Single-Molecule Long-Read Sequencing Reveals the Diversity of Full-Length Transcripts in Leaves of Gnetum (Gnetales)



Abstract
1. Introduction
2. Results
2.1. Transcriptome from PacBio Sequel Sequencing
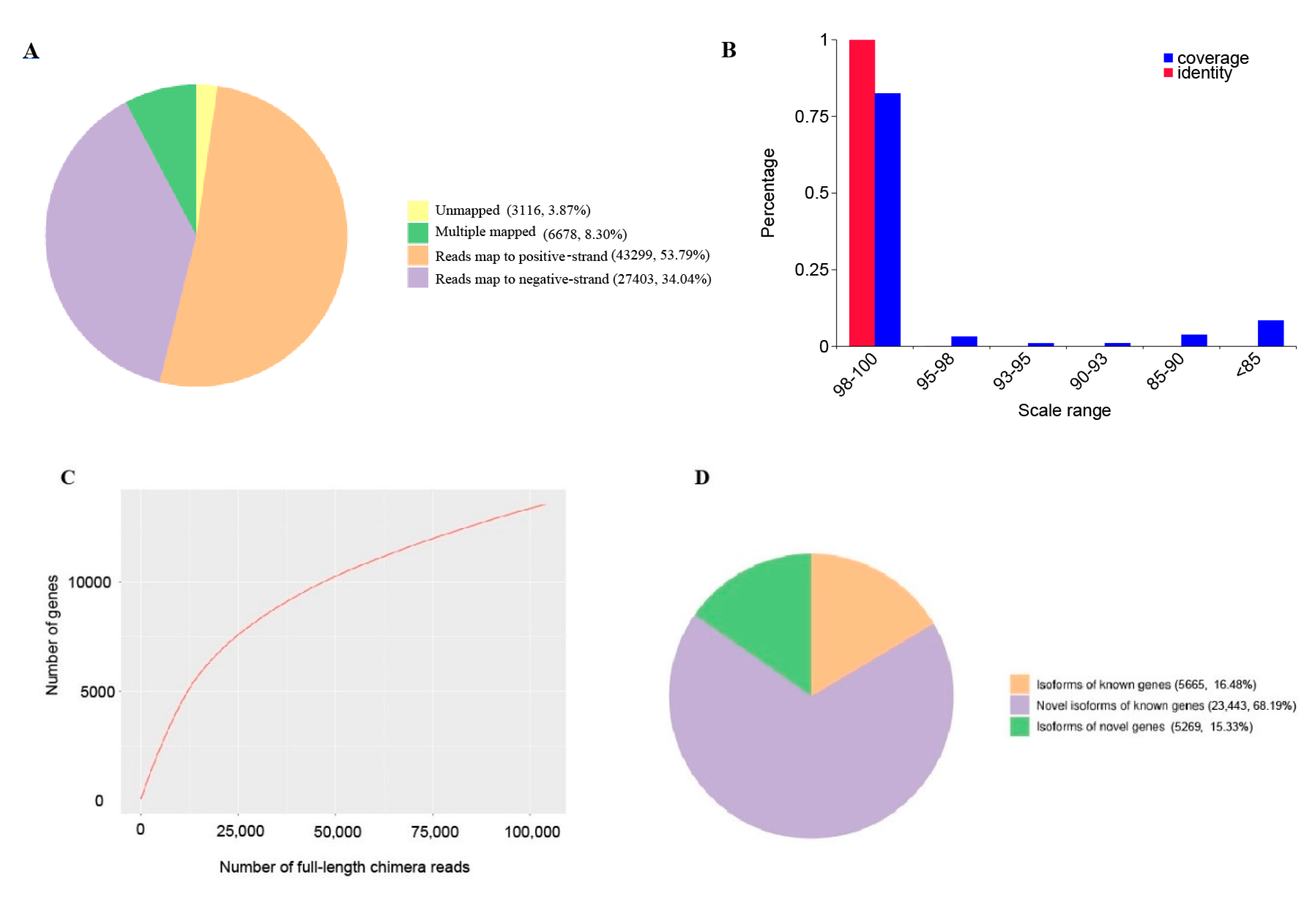
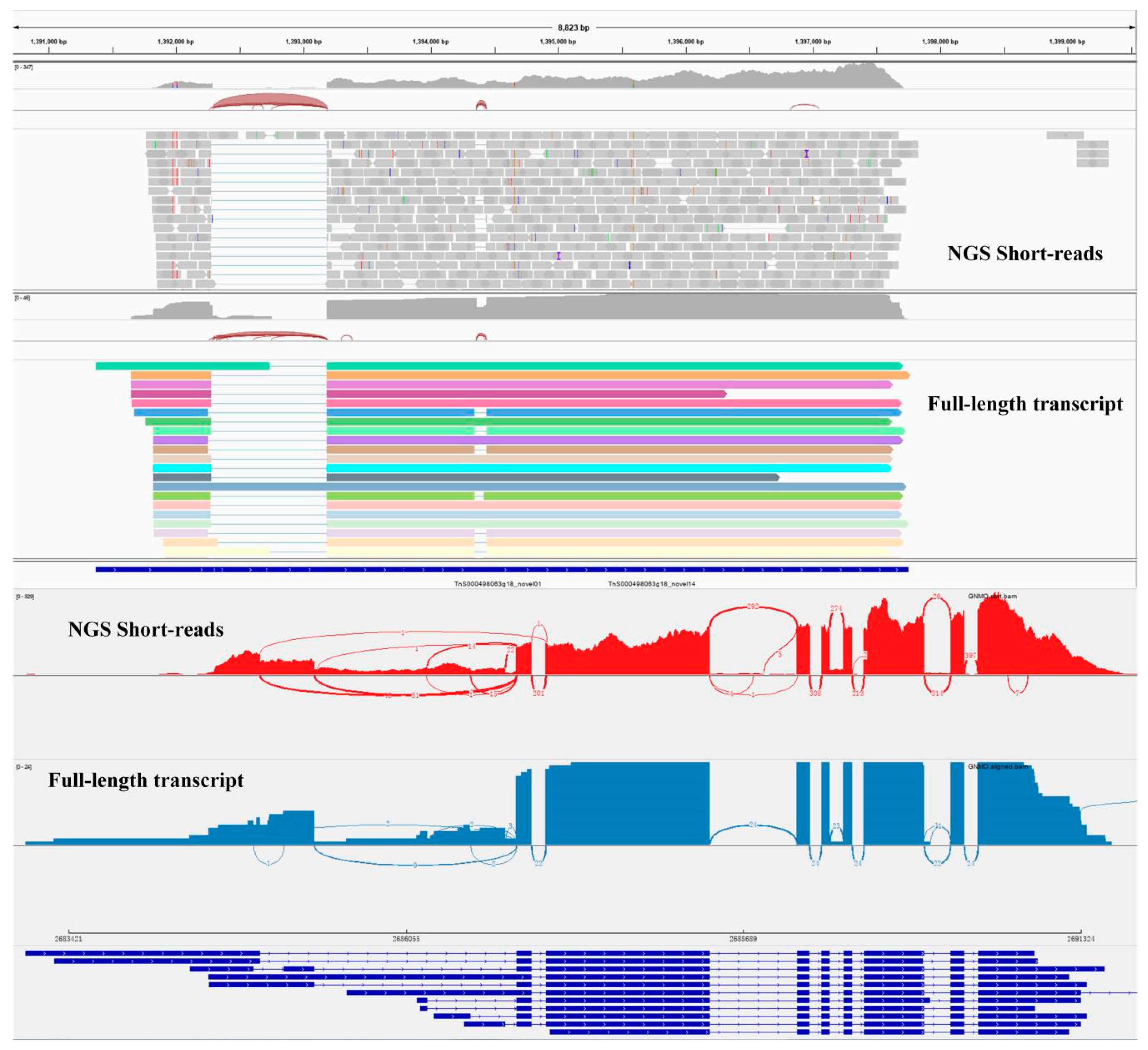
2.2. Transcript Structure Analysis
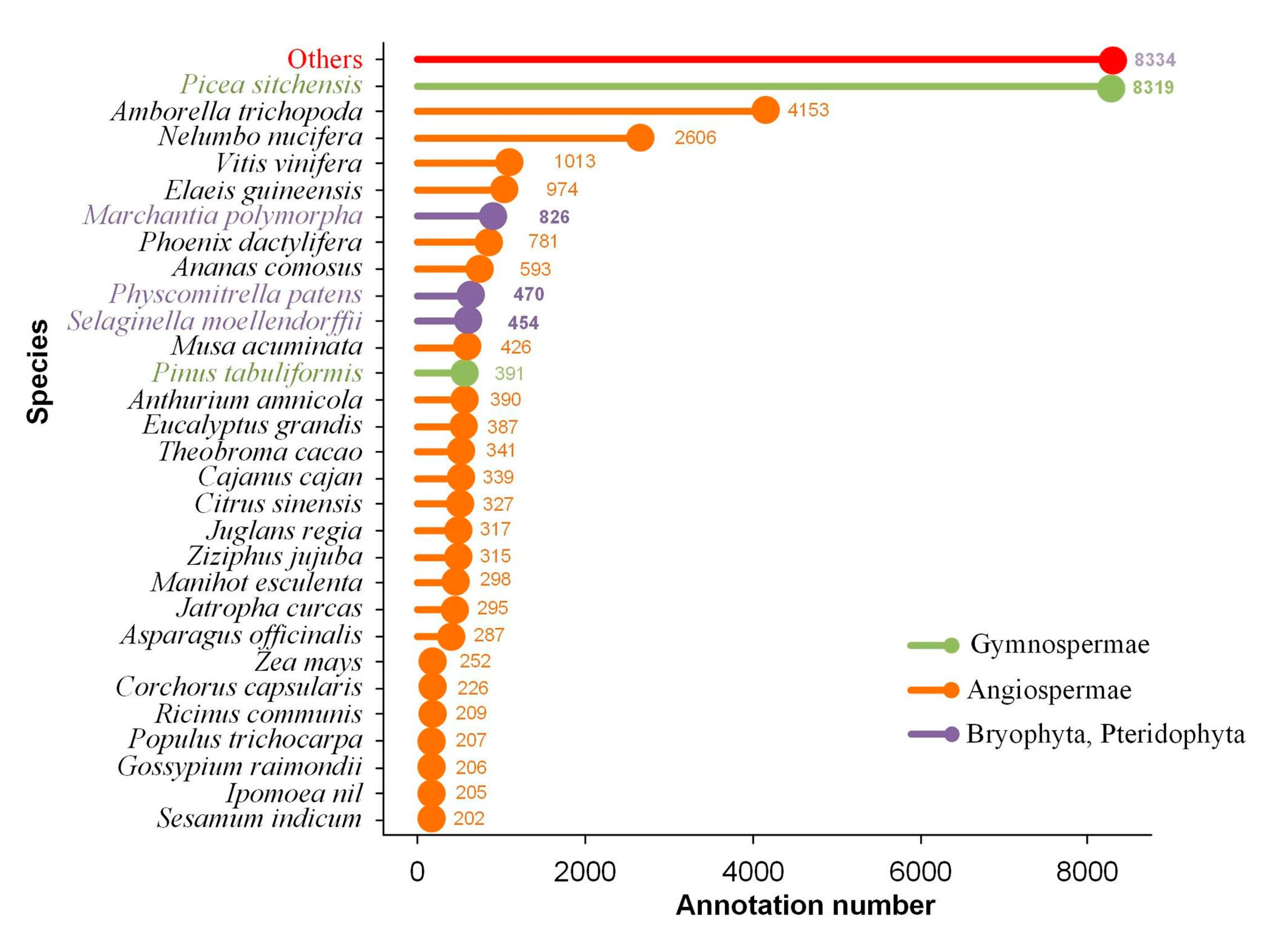
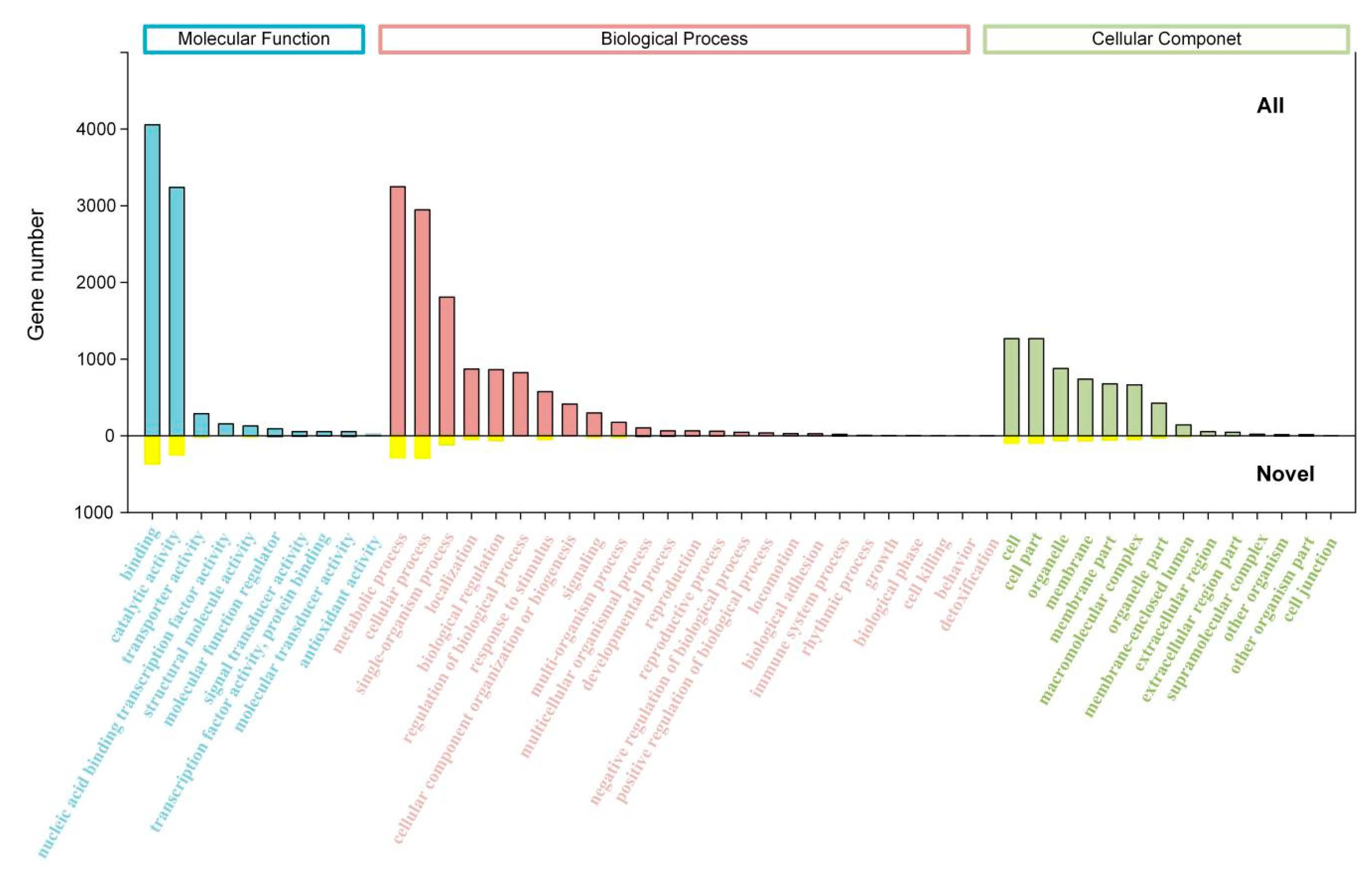
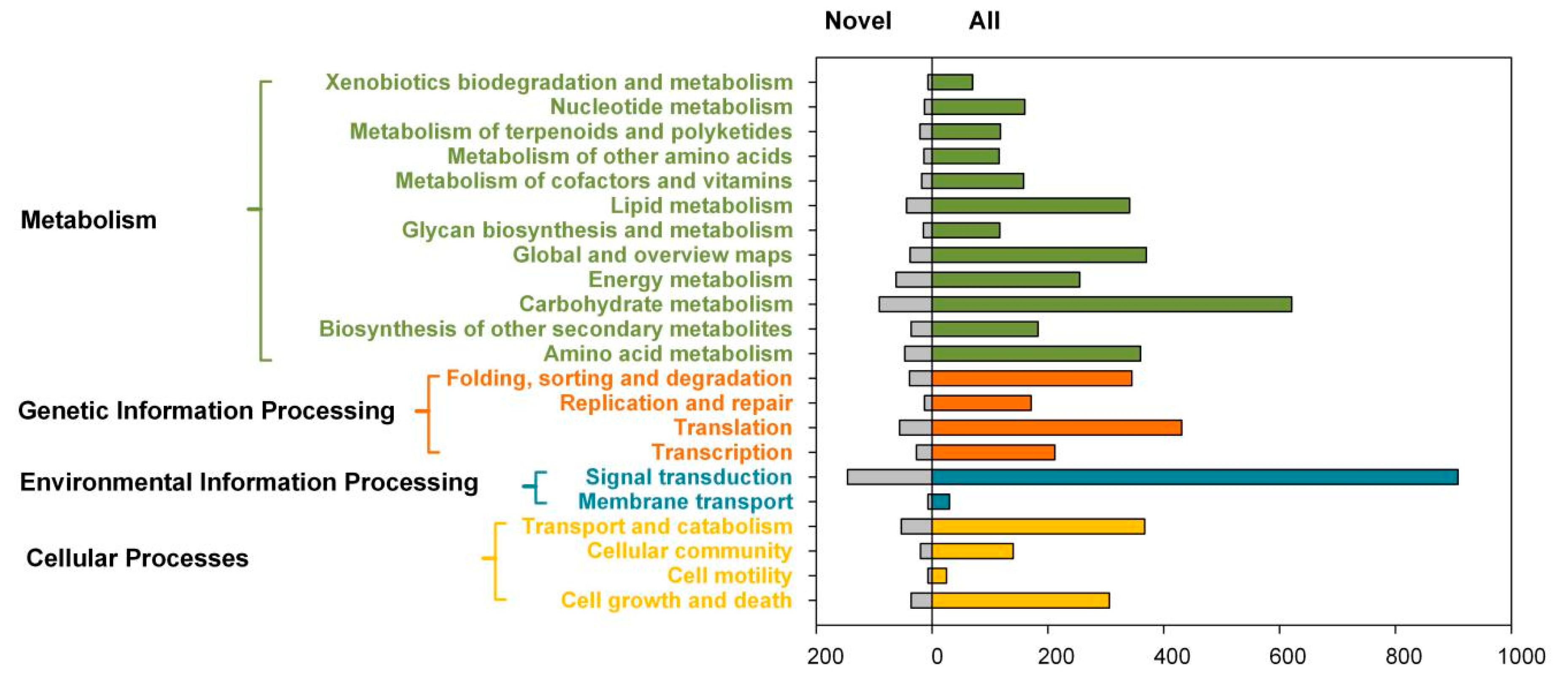
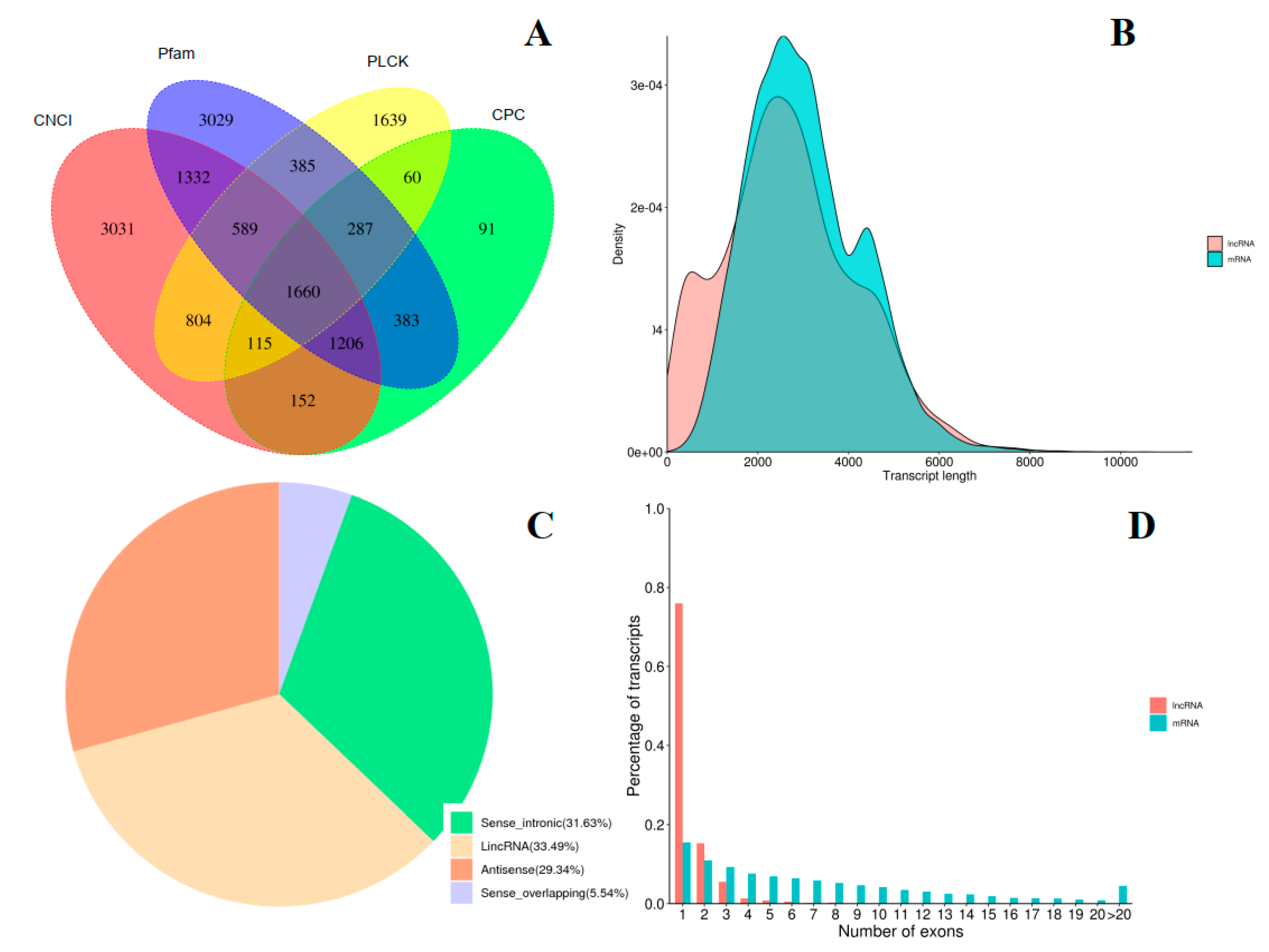
2.3. Transcript Annotation and Classification
2.4. Identification of LncRNAs and Fusion Genes
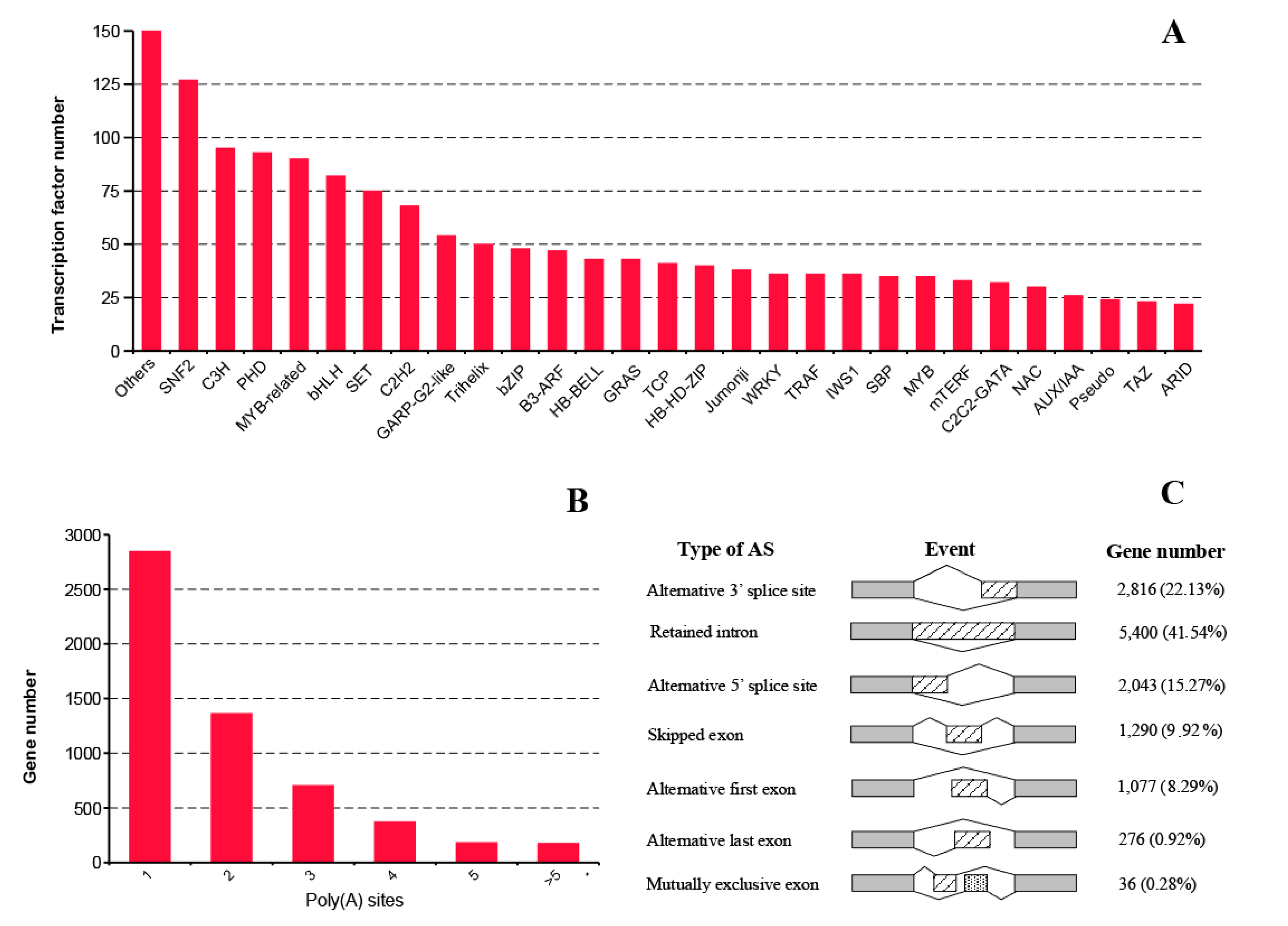
2.5. Transcription Factor, Alternative Polyadenylation, and Alternative Splicing Analyses
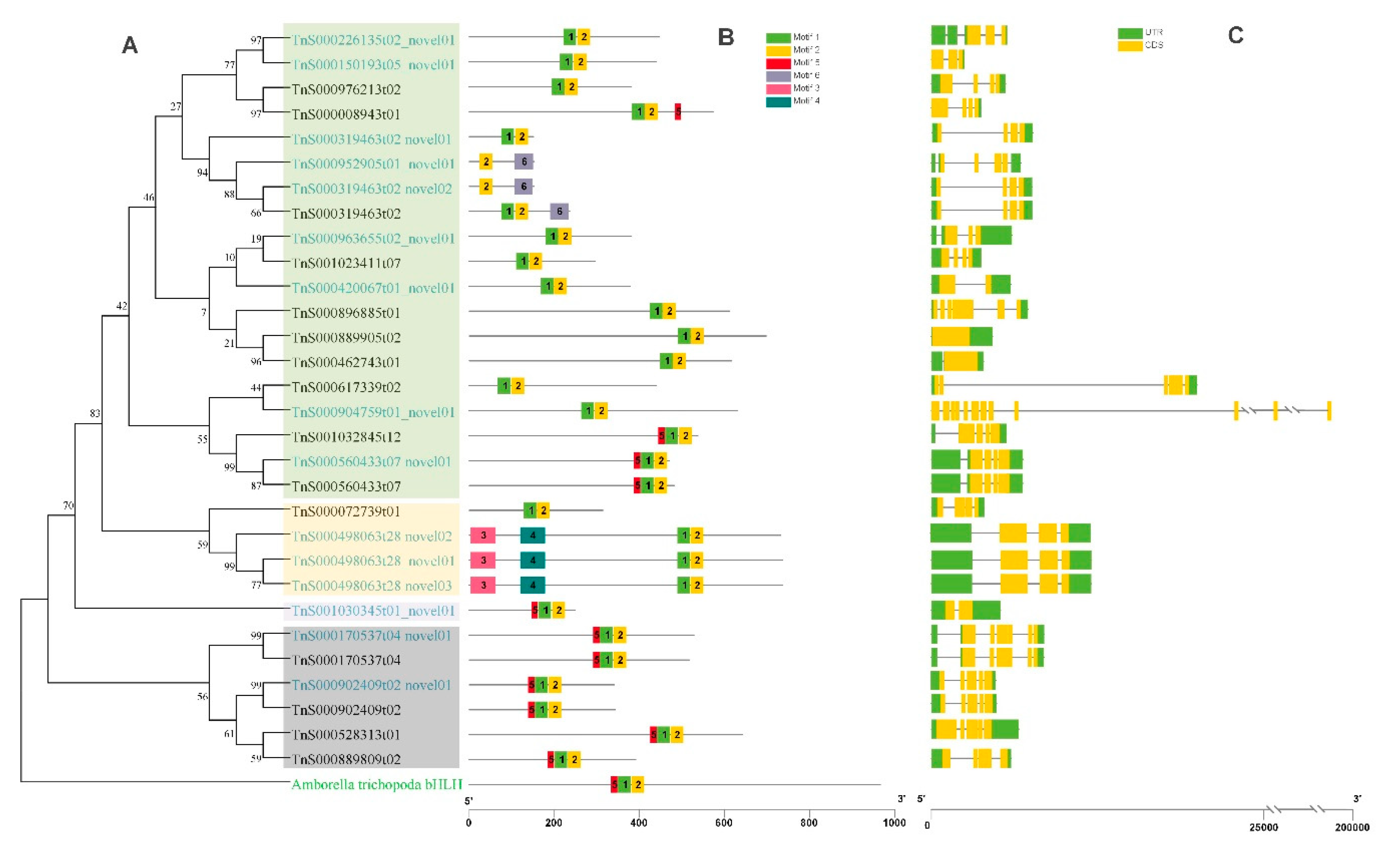
2.6. Phylogenetic Analysis of bHLH Genes in G. luofuense
3. Discussion
4. Materials and Methods
4.1. Samples Selection and RNA Extraction
4.2. Sample Preparation and PacBio Sequel Sequencing
4.3. PacBio Sequencing Data Processing
4.4. Structure Analysis and Annotation
4.5. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Deng, N.; Hou, C.; Liu, C.; Li, M.; Bartish, I.; Tian, Y.; Chen, W.; Du, C.; Jiang, Z.; Shi, S. Significance of photosynthetic characters in the evolution of Asian Gnetum (Gnetales). Front. Plant Sci. 2019, 10, 30. [Google Scholar] [CrossRef]
- Doyle, J.A. Molecular and fossil evidence on the origin of angiosperms. Rev. Earth Planet. Sci. 2012, 40, 301–326. [Google Scholar] [CrossRef]
- Wan, T.; Liu, Z.M.; Li, L.F.; Leitch, A.R.; Leitch, I.J.; Lohaus, R.; Liu, Z.J.; Xin, H.P.; Gong, Y.B.; Liu, Y. A genome for gnetophytes and early evolution of seed plants. Nat. Plants 2018, 4, 82. [Google Scholar] [CrossRef] [PubMed]
- Ickert-Bond, S.M.; Renner, S.S. The Gnetales: Recent insights on their morphology, reproductive biology, chromosome numbers, biogeography, and divergence times. J. Syst. Evol. 2016, 54, 1–16. [Google Scholar] [CrossRef]
