Pseudoxanthoma Elasticum, Kidney Stones and Pyrophosphate: From a Rare Disease to Urolithiasis and Vascular Calcifications

{kind=link}
Abstract
1. Introduction: Pseudoxanthoma Elasticum and Related Diseases
1.1. Clinical Manifestations
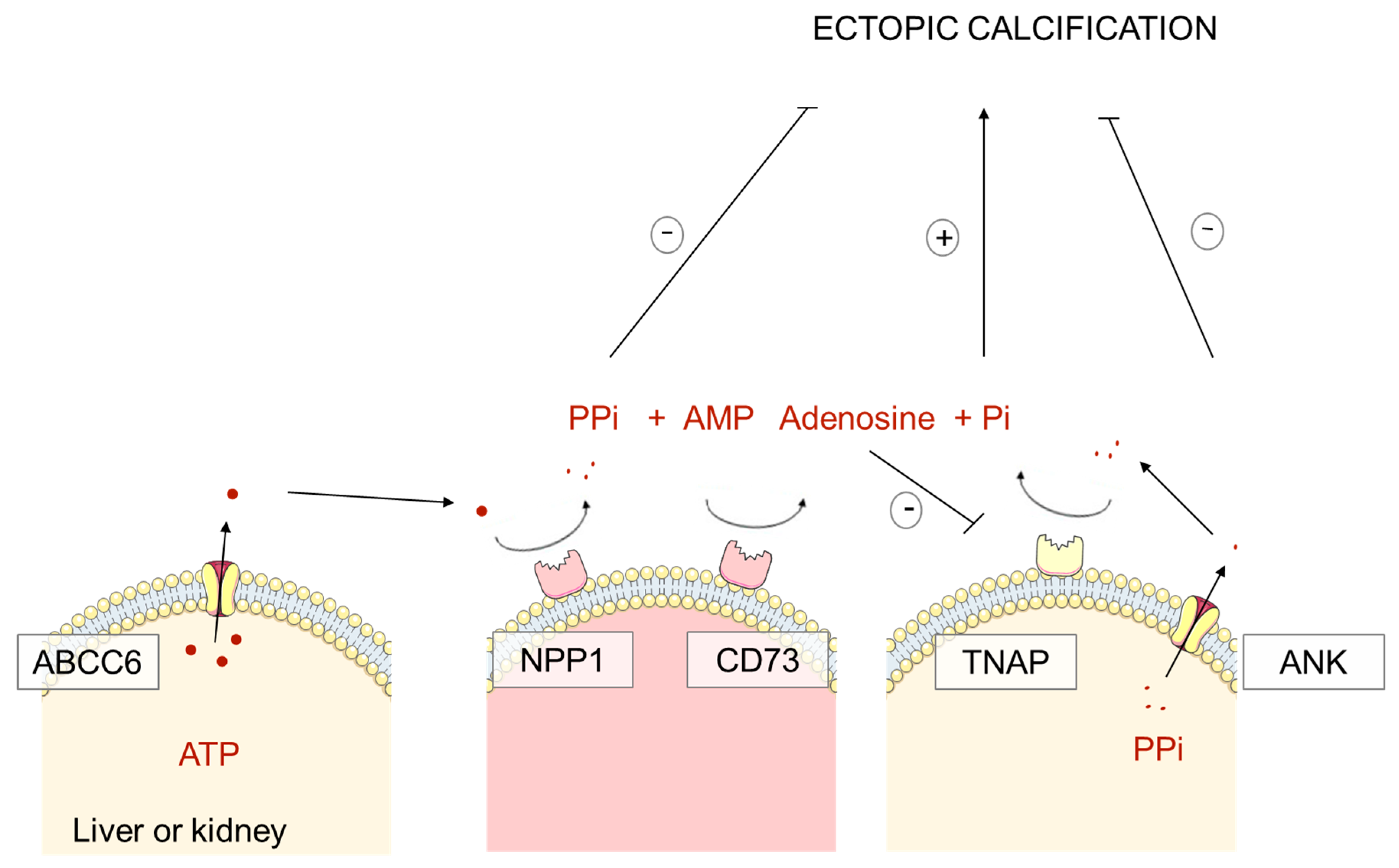
1.2. Pathophysiology of PXE, GACI and ACDC: Pyrophosphate Deficiency
2. Pseudoxanthoma Elasticum and Kidney Calcifications
2.1. Nephrocalcinosis and Kidney Stones: Preliminary Reports
2.2. High Prevalence of Kidney Stones in PXE Patients
2.3. Abcc6−/− Mice: A Murine Model of Randall’s Plaque
3. The Mystery of Randall’s Plaque Formation: A Role for Calcification Inhibitors and Pyrophosphate?
3.1. Randall’s Plaque: the Origin of Renal Calculi
3.2. Determinants of Randall’s Plaque Formation
3.3. Randall’s Plaque and Calcification Inhibitors
3.4. The Abcc6−/− Murine Model: A Tool to Identify Randall’s Plaque Determinants and New Therapeutics
4. Pyrophosphate Deficiency, the Common Link between Vascular Calcifications, Kidney Stones and Chronic Kidney Disease?
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PXE | Pseudoxanthoma elasticum |
| VEGF | Vascular Endothelial Growth Factor |
| GACI | Generalized arterial calcification of infancy |
| (E)NPP1 | ectonucleotide pyrophosphatase phosphodiesterase |
| ACDC | Arterial calcification due to deficiency of CD73 |
| PPi | Pyrophosphate |
| CT | Computed Tomography |
| CKD | Chronic kidney disease |
| TNAP | Tissue nonspecific alkaline phosphatase |
References
- Le Saux, O.; Urban, Z.; Tschuch, C.; Csiszar, K.; Bacchelli, B.; Quaglino, D.; Pasquali-Ronchetti, I.; Pope, F.M.; Richards, A.; Terry, S.; et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Beck, K.; Sachsinger, C.; Silvestri, C.; Treiber, C.; Göring, H.H.; Johnson, E.W.; De Paepe, A.; Pope, F.M.; Pasquali-Ronchetti, I.; et al. A spectrum of ABCC6 mutations is responsible for pseudoxanthoma elasticum. Am. J. Hum. Genet. 2001, 69, 749–764. [Google Scholar] [CrossRef]
- Neidner, K.H. History. Clin. Dermatol. 1988, 6, 1–4. [Google Scholar] [CrossRef]
- Uitto, J.; Li, Q.; Jiang, Q. Pseudoxanthoma Elasticum: Molecular Genetics and Putative Pathomechanisms. J. Investig. Dermatol. 2010, 130, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Lefthériotis, G.; Omarjee, L.; Le Saux, O.; Henrion, D.; Abraham, P.; Prunier, F.; Willoteaux, S.; Martin, L. The vascular phenotype in Pseudoxanthoma elasticum and related disorders: Contribution of a genetic disease to the understanding of vascular calcification. Front. Genet. 2013, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Naouri, M.; Boisseau, C.; Bonicel, P.; Daudon, P.; Bonneau, D.; Chassaing, N.; Martin, L. Manifestations of pseudoxanthoma elasticum in childhood. Br. J. Dermatol. 2009, 161, 635–639. [Google Scholar] [CrossRef]
- Hosen, M.J.; Lamoen, A.; De Paepe, A.; Vanakker, O.M. Histopathology of Pseudoxanthoma Elasticum and Related Disorders: Histological Hallmarks and Diagnostic Clues. Scientifica 2012, 2012, 598262. [Google Scholar] [CrossRef]
