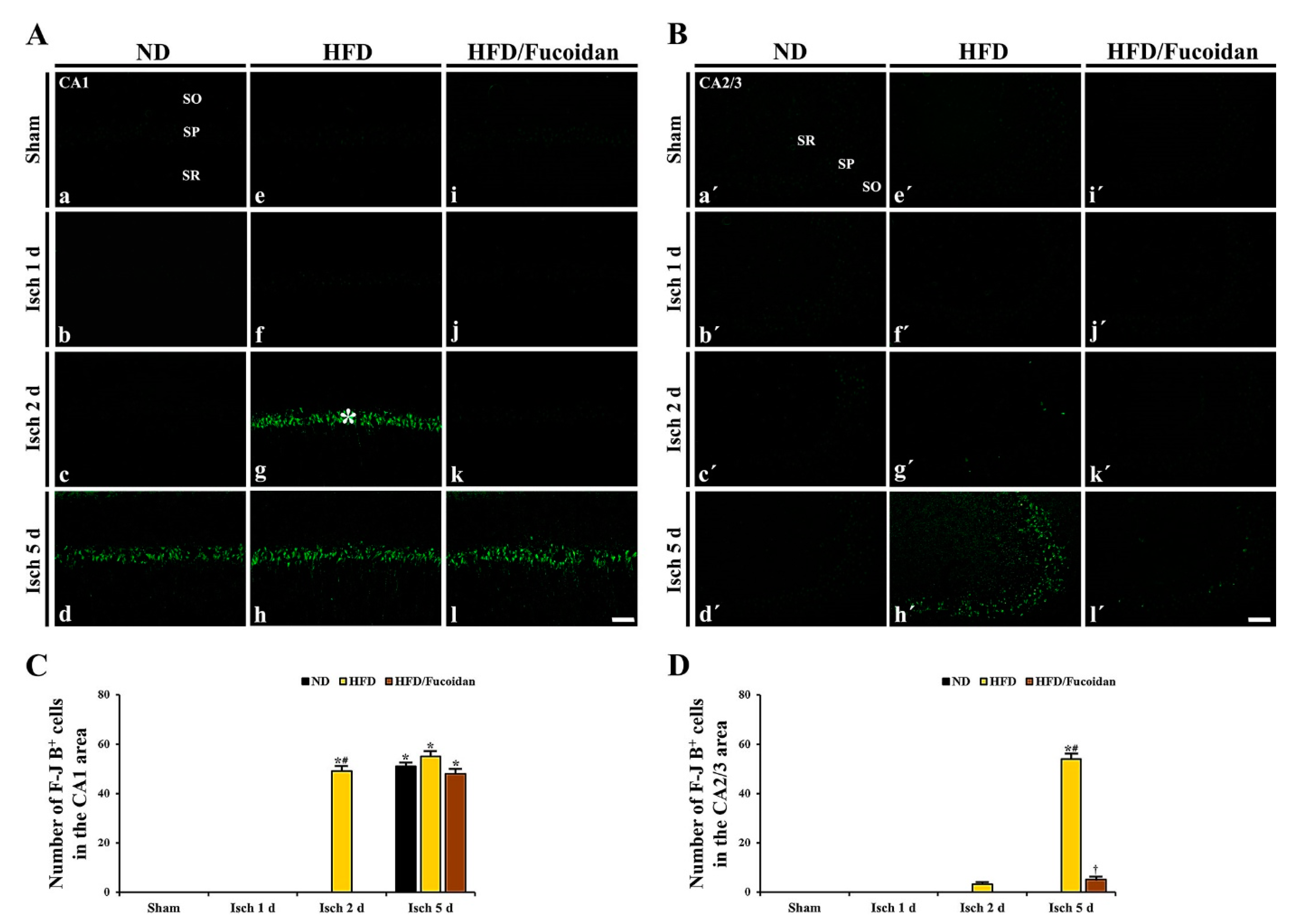
2.3.2. F-J B+ Cells
2.4. Inhibitory Effect of Fucoidan on Oxidative Stress in HFD-Ischemia Group
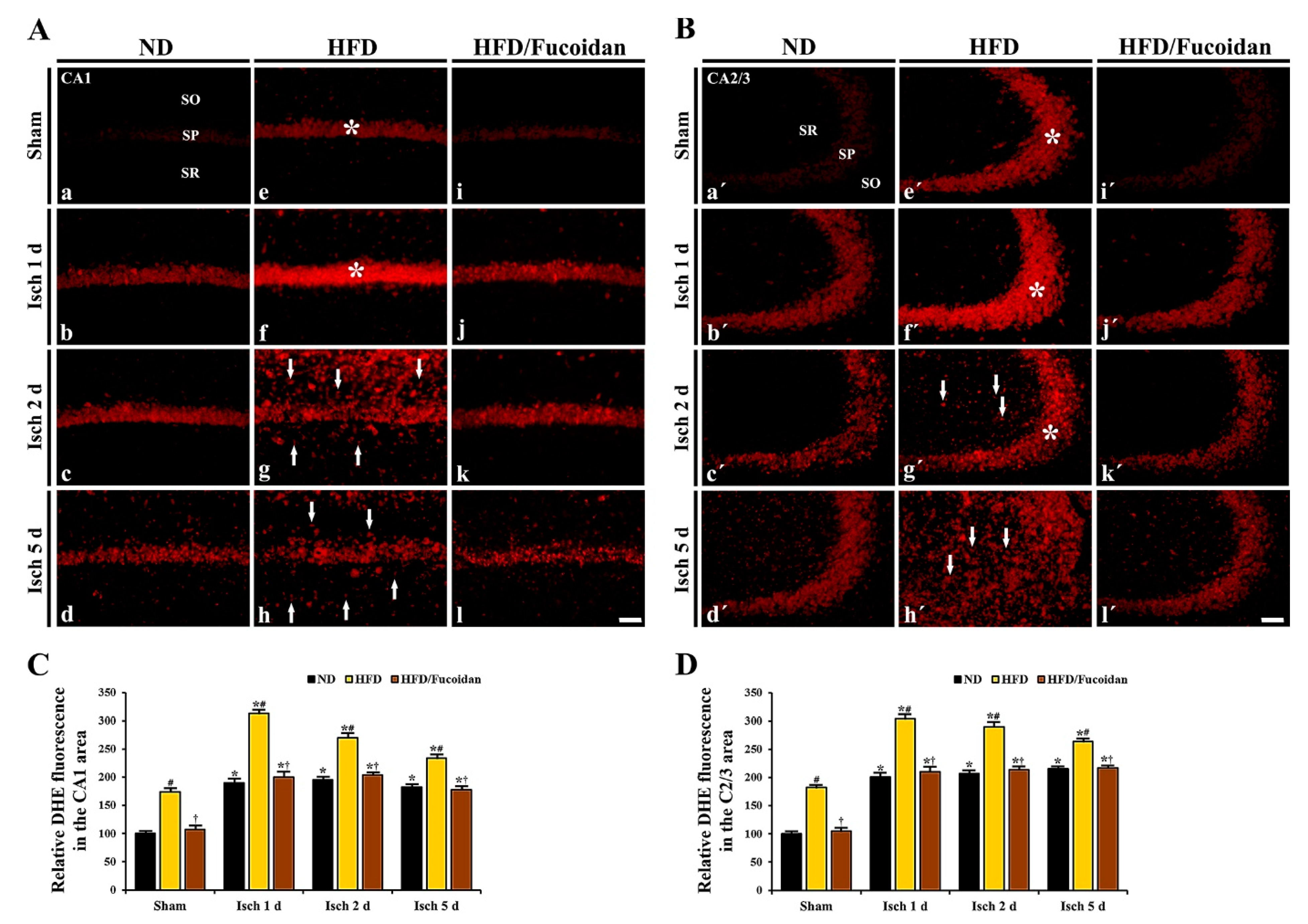
2.4.1. DHE Fluorescence
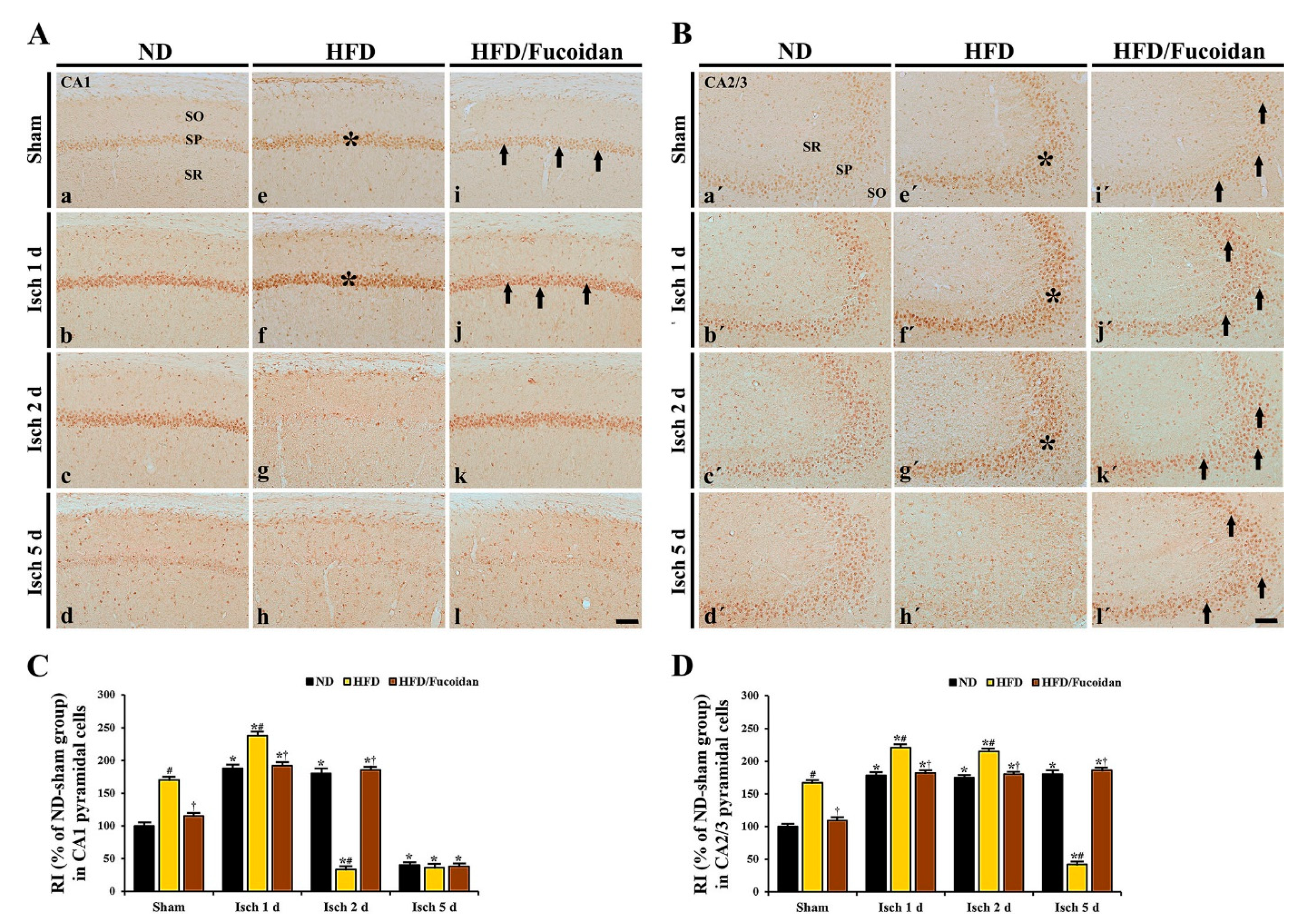
2.4.2. 8-OHG Immunoreactivity
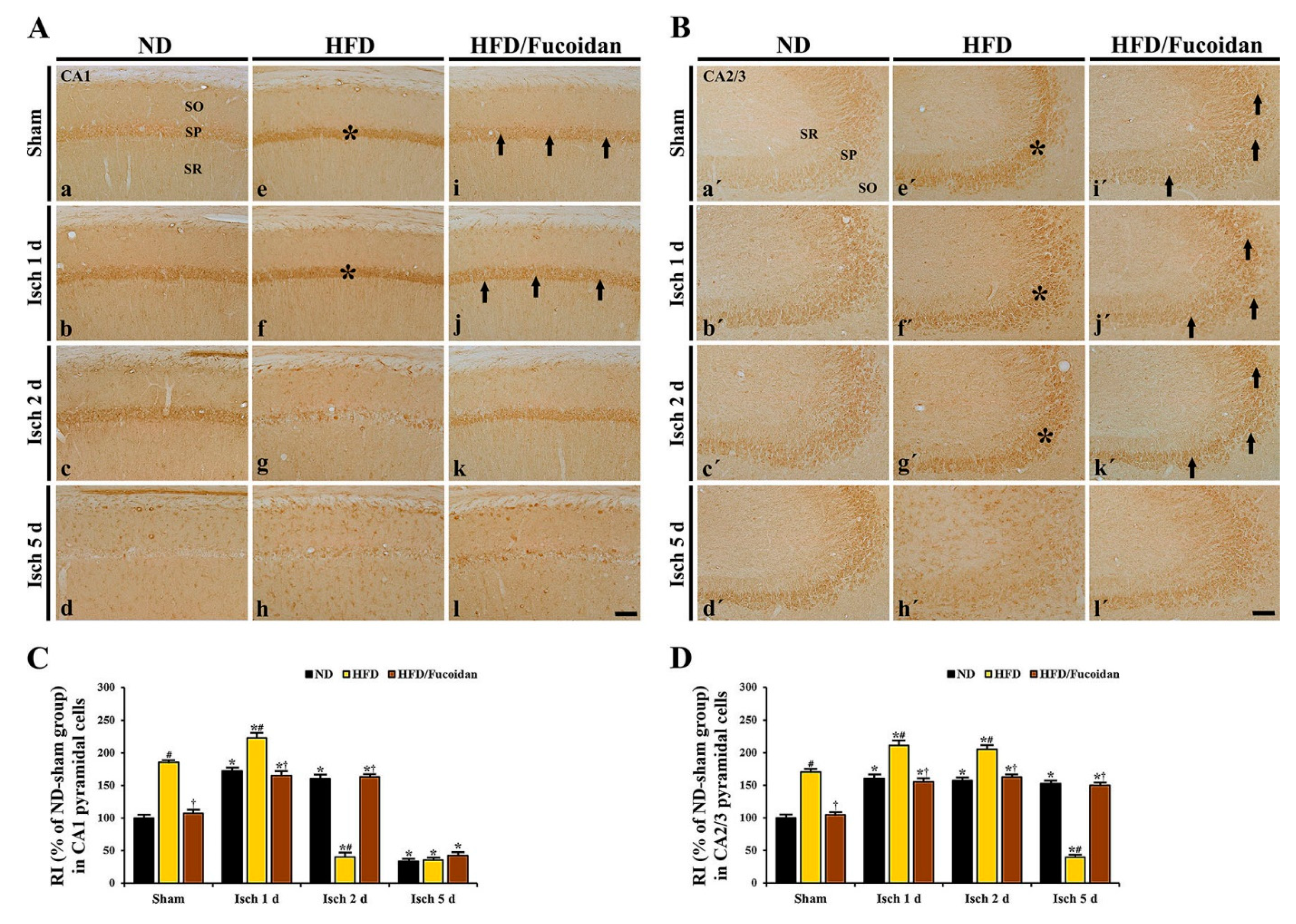
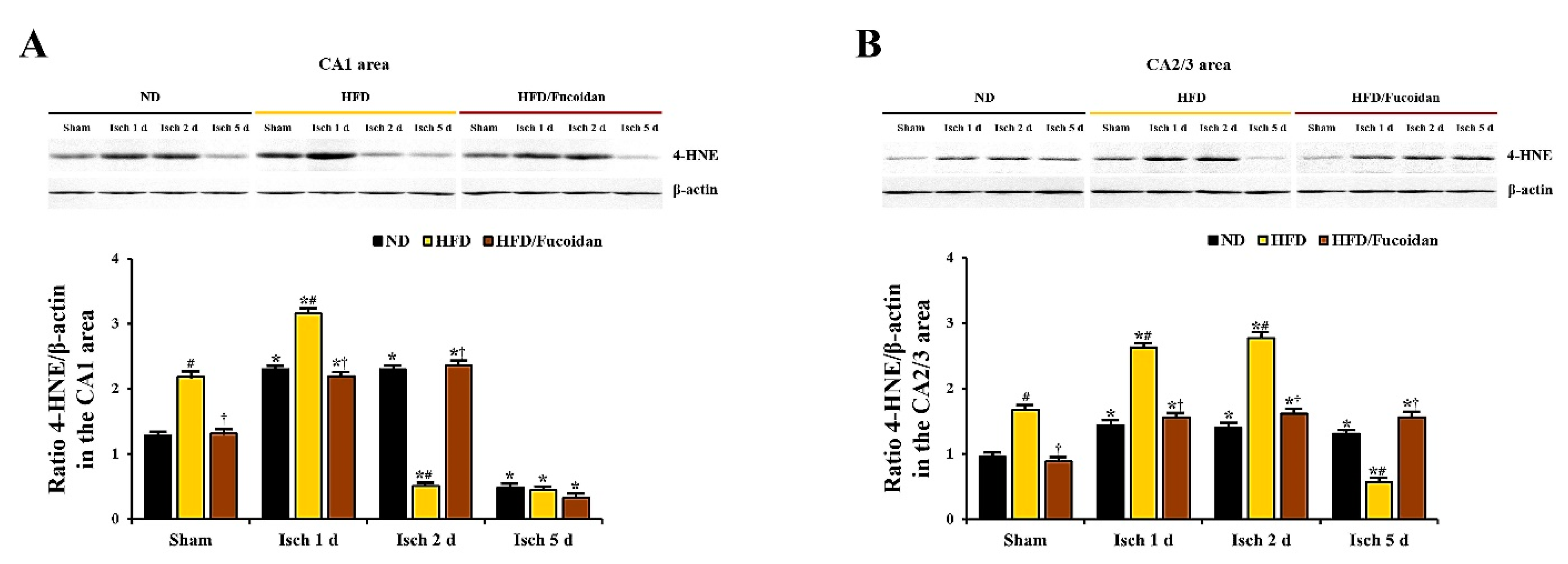
2.4.3. 4-HNE Immunoreactivity and Protein Level
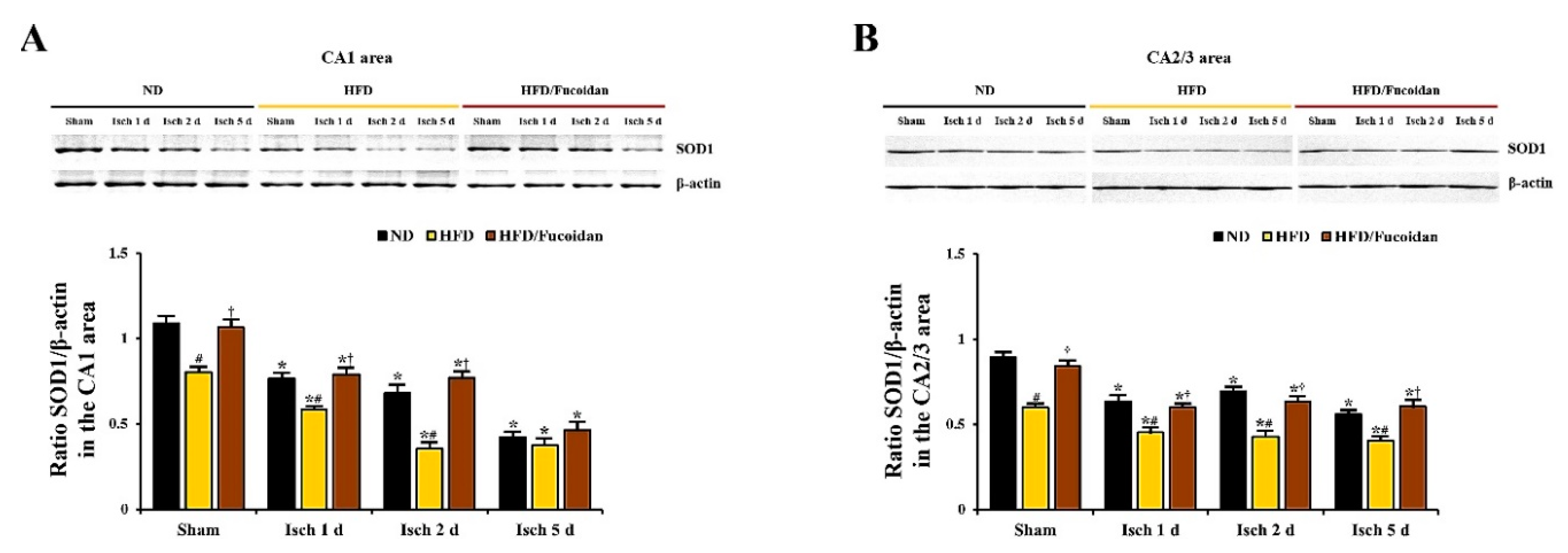
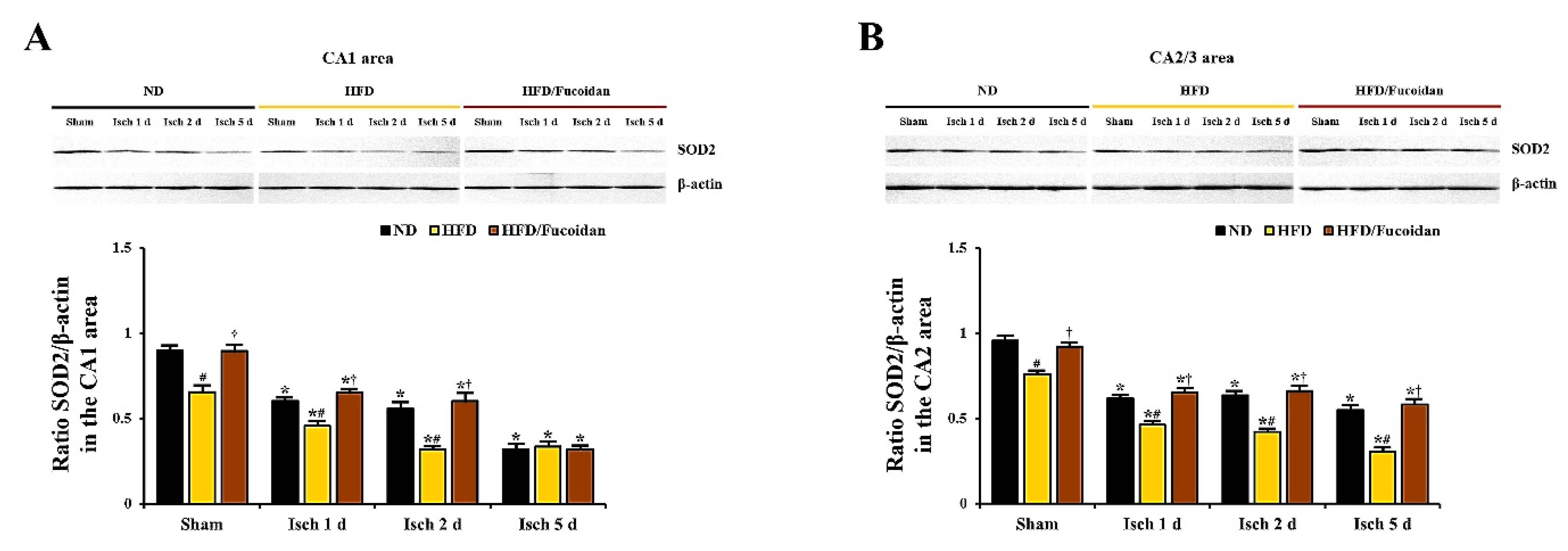
2.5. Enhancement of Antioxidants (SODs) by Fucoidan in HFD-Ischemia Group
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
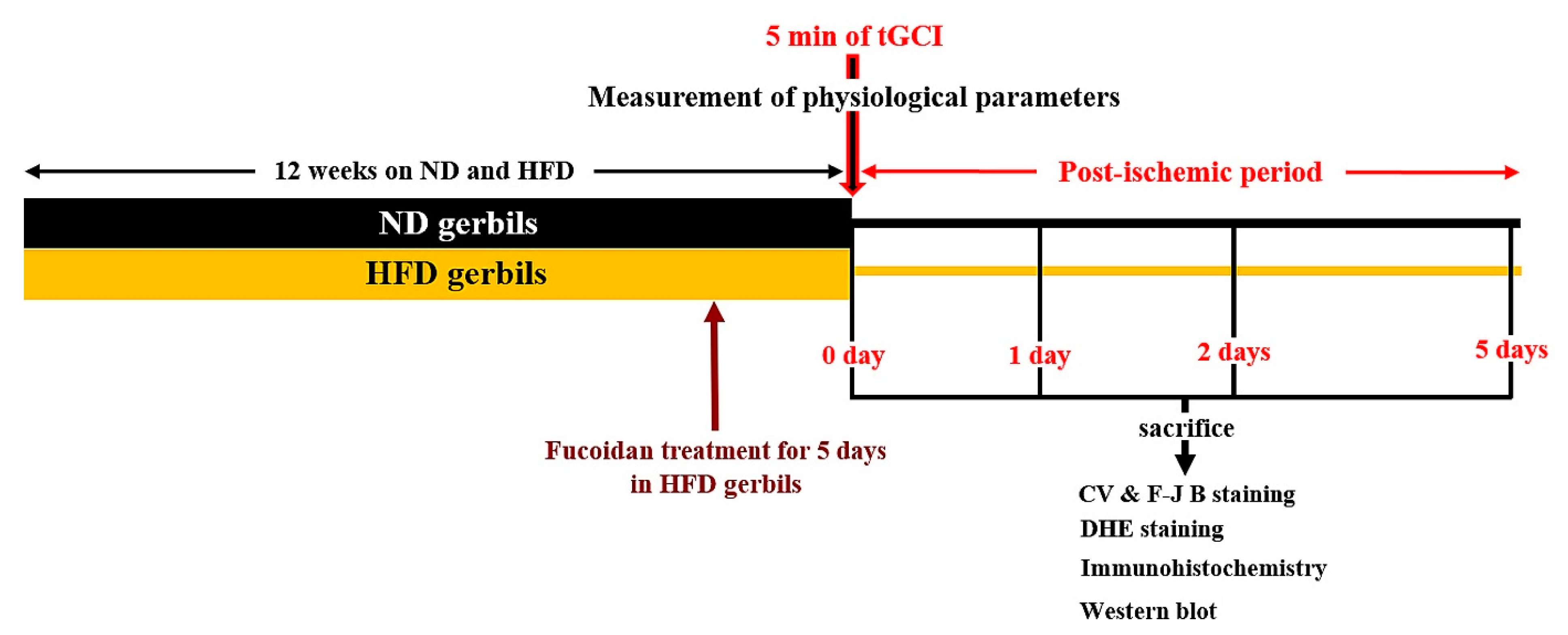
4.2. Experimental Design and Fucoidan Administration
4.3. Analysis of Body Weight, Glucose Level and Lipid Profiles
4.4. Induction of tGCI
4.5. Preparation of Histological Sections
4.6. Cresyl Violet (CV) Staining
4.7. Fluoro-Jade B (F-J B) Histofluorescence Staining
4.8. Dihydroethidium (DHE) Fluorescence Staining
4.9. Immunohistochemistry
4.10. Western Blot Analysis
4.11. Data Analysis
4.12. Statistical Analysis
Author Contributions
Funding
References
- Kawai, K.; Nitecka, L.; Ruetzler, C.A.; Nagashima, G.; Joo, F.; Mies, G.; Nowak, T.S., Jr.; Saito, N.; Lohr, J.M.; Klatzo, I. Global cerebral ischemia associated with cardiac arrest in the rat: I. Dynamics of early neuronal changes. J. Cereb. Blood Flow Metab. 1992, 12, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.T.; Cohan, C.H.; Dave, K.R.; Wright, C.B.; Perez-Pinzon, M.A. Global cerebral ischemia: Synaptic and cognitive dysfunction. Curr. Drug Targets 2013, 14, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Park, J.H.; Ahn, J.H.; Kim, I.H.; Cho, J.H.; Choi, J.H.; Yoo, K.Y.; Lee, C.H.; Hwang, I.K.; Cho, J.H.; et al. New GABAergic Neurogenesis in the Hippocampal CA1 Region of a Gerbil Model of Long-Term Survival after Transient Cerebral Ischemic Injury. Brain Pathol. 2016, 26, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Nitatori, T.; Sato, N.; Waguri, S.; Karasawa, Y.; Araki, H.; Shibanai, K.; Kominami, E.; Uchiyama, Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J. Neurosci. 1995, 15, 1001–1011. [Google Scholar] [CrossRef]