- Gong, Y.B.; Yang, M.; Vamosi, J.C.; Yang, H.M.; Mu, W.X.; Li, J.K.; Wan, T. Wind or insect pollination? Ambophily in a subtropical gymnosperm Gnetum parvifolium (Gnetales). Plant Species Biol. 2016, 31, 272–279. [Google Scholar] [CrossRef]
- Sarah, M. Phylogenetic relationships among seed plants: Persistent questions and the limits of molecular data. Am. J. Bot. 2009, 96, 228–236. [Google Scholar]
- Price, R.A. Systematics of the Gnetales: A review of morphological and molecular evidence. Int. J. Plant Sci. 1996, 157, S40–S49. [Google Scholar] [CrossRef]
- Crane, P.R. Phylogenetic analysis of seed plants and the origin of angiosperms. Ann. Mo. Bot. Gard. 1985, 72, 716–793. [Google Scholar] [CrossRef]
- Bojian, Z.; Takahiro, Y.; Yang, Z.; Masami, H. The position of gnetales among seed plants: Overcoming pitfalls of chloroplast phylogenomics. Mol. Biol. Evol. 2010, 27, 2855–2863. [Google Scholar]
- Chen, H.; Wikström, N.; Strijk, J.S.; Rydin, C. Resolving phylogenetic relationships and species delimitations in closely related gymnosperms using high-throughput NGS, Sanger sequencing and morphology. Plant Syst. Evol. 2016, 302, 1345–1365. [Google Scholar]
- Hou, C.; Humphreys, A.M.; Thureborn, O.; Rydin, C. New insights into the evolutionary history of Gnetum (Gnetales). Taxon 2015, 64, 239–253. [Google Scholar] [CrossRef]
- Deng, N.; Liu, C.; Chang, E.; Ji, J.; Yao, X.; Yue, J.; Bartish, I.; Chen, L.; Jiang, Z.; Shi, S. High temperature and UV-C treatments affect stilbenoid accumulation and related gene expression levels in Gnetum parvifolium. Electron. J. Biotechnol. 2016, 25, 43–49. [Google Scholar] [CrossRef]