- Audo, I.; Vanakker, O.M.; Smith, A.; Leroy, B.P.; Robson, A.G.; Jenkins, S.A.; Coucke, P.J.; Bird, A.C.; Paepe, A.D.; Holder, G.E.; et al. Pseudoxanthoma Elasticum with Generalized Retinal Dysfunction, a Common Finding? Investig. Ophthalmol. Vis. Sci. 2007, 48, 4250–4256. [Google Scholar] [CrossRef]
- Ebran, J.-M.; Milea, D.; Trelohan, A.; Bonicel, P.; Hamel, J.-F.; Leftheriotis, G.; Martin, L. New insights into the visual prognosis of pseudoxanthoma elasticum. Br. J. Ophthalmol. 2014, 98, 142–143. [Google Scholar] [CrossRef][Green Version]
- De Vilder, E.Y.G.; Cardoen, S.; Hosen, M.J.; Le Saux, O.; De Zaeytijd, J.; Leroy, B.P.; De Reuck, J.; Coucke, P.J.; De Paepe, A.; Hemelsoet, D.; et al. Pathogenic variants in the ABCC6 gene are associated with an increased risk for ischemic stroke. Brain Pathol. Zurich Switz. 2018, 28, 822–831. [Google Scholar] [CrossRef]
- Legrand, A.; Cornez, L.; Samkari, W.; Mazzella, J.-M.; Venisse, A.; Boccio, V.; Auribault, K.; Keren, B.; Benistan, K.; Germain, D.P.; et al. Mutation spectrum in the ABCC6 gene and genotype-phenotype correlations in a French cohort with pseudoxanthoma elasticum. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Prunier, F.; Terrien, G.; Le Corre, Y.; Apana, A.L.Y.; Bière, L.; Kauffenstein, G.; Furber, A.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Le Saux, O.; et al. Pseudoxanthoma elasticum: Cardiac findings in patients and Abcc6-deficient mouse model. PLoS ONE 2013, 8, e68700. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Boutouyrie, P.; Laloux, B.; Laurent, S. Arterial Remodeling and Stiffness in Patients With Pseudoxanthoma Elasticum. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P. Pseudoxanthoma elasticum. Orphanet J. Rare Dis. 2017, 12, 85. [Google Scholar] [CrossRef] [PubMed]
- Leftheriotis, G.; Kauffenstein, G.; Hamel, J.F.; Abraham, P.; Le Saux, O.; Willoteaux, S.; Henrion, D.; Martin, L. The contribution of arterial calcification to peripheral arterial disease in pseudoxanthoma elasticum. PLoS ONE 2014, 9, e96003. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, G.; de Jong, P.A.; Bartstra, J.W.; Lagerweij, S.J.; Lam, M.G.; Ossewaarde-van Norel, J.; Risseeuw, S.; van Leeuwen, R.; Imhof, S.M.; Verhaar, H.J.; et al. Etidronate for Prevention of Ectopic Mineralization in Patients With Pseudoxanthoma Elasticum. J. Am. Coll. Cardiol. 2018, 71, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.; le Saux, O.; Pomozi, V.; Aherrahrou, R.; Kriesen, R.; Stölting, S.; Liebers, A.; Kessler, T.; Schunkert, H.; Erdmann, J.; et al. Etidronate prevents dystrophic cardiac calcification by inhibiting macrophage aggregation. Sci. Rep. 2018, 8, 5812. [Google Scholar] [CrossRef]
- Ruf, N.; Uhlenberg, B.; Terkeltaub, R.; Nürnberg, P.; Rutsch, F. The mutational spectrum of ENPP1 as arising after the analysis of 23 unrelated patients with generalized arterial calcification of infancy (GACI). Hum. Mutat. 2005, 25, 98. [Google Scholar] [CrossRef]