- Harukuni, I.; Bhardwaj, A. Mechanisms of brain injury after global cerebral ischemia. Neurol. Clin. 2006, 24, 1–21. [Google Scholar] [CrossRef]
- Chan, P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001, 21, 2–14. [Google Scholar] [CrossRef]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta 2010, 1802, 92–99. [Google Scholar] [CrossRef]
- Ahmed, E.; Donovan, T.; Yujiao, L.; Zhang, Q. Mitochondrial Targeted Antioxidant in Cerebral Ischemia. J. Neurol. Neurosci. 2015, 6, 17. [Google Scholar] [CrossRef]
- Lee, J.C.; Won, M.H. Neuroprotection of antioxidant enzymes against transient global cerebral ischemia in gerbils. Anat. Cell Biol. 2014, 47, 149–156. [Google Scholar] [CrossRef]
- Mitchell, A.B.; Cole, J.W.; McArdle, P.F.; Cheng, Y.C.; Ryan, K.A.; Sparks, M.J.; Mitchell, B.D.; Kittner, S.J. Obesity increases risk of ischemic stroke in young adults. Stroke 2015, 46, 1690–1692. [Google Scholar] [CrossRef]
- Morrison, C.D.; Pistell, P.J.; Ingram, D.K.; Johnson, W.D.; Liu, Y.; Fernandez-Kim, S.O.; White, C.L.; Purpera, M.N.; Uranga, R.M.; Bruce-Keller, A.J.; et al. High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: Implications for decreased Nrf2 signaling. J. Neurochem. 2010, 114, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Skolarus, L.E.; Sanchez, B.N.; Levine, D.A.; Baek, J.; Kerber, K.A.; Morgenstern, L.B.; Smith, M.A.; Lisabeth, L.D. Association of body mass index and mortality after acute ischemic stroke. Circ. Cardiovasc. Qual. Outcomes 2014, 7, 64–69. [Google Scholar] [CrossRef]
- Sun, W.; Huang, Y.; Xian, Y.; Zhu, S.; Jia, Z.; Liu, R.; Li, F.; Wei, J.W.; Wang, J.G.; Liu, M.; et al. Association of body mass index with mortality and functional outcome after acute ischemic stroke. Sci. Rep. 2017, 7, 2507. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Zhang, J.; Feng, C.; Xiong, L.; Zuo, Z. Critical role of matrix metalloprotease-9 in chronic high fat diet-induced cerebral vascular remodelling and increase of ischaemic brain injury in micedagger. Cardiovasc. Res. 2014, 103, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Maysami, S.; Haley, M.J.; Gorenkova, N.; Krishnan, S.; McColl, B.W.; Lawrence, C.B. Prolonged diet-induced obesity in mice modifies the inflammatory response and leads to worse outcome after stroke. J. Neuroinflamm. 2015, 12, 140. [Google Scholar] [CrossRef]