- Deng, N.; Chang, E.; Li, M.; Ji, J.; Yao, X.; Banish, I.V.; Liu, J.; Ma, J.; Chen, L.; Jiang, Z.; et al. Transcriptome characterization of Gnetum parvifolium reveals candidate genes involved in important secondary metabolic pathways of flavonoids and stilbenoids. Front. Plant Sci. 2016, 7, 174. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Iliya, I.; Ito, T.; Furusawa, M.; Nakaya, K.I.; Iinuma, M.; Shirataki, Y.; Matsuura, N.; Ubukata, M.; Murata, J. Stilbenoids in lianas of Gnetum parvifolium. Chem. Pharm. Bull. 2001, 39, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Markgraf, F. Monographie der Gattung Gnetum. Bull. Jar. Bot. Buitenz. 1930, 10, 407–511. [Google Scholar]
- Feild, T.S.; Lawong, B. Xylem hydraulic and photosynthetic function of Gnetum (Gnetales) species from Papua New Guinea. New Phytol. 2008, 177, 665–675. [Google Scholar] [CrossRef]
- Rudall, P.J.; Rice, C.L. Epidermal patterning and stomatal development in Gnetales. Ann. Bot. 2019, 124, 149–164. [Google Scholar] [CrossRef]
- Yawen, L.; Xu, L.; Kunwu, L.; Hongtao, L.; Chentao, L. Multiple bHLH proteins form heterodimers to mediate CRY2-dependent regulation of flowering-time in Arabidopsis. PLoS Genet. 2013, 9, e1003861. [Google Scholar]
- Lynn Jo, P.; Sloan, D.B.; Bogenschutz, N.L.; Torii, K.U. Termination of asymmetric cell division and differentiation of stomata. Nature 2007, 445, 501–505. [Google Scholar]
- Kyoko, O.I.; Bergmann, D.C. Arabidopsis FAMA controls the final proliferation/differentiation switch during stomatal development. Plant Cell 2006, 18, 2493–2505. [Google Scholar]
- Gabriela, T.O.; Enamul, H.; Quail, P.H. The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell 2003, 15, 1749–1770. [Google Scholar]
- Thatcher, S.R.; Zhou, W.; Leonard, A.; Wang, B.B.; Beatty, M.; Zastrow-Hayes, G.; Zhao, X.; Baumgarten, A.; Li, B. Genome-wide analysis of alternative splicing in Zea mays: Landscape and genetic regulation. Plant Cell 2014, 26, 3472. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Will, C.L.; Reinhard, L. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, X.; Wang, S.; Tan, C.; Zhou, G.; Li, C. Involvement of alternative splicing in barley seed germination. PLoS ONE 2016, 11, e0152824. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Tang, F.; Zhu, H. Alternative splicing in plant immunity. Int. J. Mol. Sci. 2014, 15, 10424–10445. [Google Scholar] [CrossRef] [PubMed]