- Nitschke, Y.; Baujat, G.; Botschen, U.; Wittkampf, T.; du Moulin, M.; Stella, J.; Le Merrer, M.; Guest, G.; Lambot, K.; Tazarourte-Pinturier, M.-F.; et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am. J. Hum. Genet. 2012, 90, 25–39. [Google Scholar] [CrossRef]
- St Hilaire, C.; Ziegler, S.G.; Markello, T.C.; Brusco, A.; Groden, C.; Gill, F.; Carlson-Donohoe, H.; Lederman, R.J.; Chen, M.Y.; Yang, D.; et al. NT5E mutations and arterial calcifications. N. Engl. J. Med. 2011, 364, 432–442. [Google Scholar] [CrossRef]
- Nitschke, Y.; Rutsch, F. Inherited Arterial Calcification Syndromes: Etiologies and Treatment Concepts. Curr. Osteoporos. Rep. 2017, 15, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Favre, G.; Laurain, A.; Aranyi, T.; Szeri, F.; Fulop, K.; Le Saux, O.; Duranton, C.; Kauffenstein, G.; Martin, L.; Lefthériotis, G. The ABCC6 Transporter: A New Player in Biomineralization. Int. J. Mol. Sci. 2017, 18, 1941. [Google Scholar] [CrossRef] [PubMed]
- Pomozi, V.; Le Saux, O.; Brampton, C.; Apana, A.; Iliás, A.; Szeri, F.; Martin, L.; Monostory, K.; Paku, S.; Sarkadi, B.; et al. ABCC6 is a basolateral plasma membrane protein. Circ. Res. 2013, 112, e148–e151. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Bunda, S.; VanWart, C.M.; Douet, V.; Got, L.; Martin, L.; Hinek, A. Serum factors from pseudoxanthoma elasticum patients alter elastic fiber formation in vitro. J. Investig. Dermatol. 2006, 126, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Vanakker, O.M.; Martin, L.; Schurgers, L.J.; Quaglino, D.; Costrop, L.; Vermeer, C.; Pasquali-Ronchetti, I.; Coucke, P.J.; De Paepe, A. Low serum vitamin K in PXE results in defective carboxylation of mineralization inhibitors similar to the GGCX mutations in the PXE-like syndrome. Lab. Investig. J. Tech. Methods Pathol. 2010, 90, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Nigwekar, S.U.; Bloch, D.B.; Nazarian, R.M.; Vermeer, C.; Booth, S.L.; Xu, D.; Thadhani, R.I.; Malhotra, R. Vitamin K-Dependent Carboxylation of Matrix Gla Protein Influences the Risk of Calciphylaxis. J. Am. Soc. Nephrol. JASN 2017, 28, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Munroe, P.B.; Olgunturk, R.O.; Fryns, J.P.; Van Maldergem, L.; Ziereisen, F.; Yuksel, B.; Gardiner, R.M.; Chung, E. Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat. Genet. 1999, 21, 142–144. [Google Scholar] [CrossRef]
- Brampton, C.; Yamaguchi, Y.; Vanakker, O.; Van Laer, L.; Chen, L.-H.; Thakore, M.; De Paepe, A.; Pomozi, V.; Szabó, P.T.; Martin, L.; et al. Vitamin K does not prevent soft tissue mineralization in a mouse model of pseudoxanthoma elasticum. Cell Cycle Georget. Tex 2011, 10, 1810–1820. [Google Scholar] [CrossRef]
- Gorgels, T.G.M.F.; Waarsing, J.H.; Herfs, M.; Versteeg, D.; Schoensiegel, F.; Sato, T.; Schlingemann, R.O.; Ivandic, B.T.; Vermeer, C.; Schurgers, L.J.; et al. Vitamin K supplementation increases vitamin K tissue levels but fails to counteract ectopic calcification in a mouse model for pseudoxanthoma elasticum. J. Mol. Med. 2011, 89, 1125–1135. [Google Scholar] [CrossRef]