- Song, M.; Ahn, J.H.; Kim, H.; Kim, D.W.; Lee, T.K.; Lee, J.C.; Kim, Y.M.; Lee, C.H.; Hwang, I.K.; Yan, B.C.; et al. Chronic high-fat diet-induced obesity in gerbils increases pro-inflammatory cytokines and mTOR activation, and elicits neuronal death in the striatum following brief transient ischemia. Neurochem. Int. 2018, 121, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Gaire, B.P. Herbal Medicine in Ischemic Stroke: Challenges and Prospective. Chin. J. Integr. Med. 2018, 24, 243–246. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Z.; Chen, X.; You, H.; Xue, J.; Cai, D.; Zheng, Y.; Xu, Y.; Luo, D. Low molecular-weight fucoidan protects against hindlimb ischemic injury in type 2 diabetic mice through enhancing endothelial nitric oxide synthase phosphorylation. J. Diabetes 2018, 10, 820–834. [Google Scholar] [CrossRef]
- Park, H.Y.; Han, M.H.; Park, C.; Jin, C.Y.; Kim, G.Y.; Choi, I.W.; Kim, N.D.; Nam, T.J.; Kwon, T.K.; Choi, Y.H. Anti-inflammatory effects of fucoidan through inhibition of NF-kappaB, MAPK and Akt activation in lipopolysaccharide-induced BV2 microglia cells. Food Chem. Toxicol. 2011, 49, 1745–1752. [Google Scholar] [CrossRef]
- Phull, A.R.; Majid, M.; Haq, I.U.; Khan, M.R.; Kim, S.J. In vitro and in vivo evaluation of anti-arthritic, antioxidant efficacy of fucoidan from Undaria pinnatifida (Harvey) Suringar. Int. J. Biol. Macromol. 2017, 97, 468–480. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, F.; Hu, J.; Zhang, L.; Xue, C.; Zhang, Z.; Li, B. Antithrombotic activity of oral administered low molecular weight fucoidan from Laminaria Japonica. Thromb. Res. 2016, 144, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Che, N.; Ma, Y.; Xin, Y. Protective Role of Fucoidan in Cerebral Ischemia-Reperfusion Injury through Inhibition of MAPK Signaling Pathway. Biomol. Ther. 2017, 25, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ahn, J.H.; Song, M.; Kim, D.W.; Lee, T.-K.; Lee, J.-C.; Kim, Y.-M.; Kim, J.