- Estelle, R.; Cabrito, T.R.; Batista, R.A.; Hussein, M.A.M.; Teixeira, M.C.; Alekos, A.; Isabel, S.C.; Paula, D. Intron retention in the 5′UTR of the novel ZIF2 transporter enhances translation to promote zinc tolerance in Arabidopsis. PLoS Genet. 2014, 10, e1004375. [Google Scholar]
- Blencowe, B.J. Alternative splicing: New insights from global analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef]
- Jun, L.; Huan, W.; Nam-Hai, C. Long noncoding RNA transcriptome of plants. Plant. Biotechnol. J. 2015, 13, 319–328. [Google Scholar]
- Ariel, F.; Romero-Barrios, N.; Jégu, T.; Benhamed, M.; Crespi, M. Battles and hijacks: Noncoding transcription in plants. Trends Plant Sci. 2015, 20, 362–371. [Google Scholar] [CrossRef]
- Bernard, E.; Jacob, L.; Mairal, J.; Vert, J.P. Efficient RNA isoform identification and quantification from RNA-Seq data with network flows. Bioinformatics 2013, 30, 2447–2455. [Google Scholar] [CrossRef]
- Tilgner, H.; Raha, D.; Habegger, L.; Mohiuddin, M.; Gerstein, M.; Snyder, M. Accurate identification and analysis of human mRNA isoforms using deep long read sequencing. G3-Genes Genom. Genet. 2013, 3, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Donald, S.; Hagen, T.; Fabian, G.; Michael, S. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar]
- Cheng, B.; Furtado, A.; Henry, R.J. Long-read sequencing of the coffee bean transcriptome reveals the diversity of full-length transcripts. Gigascience 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Abdelghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Benhur, A.; Reddy, A.S.N. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Biotechnol. 2016, 7, 11706. [Google Scholar]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef]