- Jansen, R.S.; Küçükosmanoglu, A.; de Haas, M.; Sapthu, S.; Otero, J.A.; Hegman, I.E.M.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Borst, P.; van de Wetering, K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 2013, 110, 20206–20211. [Google Scholar] [CrossRef]
- Jansen, R.S.; Duijst, S.; Mahakena, S.; Sommer, D.; Szeri, F.; Váradi, A.; Plomp, A.; Bergen, A.A.; Oude Elferink, R.P.J.; Borst, P.; et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1985–1989. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, H.; Bisaz, S. Mechanism of calcification: Inhibitory role of pyrophosphate. Nature 1962, 195, 911. [Google Scholar] [CrossRef] [PubMed]
- Pomozi, V.; Brampton, C.; van de Wetering, K.; Zoll, J.; Calio, B.; Pham, K.; Owens, J.B.; Marh, J.; Moisyadi, S.; Váradi, A.; et al. Pyrophosphate Supplementation Prevents Chronic and Acute Calcification in ABCC6-Deficient Mice. Am. J. Pathol. 2017, 187, 1258–1272. [Google Scholar] [CrossRef] [PubMed]
- Pomozi, V.; Julian, C.B.; Zoll, J.; Pham, K.; Kuo, S.; Tőkési, N.; Martin, L.; Váradi, A.; Le Saux, O. Dietary Pyrophosphate Modulates Calcification in a Mouse Model of Pseudoxanthoma Elasticum: Implication for Treatment of Patients. J. Investig. Dermatol. 2019, 139, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Dedinszki, D.; Szeri, F.; Kozák, E.; Pomozi, V.; Tőkési, N.; Mezei, T.R.; Merczel, K.; Letavernier, E.; Tang, E.; Le Saux, O.; et al. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol. Med. 2017, 9, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Fabre, B.; Bayle, P.; Bazex, J.; Durand, D.; Lamant, L.; Chassaing, N. Pseudoxanthoma elasticum and nephrolithiasis. J. Eur. Acad. Dermatol. Venereol. 2005, 19, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Chraïbi, R.; Ismaili, N.; Belgnaoui, F.; Akallal, N.; Bouhllab, J.; Senouci, K.; Hassam, B. Pseudoxanthoma elasticum and nephrocalcinosis. Ann. Dermatol. Venereol. 2007, 134, 764–766. [Google Scholar] [CrossRef]
- Gayen, T.; Das, A.; Roy, S.; Biswas, S.; Shome, K.; Chowdhury, S.N. Pseudoxanthoma elasticum and nephrocalcinosis: Incidental finding or an infrequent manifestation? Indian Dermatol. Online J. 2014, 5, 176–178. [Google Scholar]
- Seeger, H.; Mohebbi, N. Pseudoxanthoma elasticum and nephrocalcinosis. Kidney Int. 2016, 89, 1407. [Google Scholar] [CrossRef]
- Li, Q.; Chou, D.W.; Price, T.P.; Sundberg, J.P.; Uitto, J. Genetic modulation of nephrocalcinosis in mouse models of ectopic mineralization: The Abcc6(tm1Jfk) and Enpp1(asj) mutant mice. Lab. Investig. J. Tech. Methods Pathol. 2014, 94, 623–632. [Google Scholar] [CrossRef]