-D.; Cho, J.H.; Hwang, I.K. Pretreated fucoidan confers neuroprotection against transient global cerebral ischemic injury in the gerbil hippocampal CA1 area via reducing of glial cell activation and oxidative stress. Biomed. Pharmacother. 2019, 109, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.H.; Yan, B.C.; Cho, G.S.; Kim, W.K.; Lee, C.H.; Cho, J.H.; Kim, M.; Kang, I.J.; Won, M.H.; Lee, J.C. Neuroprotective effect of fucoidin on lipopolysaccharide accelerated cerebral ischemic injury through inhibition of cytokine expression and neutrophil infiltration. J. Neurol. Sci. 2012, 318, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, T.; Chen, X.; You, H.; Zhang, Q.; Xue, J.; Zheng, Y.; Luo, D. Low molecular weight fucoidan ameliorates hindlimb ischemic injury in type 2 diabetic rats. J. Ethnopharmacol. 2018, 210, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Clifton, G.L.; Taft, W.C.; Blair, R.E.; Choi, S.C.; DeLorenzo, R.J. Conditions for pharmacologic evaluation in the gerbil model of forebrain ischemia. Stroke 1989, 20, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Ventura, L.L.; Fortes, N.C.; Santiago, H.C.; Caliari, M.V.; Gomes, M.A.; Oliveira, D.R. Obesity-induced diet leads to weight gain, systemic metabolic alterations, adipose tissue inflammation, hepatic steatosis, and oxidative stress in gerbils (Meriones unguiculatus). PeerJ 2017, 5, e2967. [Google Scholar] [CrossRef] [PubMed]
- Du, X.Y.; Zhu, X.D.; Dong, G.; Lu, J.; Wang, Y.; Zeng, L.; Zhao, T.Y.; Ye, H.H.; Li, R.S.; Bai, J.Y.; et al. Characteristics of circle of Willis variations in the mongolian gerbil and a newly established ischemia-prone gerbil group. ILAR J. 2011, 52, E1–E7. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.S.; Machado, J.M.; Perez-Saad, H.; Coro-Antich, R.M.; Berlanga-Acosta, J.A.; Salgueiro, S.R.; Illera, G.G.; Alba, J.S.; del Barco, D.G. Global brain ischemia in Mongolian gerbils: Assessing the level of anastomosis in the cerebral circle of Willis. Acta Neurobiol. Exp. 2012, 72, 377–384. [Google Scholar]
- Majid, A. Neuroprotection in stroke: Past, present, and future. ISRN Neurol. 2014, 2014, 515716. [Google Scholar] [CrossRef]