- Wang, M.; Wang, P.; Liang, F.; Ye, Z.; Li, J.; Shen, C.; Pei, L.; Wang, F.; Hu, J.; Tu, L. A global survey of alternative splicing in allopolyploid cotton: Landscape, complexity and regulation. New Phytol. 2017, 217, 163. [Google Scholar] [CrossRef]
- Makita, Y.; Kawashima, M.; Lau, N.S.; Othman, A.S.; Matsui, M. Construction of Pará rubber tree genome and multi-transcriptome database accelerates rubber researches. BMC Genom. 2018, 19, 922. [Google Scholar] [CrossRef]
- Kin Fai, A.; Underwood, J.G.; Lawrence, L.; Wing Hung, W. Improving PacBio long read accuracy by short read alignment. PLoS ONE 2012, 7, e46679. [Google Scholar]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Jun, L.; Choonkyun, J.; Jun, X.; Huan, W.; Shulin, D.; Lucia, B.; Catalina, A.H.; Nam-Hai, C. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar]
- He, F.; Liu, Q.; Zheng, L.; Cui, Y.; Shen, Z.; Zheng, L. RNA-Seq analysis of rice roots rveals the involvement of post-transcriptional regulation in response to cadmium stress. Front. Plant Sci. 2015, 6, 1136. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Sun, L.; Luo, H.; Ma, Q.; Zhao, Y.; Pei, D. Genome-wide identification and characterization of long non-coding RNAs from mulberry (Morus notabilis) RNA-seq Data. Genes 2016, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Kwenda, S.; Birch, P.R.J.; Moleleki, L.N. Genome-wide identification of potato long intergenic noncoding RNAs responsive to Pectobacterium carotovorum subspecies brasiliense infection. BMC Genom. 2016, 17, 614. [Google Scholar] [CrossRef] [PubMed]
- Yuqiu, W.; Xiuduo, F.; Fang, L.; Guangming, H.; William, T.; Danmeng, Z.; Xing Wang, D. Arabidopsis noncoding RNA mediates control of photomorphogenesis by red light. Proc. Natl. Acad. Sci. USA 2014, 111, 10359. [Google Scholar]
