Optimized Xenograft Protocol for Chronic Lymphocytic Leukemia Results in High Engraftment Efficiency for All CLL Subgroups

 and
and
Abstract
1. Introduction
2. Results
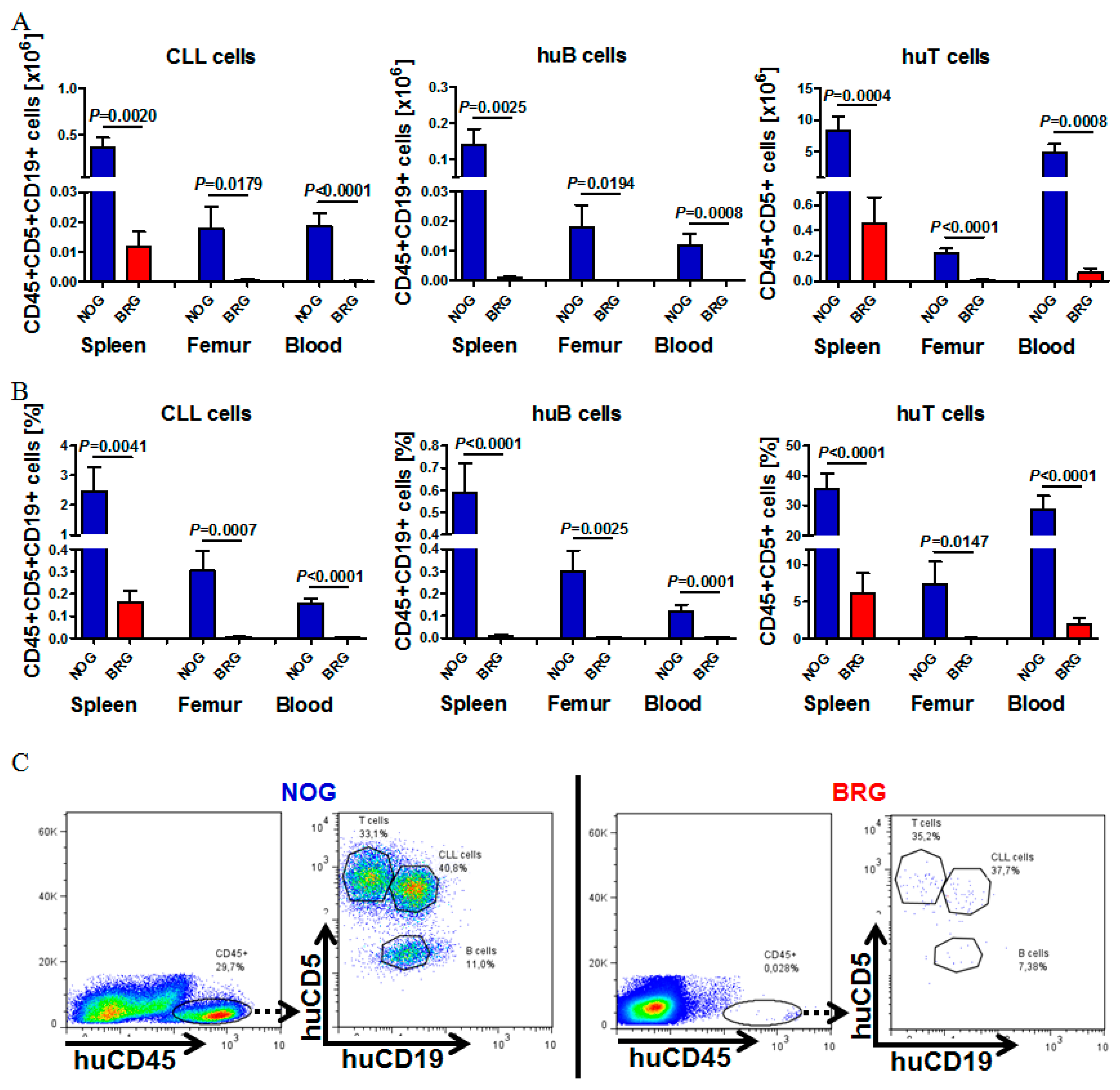
2.1. Reliable CLL Engraftment is Detectable in NOG, but Not BRG Mice
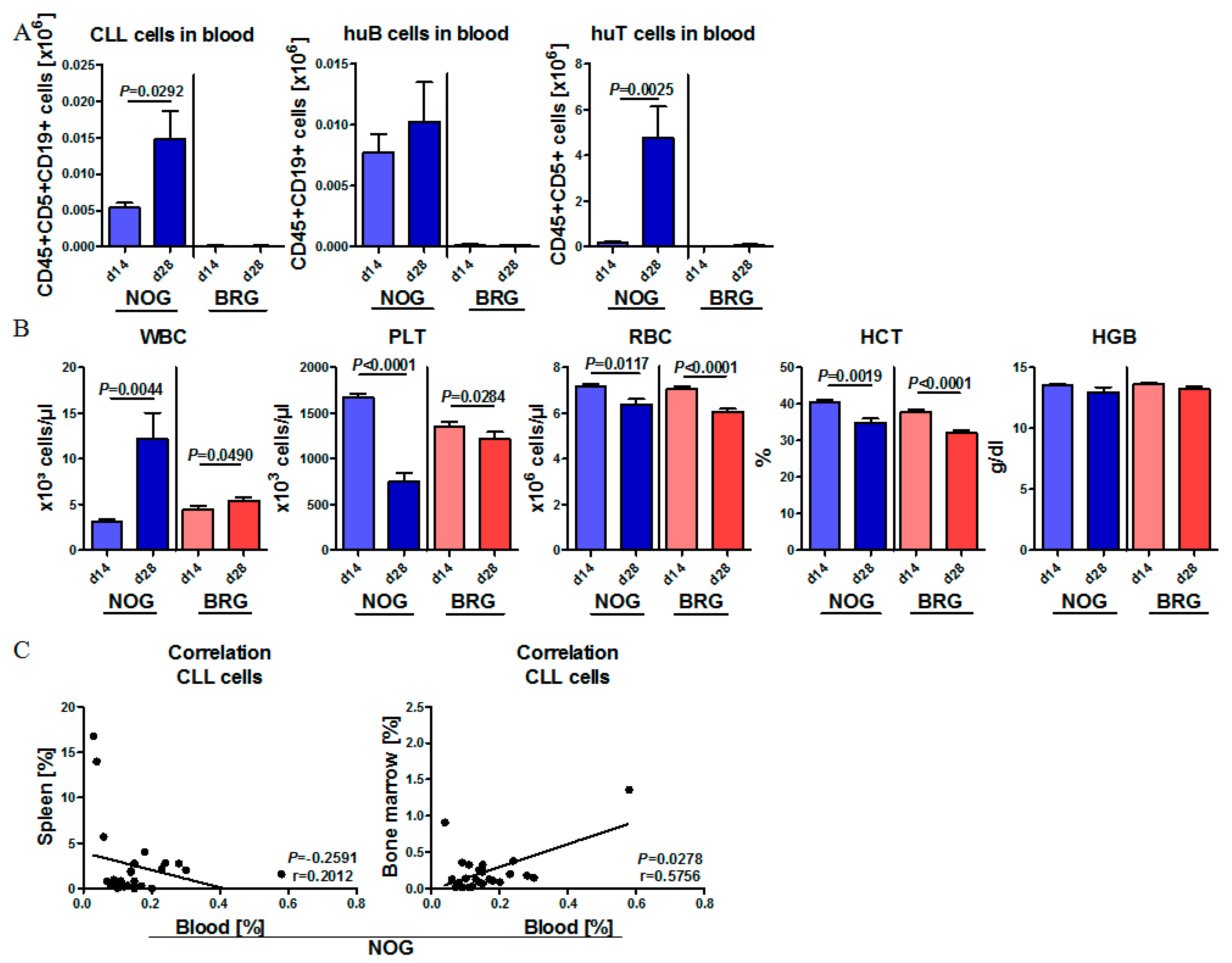
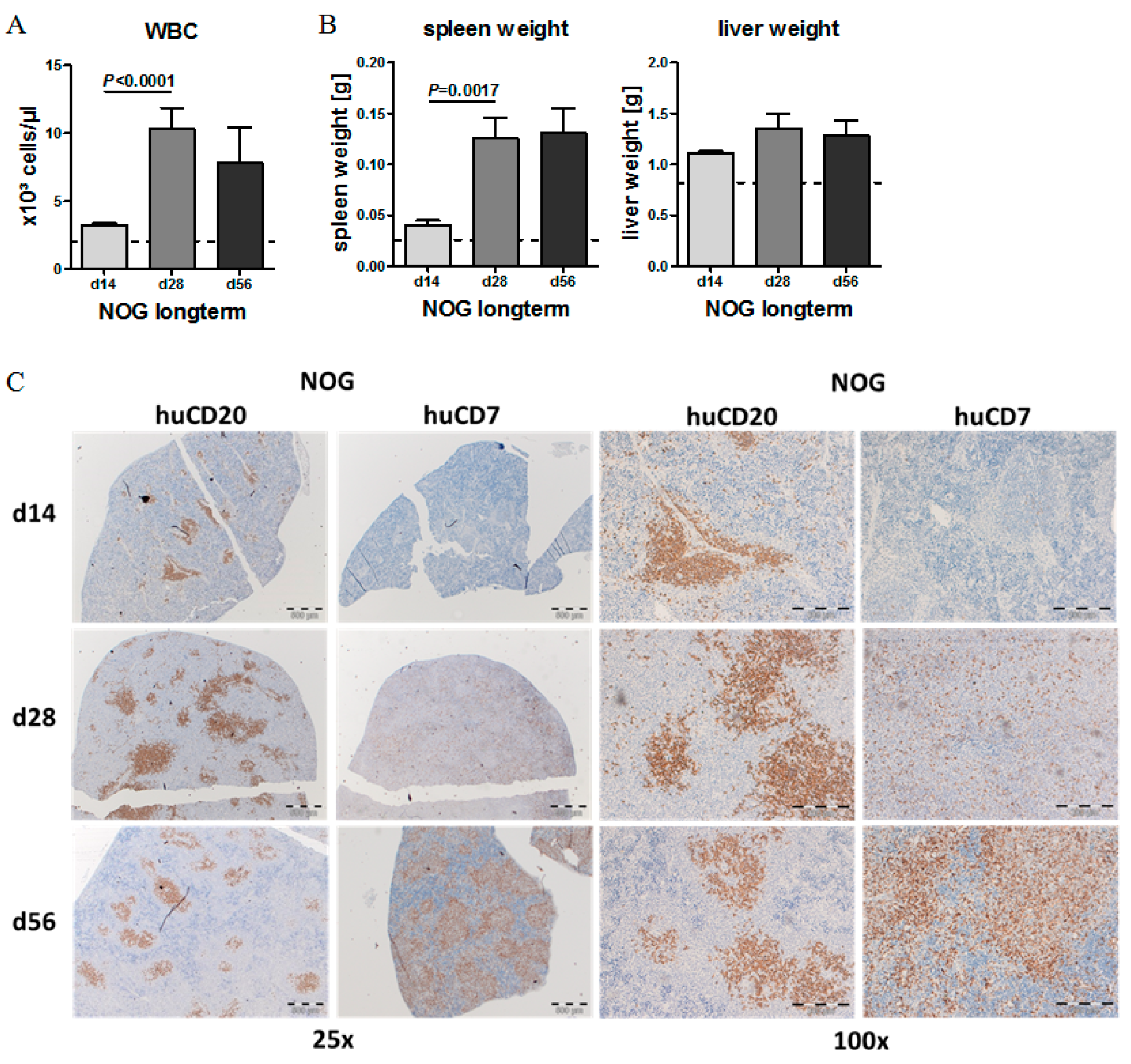
2.2. Human CLL Cells and T Cells Expand during the First Weeks of Engraftment in NOG Mice, but Not in BRG Mice
2.3. CLL Cells Rebuild Lymphoid Follicles in the Spleen of NOG Mice, but Not BRG Mice
2.4. Phenotypic Time Course of CLL Engraftment
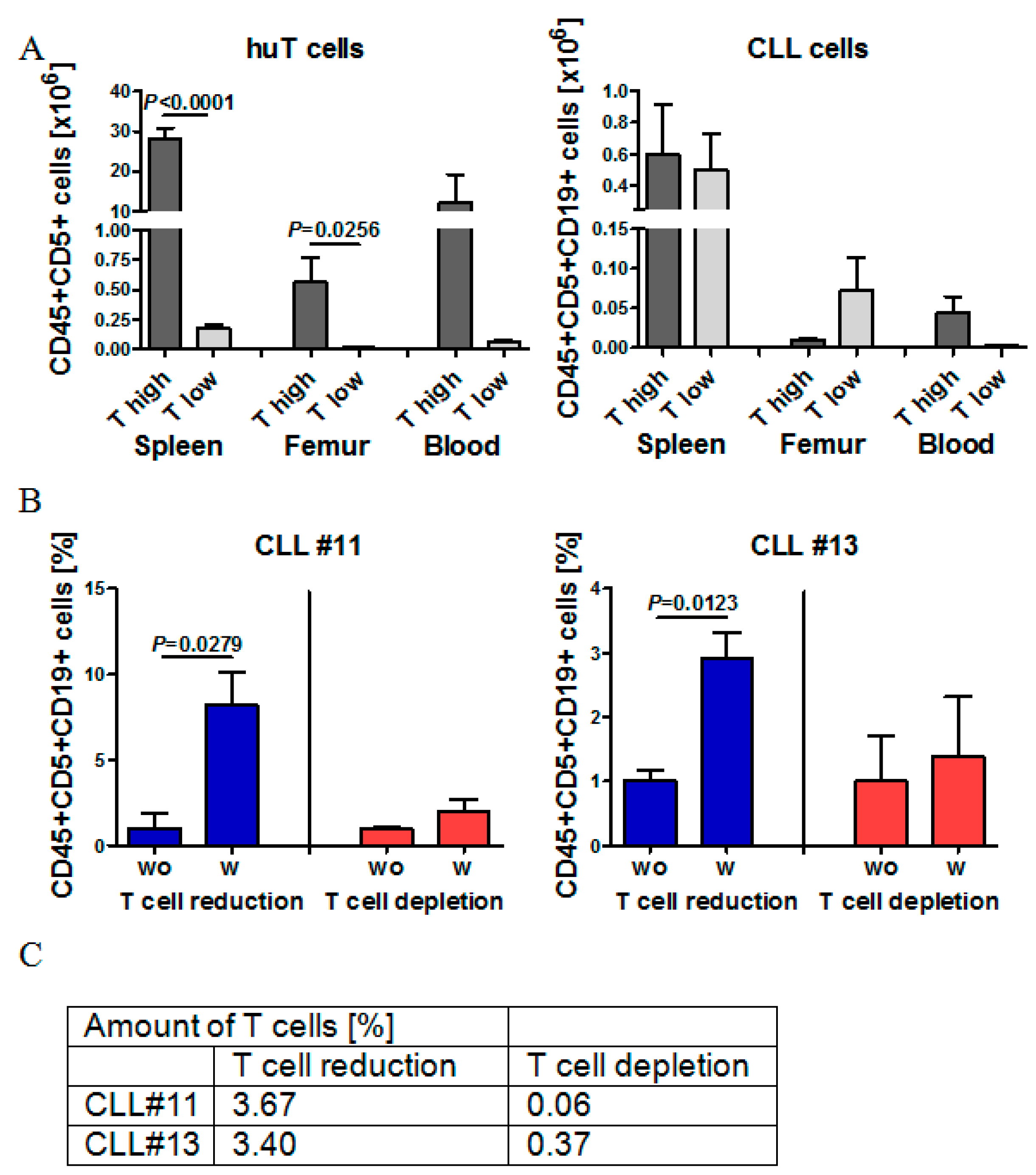
2.5. Effect of Autologous T Cell Quantity on CLL Cell Engraftment in NOG Mice
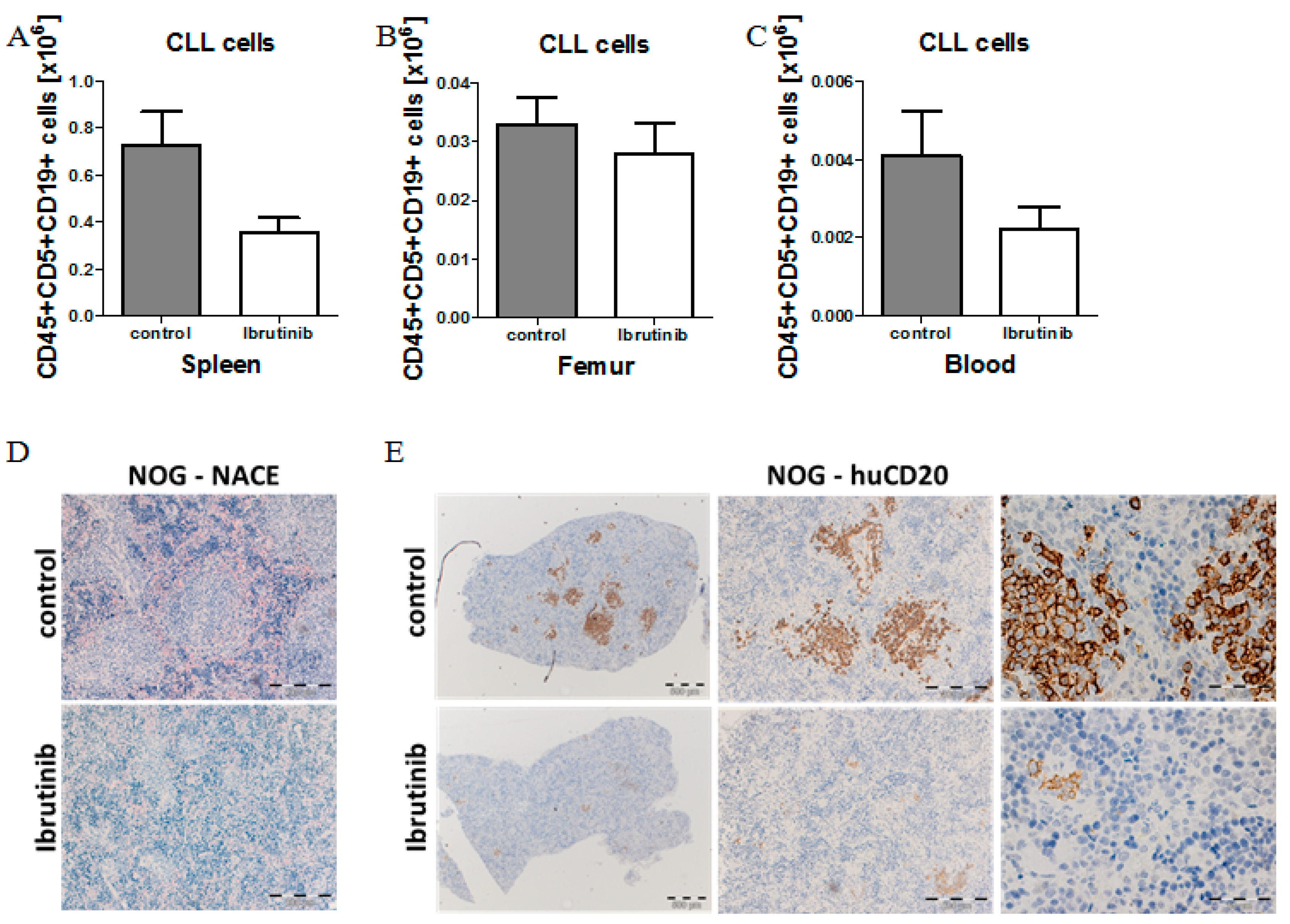
2.6. Model Validation Using Ibrutinib Treatment to Block CLL Expansion in Vivo
3. Discussion
4. Materials and Methods
4.1. CLL Patient Samples
4.2. Transplantation of BRG and NOG Mice
4.3. Histology and Immunohistochemistry (IHC)
4.4. T Cell Reduction or Depletion
4.5. Ibrutinib Treatment
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CLL | Chronic lymphocytic leukemia |
| PB | Peripheral blood |
| BM | Bone marrow |
| PBMC | Peripheral blood mononuclear cell |
References