- Ankolekar, S.; Rewell, S.; Howells, D.W.; Bath, P.M. The influence of stroke risk factors and comorbidities on assessment of stroke therapies in humans and animals. Int. J. Stroke 2012, 7, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, A.A.; Rabie, T.; Sutherland, B.A.; Papadakis, M.; Hadley, G.; Cai, R.; Buchan, A.M. Importance of preclinical research in the development of neuroprotective strategies for ischemic stroke. JAMA Neurol. 2014, 71, 634–639. [Google Scholar] [CrossRef] [PubMed]
- ElAli, A.; Doeppner, T.R.; Zechariah, A.; Hermann, D.M. Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: Role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9, and RhoA overactivation. Stroke 2011, 42, 3238–3244. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Prakash, R.; Chawla, D.; Du, W.; Didion, S.P.; Filosa, J.A.; Zhang, Q.; Brann, D.W.; Lima, V.V.; Tostes, R.C.; et al. Early effects of high-fat diet on neurovascular function and focal ischemic brain injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R1001–R1008. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.H.; Yan, B.C.; Chen, B.H.; Park, J.H.; Ahn, J.H.; Kim, I.H.; Lee, J.C.; Park, Y.S.; Kim, M.J.; Lee, Y.L.; et al. Accelerated and exacerbated effects of high dietary fat on neuronal damage induced by transient cerebral ischemia in the gerbil septum. Endocrinol. Metab. 2014, 29, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.C.; Park, J.H.; Ahn, J.H.; Kim, I.H.; Lee, J.C.; Yoo, K.Y.; Choi, J.H.; Hwang, I.K.; Cho, J.H.; Kwon, Y.G.; et al. Effects of high-fat diet on neuronal damage, gliosis, inflammatory process and oxidative stress in the hippocampus induced by transient cerebral ischemia. Neurochem. Res. 2014, 39, 2465–2478. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kastner, R.; Freund, T.F. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 1991, 40, 599–636. [Google Scholar] [CrossRef]
- Yu, D.K.; Yoo, K.Y.; Shin, B.N.; Kim, I.H.; Park, J.H.; Lee, C.H.; Choi, J.H.; Cho, Y.J.; Kang, I.J.; Kim, Y.M.; et al. Neuronal damage in hippocampal subregions induced by various durations of transient cerebral ischemia in gerbils using Fluoro-Jade B histofluorescence. Brain Res. 2012, 1437, 50–57. [Google Scholar] [CrossRef]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef]