- Amor, B.B.; Wirth, S.; Merchan, F.; Laporte, P.; D’Aubentoncarafa, Y.; Hirsch, J.; Maizel, A.; Mallory, A.; Lucas, A.; Deragon, J.M. Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 2009, 19, 57. [Google Scholar] [CrossRef]
- Liu, X.; Li, D.; Zhang, D.; Yin, D.; Zhao, Y.; Ji, C.; Zhao, X.; Li, X.; He, Q.; Chen, R. A novel antisense long noncoding RNA, twisted leaf, maintains leaf blade flattening by regulating its associated sense R2R3-MYB gene in rice. New Phytol. 2018, 218, 774–788. [Google Scholar] [CrossRef]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef]
- Li, Y.; Dai, C.; Hu, C.; Liu, Z.; Kang, C. Global identification of alternative splicing via comparative analysis of SMRT- and Illumina-based RNA-seq in strawberry. Plant J. 2017, 90, 164. [Google Scholar] [CrossRef]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653. [Google Scholar] [CrossRef]
- Atchley, W.R.; Fitch, W.M. A natural classification of the basic helix-loop-helix class of transcription factors. Proc. Natl. Acad. Sci. USA 1997, 94, 5172–5176. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, B.; Atchley, W.R. Evolution of bHLH transcription factors: Modular evolution by domain shuffling? Mol. Biol. Evol. 1999, 16, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Furtado, A. RNA extraction from developing or mature wheat seeds. Methods Mol. Biol. 2014, 1099, 23. [Google Scholar] [PubMed]
- Leena, S.; Eric, R. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar]
- Wu, T.; Watanabe, C. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859. [Google Scholar] [CrossRef]
- Shimizu, K.; Adachi, J.; Muraoka, Y. ANGLE: A sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinform. Comput. Biol. 2011, 4, 649–664. [Google Scholar] [CrossRef]
- Liang, S.; Haitao, L.; Dechao, B.; Guoguang, Z.; Kuntao, Y.; Changhai, Z.; Yuanning, L.; Runsheng, C.; Yi, Z. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar]