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Munk Pedersen, I.; Reed, J. Microenvironmental interactions and survival of CLL B-cells. Leuk. Lymphoma 2004, 45, 2365–2372. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; Caligaris-Cappio, F. The origin of B-cell chronic lymphocytic leukemia. Semin. Oncol. 2006, 33, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Tzankov, A.; Fong, D. Hodgkin’s disease variant of Richter’s syndrome clonally related to chronic lymphocytic leukemia arises in ZAP-70 negative mutated CLL. Med. Hypotheses 2006, 66, 577–579. [Google Scholar] [CrossRef]
- Rozman, C.; Montserrat, E. Chronic lymphocytic leukemia. N. Engl. J. Med. 1995, 333, 1052–1057. [Google Scholar] [CrossRef]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef]
- Rassenti, L.Z.; Huynh, L.; Toy, T.L.; Chen, L.; Keating, M.J.; Gribben, J.G.; Neuberg, D.S.; Flinn, I.W.; Rai, K.R.; Byrd, J.C.; et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2004, 351, 893–901. [Google Scholar] [CrossRef]
- Wiestner, A.; Rosenwald, A.; Barry, T.S.; Wright, G.; Davis, R.E.; Henrickson, S.E.; Zhao, H.; Ibbotson, R.E.; Orchard, J.A.; Davis, Z.; et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood 2003, 101, 4944–4951. [Google Scholar] [CrossRef]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.J.; Montserrat, E.; Rai, K.R.; et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008, 111, 5446–5456. [Google Scholar] [CrossRef]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, S.; Oleś, M.; Lu, J.; Sellner, L.; Anders, S.; Velten, B.; Wu, B.; Hüllein, J.; da Silva Liberio, M.; Walther, T.; et al. Drug-perturbation-based stratification of blood cancer. J. Clin. Invest. 