- Letavernier, E.; Kauffenstein, G.; Huguet, L.; Navasiolava, N.; Bouderlique, E.; Tang, E.; Delaitre, L.; Bazin, D.; de Frutos, M.; Gay, C.; et al. ABCC6 Deficiency Promotes Development of Randall Plaque. J. Am. Soc. Nephrol. 2018, 29, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Gorgels, T.G.M.F.; Hu, X.; Scheffer, G.L.; van der Wal, A.C.; Toonstra, J.; de Jong, P.T.V.M.; van Kuppevelt, T.H.; Levelt, C.N.; de Wolf, A.; Loves, W.J.P.; et al. Disruption of Abcc6 in the mouse: Novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum. Mol. Genet. 2005, 14, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Randall, A. THE ORIGIN AND GROWTH OF RENAL CALCULI. Ann. Surg. 1937, 105, 1009–1027. [Google Scholar] [CrossRef] [PubMed]
- Vermooten, V. The incidence and significance of the deposition of calcium plaques in the renal papilla as observed in the Caucasian and Bantu population in South Africa. J. Urol. 1941, 193–196. [Google Scholar] [CrossRef]
- Low, R.K.; Stoller, M.L. Endoscopic mapping of renal papillae for Randall’s plaques in patients with urinary stone disease. J. Urol. 1997, 158, 2062–2064. [Google Scholar] [CrossRef]
- Matlaga, B.R.; Williams, J.C.; Kim, S.C.; Kuo, R.L.; Evan, A.P.; Bledsoe, S.B.; Coe, F.L.; Worcester, E.M.; Munch, L.C.; Lingeman, J.E. Endoscopic evidence of calculus attachment to Randall’s plaque. J. Urol. 2006, 175, 1720–1724. [Google Scholar] [CrossRef]
- Khan, S.R.; Pearle, M.S.; Robertson, W.G.; Gambaro, G.; Canales, B.K.; Doizi, S.; Traxer, O.; Tiselius, H.-G. Kidney stones. Nat. Rev. Dis. Primer 2016, 2, 16008. [Google Scholar] [CrossRef]
- Daudon, M.; Bazin, D.; Letavernier, E. Randall’s plaque as the origin of calcium oxalate kidney stones. Urolithiasis 2015, 43, 5–11. [Google Scholar] [CrossRef]
- Letavernier, E.; Bazin, D.; Daudon, M. Randall’s plaque and kidney stones: Recent advances and future challenges. Comptes Rendus Chim. 2016, 19, 1456–1460. [Google Scholar] [CrossRef]
- Daudon, M.; Letavernier, E.; Frochot, V.; Haymann, J.-P.; Bazin, D.; Jungers, P. Respective influence of calcium and oxalate urine concentration on the formation of calcium oxalate monohydrate or dihydrate crystals. Comptes Rendus Chim. 2016, 19, 1504–1513. [Google Scholar] [CrossRef]
- Ziemba, J.B.; Matlaga, B.R. Epidemiology and economics of nephrolithiasis. Investig. Clin. Urol. 2017, 58, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Letavernier, E.; Vandermeersch, S.; Traxer, O.; Tligui, M.; Baud, L.; Ronco, P.; Haymann, J.-P.; Daudon, M. Demographics and characterization of 10,282 Randall plaque-related kidney stones: A new epidemic? Medicine (Baltimore) 2015, 94, e566. [Google Scholar] [CrossRef]
- Evan, A.P.; Lingeman, J.E.; Coe, F.L.; Parks, J.H.; Bledsoe, S.B.; Shao, Y.; Sommer, A.J.; Paterson, R.F.; Kuo, R.L.; Grynpas, M. Randall’s plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J. Clin. Investig. 2003, 111, 607–616. [Google Scholar] [CrossRef]
- Asplin, J.R.; Mandel, N.S.; Coe, F.L. Evidence of calcium phosphate supersaturation in the loop of Henle. Am. J. Physiol. 1996, 270, F604–F613. [Google Scholar] [CrossRef]