- Saito, A.; Maier, C.M.; Narasimhan, P.; Nishi, T.; Song, Y.S.; Yu, F.; Liu, J.; Lee, Y.S.; Nito, C.; Kamada, H.; et al. Oxidative stress and neuronal death/survival signaling in cerebral ischemia. Mol. Neurobiol. 2005, 31, 105–116. [Google Scholar] [CrossRef]
- Hajiluian, G.; Abbasalizad Farhangi, M.; Nameni, G.; Shahabi, P.; Megari-Abbasi, M. Oxidative stress-induced cognitive impairment in obesity can be reversed by vitamin D administration in rats. Nutr. Neurosci. 2018, 21, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Warner, D.S.; Sheng, H.; Batinic-Haberle, I. Oxidants, antioxidants and the ischemic brain. J. Exp. Biol. 2004, 207, 3221–3231. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Murakami, K.; Fujimura, M.; Morita-Fujimura, Y.; Gasche, Y.; Kondo, T.; Scott, R.W.; Chan, P.H. Exacerbation of delayed cell injury after transient global ischemia in mutant mice with CuZn superoxide dismutase deficiency. Stroke 1999, 30, 1962–1968. [Google Scholar] [CrossRef]
- Mehta, S.L.; Lin, Y.; Chen, W.; Yu, F.; Cao, L.; He, Q.; Chan, P.H.; Li, P.A. Manganese superoxide dismutase deficiency exacerbates ischemic brain damage under hyperglycemic conditions by altering autophagy. Transl. Stroke Res. 2011, 2, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Kindy, M.S.; Holtsberg, F.W.; St Clair, D.K.; Yen, H.C.; Germeyer, A.; Steiner, S.M.; Bruce-Keller, A.J.; Hutchins, J.B.; Mattson, M.P. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: Suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci. 1998, 18, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Kinouchi, H.; Epstein, C.J.; Mizui, T.; Carlson, E.; Chen, S.F.; Chan, P.H. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc. Natl. Acad. Sci. USA 1991, 88, 11158–11162. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Noshita, N.; Lewen, A.; Gasche, Y.; Ferrand-Drake, M.; Fujimura, M.; Morita-Fujimura, Y.; Chan, P.H. Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J. Neurosci. 