2018, 128, 427–445. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A. Nurture versus nature: The microenvironment in chronic lymphocytic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.J.; Verschuer, L.A.; Harmon, B.V.; Prentice, R.L.; Pope, J.H.; Kerr, J.F. Spontaneous programmed death (apoptosis) of B-chronic lymphocytic leukaemia cells following their culture in vitro. Br. J. Haematol. 1989, 71, 343–350. [Google Scholar] [CrossRef]
- Bichi, R.; Shinton, S.A.; Martin, E.S.; Koval, A.; Calin, G.A.; Cesari, R.; Russo, G.; Hardy, R.R.; Croce, C.M. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6955–6960. [Google Scholar] [CrossRef]
- Zapata, J.M.; Krajewska, M.; Morse, H.C.; Choi, Y.; Reed, J.C. TNF receptor-associated factor (TRAF) domain and Bcl-2 cooperate to induce small B cell lymphoma/chronic lymphocytic leukemia in transgenic mice. Proc. Natl. Acad. Sci. USA 2004, 101, 16600–16605. [Google Scholar] [CrossRef]
- Klein, U.; Lia, M.; Crespo, M.; Siegel, R.; Shen, Q.; Mo, T.; Ambesi-Impiombato, A.; Califano, A.; Migliazza, A.; Bhagat, G.; et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010, 17, 28–40. [Google Scholar] [CrossRef]
- Bertilaccio, M.T.S.; Scielzo, C.; Simonetti, G.; Ten Hacken, E.; Apollonio, B.; Ghia, P.; Caligaris-Cappio, F. Xenograft models of chronic lymphocytic leukemia: Problems, pitfalls and future directions. Leukemia 2013, 27, 534–540. [Google Scholar] [CrossRef]