- Lieske, J.C.; Norris, R.; Swift, H.; Toback, F.G. Adhesion, internalization and metabolism of calcium oxalate monohydrate crystals by renal epithelial cells. Kidney Int. 1997, 52, 1291–1301. [Google Scholar] [CrossRef]
- Verrier, C.; Bazin, D.; Huguet, L.; Stéphan, O.; Gloter, A.; Verpont, M.-C.; Frochot, V.; Haymann, J.-P.; Brocheriou, I.; Traxer, O.; et al. Topography, Composition and Structure of Incipient Randall Plaque at the Nanoscale Level. J. Urol. 2016, 196, 1566–1574. [Google Scholar] [CrossRef]
- Stoller, M.L.; Low, R.K.; Shami, G.S.; McCormick, V.D.; Kerschmann, R.L. High resolution radiography of cadaveric kidneys: Unraveling the mystery of Randall’s plaque formation. J. Urol. 1996, 156, 1263–1266. [Google Scholar] [CrossRef]
- Kuo, R.L.; Lingeman, J.E.; Evan, A.P.; Paterson, R.F.; Parks, J.H.; Bledsoe, S.B.; Munch, L.C.; Coe, F.L. Urine calcium and volume predict coverage of renal papilla by Randall’s plaque. Kidney Int. 2003, 64, 2150–2154. [Google Scholar] [CrossRef]
- Vermooten, V. The Origin and Development in the Renal Papilla of Randall’s Calcium Plaques. J. Urol. 1942, 48, 27–37. [Google Scholar] [CrossRef]
- Tournus, M.; Seguin, N.; Perthame, B.; Thomas, S.R.; Edwards, A. A model of calcium transport along the rat nephron. Am. J. Physiol. Renal Physiol. 2013, 305, F979–F994. [Google Scholar] [CrossRef][Green Version]
- Granjon, D.; Bonny, O.; Edwards, A. A model of calcium homeostasis in the rat. Am. J. Physiol. Renal Physiol. 2016, 311, F1047–F1062. [Google Scholar] [CrossRef]
- Jungers, D. Lithiase Urinaire, 2nd ed.; Lavoisier: Paris, France, 2012; pp. 145–150. ISBN 978-2-257-22597-9. [Google Scholar]
- Batlle, D.; Haque, S.K. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol. Dial. Transplant. 2012, 27, 3691–3704. [Google Scholar] [CrossRef]
- Letavernier, E.; Verrier, C.; Goussard, F.; Perez, J.; Huguet, L.; Haymann, J.-P.; Baud, L.; Bazin, D.; Daudon, M. Calcium and vitamin D have a synergistic role in a rat model of kidney stone disease. Kidney Int. 2016, 90, 809–817. [Google Scholar] [CrossRef]
- Bushinsky, D.A.; Grynpas, M.D.; Nilsson, E.L.; Nakagawa, Y.; Coe, F.L. Stone formation in genetic hypercalciuric rats. Kidney Int. 1995, 48, 1705–1713. [Google Scholar] [CrossRef]
- Russell, R.G.; Hodgkinson, A. The urinary excretion of inorganic pyrophosphate by normal subjects and patients with renal calculus. Clin. Sci. 1966, 31, 51–62. [Google Scholar]
- Letavernier, E.; Daudon, M. Vitamin D, Hypercalciuria and Kidney Stones. Nutrients 2018, 10, 366. [Google Scholar] [CrossRef]
- Bouderlique, E.; Tang, E.; Perez, J.; Coudert, A.; Bazin, D.; Verpont, M.-C.; Duranton, C.; Rubera, I.; Haymann, J.-P.; Leftheriotis, G.; et al. Vitamin D and Calcium Supplementation Accelerates Randall’s Plaque Formation in a Murine Model. Am. J. Pathol. 2019, 189, 2171–2180. [Google Scholar] [CrossRef]
- Cheungpasitporn, W.; Thongprayoon, C.; Mao, M.A.; O’Corragain, O.A.; Edmonds, P.J.; Erickson, S.B. The Risk of Coronary Heart Disease in Patients with Kidney Stones: A Systematic Review and Meta-analysis. N. Am. J. Med. Sci. 2014, 6, 580–585. [Google Scholar] [CrossRef]