- Lei, K.; Yong, Z.; Zhi-Qiang, Y.; Xiao-Qiao, L.; Shu-Qi, Z.; Liping, W.; Ge, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11. [Google Scholar] [CrossRef]
- Chen, X.; Xizeng, M.; Jiaju, H.; Yang, D.; Jianmin, W.; Shan, D.; Lei, K.; Ge, G.; Chuan-Yun, L.; Liping, W. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar]
- Heng, L. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terms | Amount |
|---|---|
| Subreads bases | 9.98 G |
| Number of subreads | 3,689,825 |
| Average length of subreads | 2750 bp |
| N50 of subreads | 3168 bp |
| Number of CCSs | 185,089 |
| Number of sequences with 5′ terminal primers | 167,590 |
| Number of sequences with 3′ terminal primers | 166,862 |
| Number of sequences with poly(A) tails | 156,554 |
| Number of full-length sequences | 143,578 |
| Number of full-length non-chimeric (flnc) reads | 139,488 |
| Average length of flnc reads | 3065 bp |
| Percentage of flnc reads | 75% |
| Number of polished consensus reads | 80,496 |
| Minimum length of consensus reads | 167 bp |
| Maximum length of consensus reads | 14,735 bp |
| Average length of consensus reads | 3223 bp |
| N50 of consensus reads | 3614 bp |
| Type | Before Correction (Pacbio Sequel) | After Correction (Pacbio Sequel) | Short Reads (Illumina) |
|---|---|---|---|
| Total nucleotide | 259,381,401 | 260,514,867 | 44,269,498 |
| Total sequence | 80,496 | 80,496 | 45,566 |
| Mean length | 3223 bp | 3237 bp | 972 bp |
| Minimum length | 167 bp | 167 bp | 201 bp |
| Maximum length | 14,735 bp | 14,734 bp | 12,325 bp |
| N50 | 3614 bp | 3629 bp | 2030 bp |
| N90 | 2102 bp | 2102 bp | 323 bp |
| Terms | PacBio Sequenced Data | Illumina Sequenced Data | ||
|---|---|---|---|---|
| Number of Reads | Percentage | Number of Reads | Percentage | |
| Total mapped | 77,380 | 96.13% | 54,289,038 | 93.01% |
| Unmapped | 3116 | 3.87% | 4,079,995 | 6.99% |
| Multiple mapped | 6678 | 8.30% | 1,691,837 | 2.90% |