- Bertilaccio, M.T.S.; Scielzo, C.; Simonetti, G.; Ponzoni, M.; Apollonio, B.; Fazi, C.; Scarfò, L.; Rocchi, M.; Muzio, M.; Caligaris-Cappio, F.; et al. A novel Rag2-/-gammac-/--xenograft model of human CLL. Blood 2010, 115, 1605–1609. [Google Scholar] [CrossRef]
- Aydin, S.; Grabellus, F.; Eisele, L.; Möllmann, M.; Hanoun, M.; Ebeling, P.; Moritz, T.; Carpinteiro, A.; Nückel, H.; Sak, A.; et al. Investigating the role of CD38 and functionally related molecular risk factors in the CLL NOD/SCID xenograft model. Eur. J. Haematol. 2011, 87, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Marcus, H.; Shimoni, A.; Ergas, D.; Canaan, A.; Dekel, B.; Ben-David, D.; David, M.; Sigler, E.; Reisner, Y.; Berrebi, A. Human/mouse radiation chimera generated from PBMC of B chronic lymphocytic leukemia patients with autoimmune hemolytic anemia produce anti-human red cell antibodies. Leukemia 1997, 11, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Shimoni, A.; Marcus, H.; Dekel, B.; Shkarchi, R.; Arditti, F.; Shvidel, L.; Shtalrid, M.; Bucher, W.; Canaan, A.; Ergas, D.; et al. Autologous T cells control B-chronic lymphocytic leukemia tumor progression in human-->mouse radiation chimera. Cancer Res. 1999, 59, 5968–5974. [Google Scholar] [PubMed]