- Reiner, A.P.; Kahn, A.; Eisner, B.H.; Pletcher, M.J.; Sadetsky, N.; Williams, O.D.; Polak, J.F.; Jacobs, D.R.; Stoller, M.L. Kidney stones and subclinical atherosclerosis in young adults: Coronary Artery Risk Development in Young Adults (CARDIA) study. J. Urol. 2011, 185. [Google Scholar] [CrossRef]
- Fabris, A.; Ferraro, P.M.; Comellato, G.; Caletti, C.; Fantin, F.; Zaza, G.; Zamboni, M.; Lupo, A.; Gambaro, G. The relationship between calcium kidney stones, arterial stiffness and bone density: Unraveling the stone-bone-vessel liaison. J. Nephrol. 2015, 28, 549–555. [Google Scholar] [CrossRef]
- Shavit, L.; Girfoglio, D.; Vijay, V.; Goldsmith, D.; Ferraro, P.M.; Moochhala, S.H.; Unwin, R. Vascular Calcification and Bone Mineral Density in Recurrent Kidney Stone Formers. Clin. J. Am. Soc. Nephrol. 2015, 10, 278–285. [Google Scholar] [CrossRef]
- Schibler, D.; Russell, R.G.; Fleisch, H. Inhibition by pyrophosphate and polyphosphate of aortic calcification induced by vitamin D3 in rats. Clin. Sci. 1968, 35, 363–372. [Google Scholar]
- Villa-Bellosta, R.; O’Neill, W.C. Pyrophosphate deficiency in vascular calcification. Kidney Int. 2018, 93, 1293–1297. [Google Scholar] [CrossRef]
- Lomashvili, K.A.; Garg, P.; Narisawa, S.; Millan, J.L.; O’Neill, W.C. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: Potential mechanism for uremic vascular calcification. Kidney Int. 2008, 73, 1024–1030. [Google Scholar] [CrossRef]
- Lau, W.L.; Liu, S.; Vaziri, N.D. Chronic kidney disease results in deficiency of ABCC6, the novel inhibitor of vascular calcification. Am. J. Nephrol. 2014, 40, 51–55. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Letavernier, E.; Bouderlique, E.; Zaworski, J.; Martin, L.; Daudon, M. Pseudoxanthoma Elasticum, Kidney Stones and Pyrophosphate: From a Rare Disease to Urolithiasis and Vascular Calcifications. Int. J. Mol. Sci. 2019, 20, 6353. https://doi.org/10.3390/ijms20246353
Letavernier E, Bouderlique E, Zaworski J, Martin L, Daudon M. Pseudoxanthoma Elasticum, Kidney Stones and Pyrophosphate: From a Rare Disease to Urolithiasis and Vascular Calcifications. International Journal of Molecular Sciences. 2019; 20(24):6353. https://doi.org/10.3390/ijms20246353
Chicago/Turabian StyleLetavernier, Emmanuel, Elise Bouderlique, Jeremy Zaworski, Ludovic Martin, and Michel Daudon. 2019. "Pseudoxanthoma Elasticum, Kidney Stones and Pyrophosphate: From a Rare Disease to Urolithiasis and Vascular Calcifications" International Journal of Molecular Sciences 20, no. 24: 6353. https://doi.org/10.3390/ijms20246353
APA StyleLetavernier, E., Bouderlique, E., Zaworski, J., Martin, L., & Daudon, M. (2019). Pseudoxanthoma Elasticum, Kidney Stones and Pyrophosphate: From a Rare Disease to Urolithiasis and Vascular Calcifications. International Journal of Molecular Sciences, 20(24), 6353. https://doi.org/10.3390/ijms20246353