2002, 22, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Park, J.H.; Yoo, K.Y.; Choi, J.H.; Hwang, I.K.; Ryu, P.D.; Kim, D.H.; Kwon, Y.G.; Kim, Y.M.; Won, M.H. Pre- and post-treatments with escitalopram protect against experimental ischemic neuronal damage via regulation of BDNF expression and oxidative stress. Exp. Neurol. 2011, 229, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, Y.Y.; Liu, X.Y.; Jia, S.W.; Zhao, J.; Cui, D.; Wang, L. Resveratrol protects neurons and the myocardium by reducing oxidative stress and ameliorating mitochondria damage in a cerebral ischemia rat model. Cell. Physiol. Biochem. 2014, 34, 854–864. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, C.; Li, T.; Wang, W.; Ye, W.; Zeng, R.; Ni, L.; Lai, Z.; Wang, X.; Liu, C. The protective effect of melatonin on brain ischemia and reperfusion in rats and humans: In vivo assessment and a randomized controlled trial. J. Pineal Res. 2018, 65, e12521. [Google Scholar] [CrossRef]
- Gao, Y.; Dong, C.; Yin, J.; Shen, J.; Tian, J.; Li, C. Neuroprotective effect of fucoidan on H2O2-induced apoptosis in PC12 cells via activation of PI3K/Akt pathway. Cell. Mol. Neurobiol. 2012, 32, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Park, J.H.; Kim, I.H.; Cho, G.S.; Ahn, J.H.; Tae, H.J.; Choi, S.Y.; Cho, J.H.; Kim, D.W.; Kwon, Y.G.; et al. Neuroprotection of ischemic preconditioning is mediated by thioredoxin 2 in the hippocampal CA1 region following a subsequent transient cerebral ischemia. Brain Pathol. 2017, 27, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.K.; Yoo, K.Y.; Kim, D.S.; Kang, T.C.; Lee, B.H.; Kim, Y.S.; Won, M.H. Chronological distribution of Rip immunoreactivity in the gerbil hippocampus during normal aging. Neurochem. Res. 2006, 31, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Radtke-Schuller, S.; Schuller, G.; Angenstein, F.; Grosser, O.S.; Goldschmidt, J.; Budinger, E. Brain atlas of the Mongolian gerbil (Meriones unguiculatus) in CT/MRI-aided stereotaxic coordinates. Brain Struct. Funct. 2016, 221, 1–272. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Park, C.W.; Ahn, J.H.; Choi, S.Y.; Shin, M.C.; Cho, J.H.; Lee, T.K.; Kim, I.H.; Cho, J.H.; Lee, J.C.; et al. Neuroprotection and reduced gliosis by pre- and post-treatments of hydroquinone in a gerbil model of transient cerebral ischemia. Chem. Biol. Interact. 2017, 278, 230–238. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | ND | HFD | HFD/Fucoidan |