- Dürig, J.; Ebeling, P.; Grabellus, F.; Sorg, U.R.; Möllmann, M.; Schütt, P.; Göthert, J.; Sellmann, L.; Seeber, S.; Flasshove, M.; et al. A novel nonobese diabetic/severe combined immunodeficient xenograft model for chronic lymphocytic leukemia reflects important clinical characteristics of the disease. Cancer Res. 2007, 67, 8653–8661. [Google Scholar] [CrossRef]
- Herman, S.E.M.; Sun, X.; McAuley, E.M.; Hsieh, M.M.; Pittaluga, S.; Raffeld, M.; Liu, D.; Keyvanfar, K.; Chapman, C.M.; Chen, J.; et al. Modeling tumor-host interactions of chronic lymphocytic leukemia in xenografted mice to study tumor biology and evaluate targeted therapy. Leukemia 2013, 27, 2311–2321. [Google Scholar] [CrossRef]
- Oldreive, C.E.; Skowronska, A.; Davies, N.J.; Parry, H.; Agathanggelou, A.; Krysov, S.; Packham, G.; Rudzki, Z.; Cronin, L.; Vrzalikova, K.; et al. T-cell number and subtype influence the disease course of primary chronic lymphocytic leukaemia xenografts in alymphoid mice. Dis. Model. Mech. 2015, 8, 1401–1412. [Google Scholar] [CrossRef]
- Bagnara, D.; Kaufman, M.S.; Calissano, C.; Marsilio, S.; Patten, P.E.M.; Simone, R.; Chum, P.; Yan, X.-J.; Allen, S.L.; Kolitz, J.E.; et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood 2011, 117, 5463–5472. [Google Scholar] [CrossRef]
- Patten, P.E.; Chen, S.-S.; Bagnara, D.; Simone, R.; Marsilio, S.; Tong, T.; Barrientos, J.C.; Kolitz, J.E.; Allen, S.L.; Rai, K.R.; et al. Engraftment of CLL-Derived T Cells in NSG Mice Is Feasible, Can Support CLL Cell Proliferation, and Eliminates the Need for Third Party Antigen Presenting Cells. Blood 2011, 118, 975. [Google Scholar] [CrossRef]