| Uniquely mapped | 70,702 | 87.83% | 52,597,201 | 90.11% |
| Uniquely mappedto positive strands | 43,299 | 53.79% | 26,287,949 | 45.04% |
| Uniquely mappedto negative strands | 27,403 | 34.04% | 26,309,252 | 45.07% |
| Database | Total Number of Annotated Isoforms | Number of Novel Genes | Number of Novel Isoforms of Known Genes | Number of Isoforms of Known Genes |
|---|---|---|---|---|
| NR | 34,170 | 3782 | 22,167 | 8221 |
| SwissProt | 28,954 | 2741 | 19,023 | 7190 |
| KEGG | 33,813 | 3659 | 22,005 | 8149 |
| KOG | 21,723 | 1925 | 14,658 | 5140 |
| GO | 22,297 | 1047 | 15,180 | 6070 |
| NT | 15,467 | 936 | 10,680 | 3851 |
| Pfam | 22,297 | 1047 | 15,180 | 6070 |
| In all databases | 8980 | 116 | 6548 | 8281 |
| At least in one database | 34,667 | 5269 | 23,443 | 2316 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, N.; Hou, C.; Ma, F.; Liu, C.; Tian, Y. Single-Molecule Long-Read Sequencing Reveals the Diversity of Full-Length Transcripts in Leaves of Gnetum (Gnetales). Int. J. Mol. Sci. 2019, 20, 6350. https://doi.org/10.3390/ijms20246350
Deng N, Hou C, Ma F, Liu C, Tian Y. Single-Molecule Long-Read Sequencing Reveals the Diversity of Full-Length Transcripts in Leaves of Gnetum (Gnetales). International Journal of Molecular Sciences. 2019; 20(24):6350. https://doi.org/10.3390/ijms20246350
Chicago/Turabian StyleDeng, Nan, Chen Hou, Fengfeng Ma, Caixia Liu, and Yuxin Tian. 2019. "Single-Molecule Long-Read Sequencing Reveals the Diversity of Full-Length Transcripts in Leaves of Gnetum (Gnetales)" International Journal of Molecular Sciences 20, no. 24: 6350. https://doi.org/10.3390/ijms20246350
APA StyleDeng, N., Hou, C., Ma, F., Liu, C., & Tian, Y. (2019). Single-Molecule Long-Read Sequencing Reveals the Diversity of Full-Length Transcripts in Leaves of Gnetum (Gnetales). International Journal of Molecular Sciences, 20(24), 6350. https://doi.org/10.3390/ijms20246350