|---|---|---|---|
| Body weight (g) | 81.2 ± 0.7 | 112.5 ± 1.3 * | 105.7 ± 0.5 * |
| Glucose (mg/dL) | 95.5 ± 2.5 | 181.2 ± 3.7 * | 170.1 ± 2.9 * |
| Triglyceride (mg/dL) | 91.6 ± 3.8 | 169.3 ± 4.2 * | 162.7 ± 3.5 * |
| Total cholesterol (mg/dL) | 101.4 ± 4.2 | 185.9 ± 5.1 * | 179.8 ± 6.2 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, J.H.; Shin, M.C.; Kim, D.W.; Kim, H.; Song, M.; Lee, T.-K.; Lee, J.-C.; Kim, H.; Cho, J.H.; Kim, Y.-M.; et al. Antioxidant Properties of Fucoidan Alleviate Acceleration and Exacerbation of Hippocampal Neuronal Death Following Transient Global Cerebral Ischemia in High-Fat Diet-Induced Obese Gerbils. Int. J. Mol. Sci. 2019, 20, 554. https://doi.org/10.3390/ijms20030554
Ahn JH, Shin MC, Kim DW, Kim H, Song M, Lee T-K, Lee J-C, Kim H, Cho JH, Kim Y-M, et al. Antioxidant Properties of Fucoidan Alleviate Acceleration and Exacerbation of Hippocampal Neuronal Death Following Transient Global Cerebral Ischemia in High-Fat Diet-Induced Obese Gerbils. International Journal of Molecular Sciences. 2019; 20(3):554. https://doi.org/10.3390/ijms20030554
Chicago/Turabian StyleAhn, Ji Hyeon, Myoung Cheol Shin, Dae Won Kim, Hyunjung Kim, Minah Song, Tae-Kyeong Lee, Jae-Chul Lee, Hyeyoung Kim, Jun Hwi Cho, Young-Myeong Kim, and et al. 2019. "Antioxidant Properties of Fucoidan Alleviate Acceleration and Exacerbation of Hippocampal Neuronal Death Following Transient Global Cerebral Ischemia in High-Fat Diet-Induced Obese Gerbils" International Journal of Molecular Sciences 20, no. 3: 554. https://doi.org/10.3390/ijms20030554
APA StyleAhn, J. H., Shin, M. C., Kim, D. W., Kim, H., Song, M., Lee, T.-K., Lee, J.-C., Kim, H., Cho, J. H., Kim, Y.-M., Kim, J.-D., Choi, S. Y., Won, M.-H., & Park, J. H. (2019). Antioxidant Properties of Fucoidan Alleviate Acceleration and Exacerbation of Hippocampal Neuronal Death Following Transient Global Cerebral Ischemia in High-Fat Diet-Induced Obese Gerbils. International Journal of Molecular Sciences, 20(3), 554. https://doi.org/10.3390/ijms20030554