- Patten, P.E.M.; Ferrer, G.; Chen, S.-S.; Simone, R.; Marsilio, S.; Yan, X.-J.; Gitto, Z.; Yuan, C.; Kolitz, J.E.; Barrientos, J.; et al. Chronic lymphocytic leukemia cells diversify and differentiate in vivo via a nonclassical Th1-dependent, Bcl-6-deficient process. JCI Insight 2016, 1, e86288. [Google Scholar] [CrossRef]
- Serra, S.; Vaisitti, T.; Audrito, V.; Bologna, C.; Buonincontri, R.; Chen, S.-S.; Arruga, F.; Brusa, D.; Coscia, M.; Jaksic, O.; et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv. 2016, 1, 47–61. [Google Scholar] [CrossRef]
- Shimoni, A.; Marcus, H.; Canaan, A.; Ergas, D.; David, M.; Berrebi, A.; Reisner, Y. A model for human B-chronic lymphocytic leukemia in human/mouse radiation chimera: Evidence for tumor-mediated suppression of antibody production in low-stage disease. Blood 1997, 89, 2210–2218. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. NOD/SCID/gamma(c)(null) mouse: An excellent recipient mouse model for engraftment of human cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.P.; Blundell, M.P.; Lopes, L.; Kinnon, C.; Di Santo, J.P.; Thrasher, A.J. Enhanced human cell engraftment in mice deficient in RAG2 and the common cytokine receptor gamma chain. Br. J. Haematol. 1998, 103, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.H.; Buggy, J.J.; Sharman, J.P.; Smith, S.M.; Boyd, T.E.; Grant, B.; Kolibaba, K.S.; Furman, R.R.; Rodriguez, S.; Chang, B.Y.; et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 88–94. [Google Scholar] [CrossRef]
- Woyach, J.A.; Johnson, A.J.; Byrd, J.C. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood 2012, 120, 1175–1184. [Google Scholar] [CrossRef]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef]
- Wiestner, A. Clinical trial data on the BTK pathway in B-cell malignancies. Clin. Adv. Hematol. Oncol. HO 2013, 11 (Suppl. 9), 6–10. [Google Scholar]
- Herman, S.E.M.; Montraveta, A.; Niemann, C.U.; Mora-Jensen, H.; Gulrajani, M.; Krantz, F.; Mantel, R.; Smith, L.L.; McClanahan, F.; Harrington, B.K.; et al. The Bruton Tyrosine Kinase (BTK) Inhibitor Acalabrutinib Demonstrates Potent On-Target Effects and Efficacy in Two Mouse Models of Chronic Lymphocytic Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2831–2841. [Google Scholar] [CrossRef]
- Piper, K.P.; Karanth, M.; McLarnon, A.; Kalk, E.; Khan, N.; Murray, J.; Pratt, G.; Moss, P.A.H. Chronic lymphocytic leukaemia cells drive the global CD4+ T cell repertoire towards a regulatory phenotype and leads to the accumulation of CD4+ forkhead box P3+ T cells. Clin. Exp. Immunol. 2011, 166, 154–163. [Google Scholar] [CrossRef]
- Riches, J.C.; Gribben, J.G. Understanding the immunodeficiency in chronic lymphocytic leukemia: Potential clinical implications. Hematol. Oncol. Clin. North Am. 2013, 27, 207–235. [Google Scholar] [CrossRef]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.O.; Yu, H.; Yue, R.; Zhao, Z.; Rios, J.J.; Naveiras, O.; Morrison, S.J. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat. Cell Biol. 2017, 19, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Wang, T.C.; Guha, C. Stromal Progenitor Cells in Mitigation of Non-Hematopoietic Radiation Injuries. Curr. Pathobiol. Rep. 2016, 4, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, M.S.; Kulkarni, R.S.; Ghode, S.S.; Limaye, L.S.; Kale, V.P. Irradiation-induced secretion of BMP4 by marrow cells causes marrow adipogenesis post-myelosuppression. Stem Cell Res. 2016, 17, 646–653. [Google Scholar] [CrossRef]
- Yu, H.; Borsotti, C.; Schickel, J.-N.; Zhu, S.; Strowig, T.; Eynon, E.E.; Frleta, D.; Gurer, C.; Murphy, A.J.; Yancopoulos, G.D.; et al. A novel humanized mouse model with significant improvement of class-switched, antigen-specific antibody production. Blood 2017, 129, 959–969. [Google Scholar] [CrossRef]
- McIntosh, B.E.; Brown, M.E.; Duffin, B.M.; Maufort, J.P.; Vereide, D.T.; Slukvin, I.I.; Thomson, J.A. Nonirradiated NOD,B6.SCID Il2rγ-/- Kit(W41/W41) (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Rep. 2015, 4, 171–180. [Google Scholar] [CrossRef]
- Cosgun, K.N.; Rahmig, S.; Mende, N.; Reinke, S.; Hauber, I.; Schäfer, C.; Petzold, A.; Weisbach, H.; Heidkamp, G.; Purbojo, A.; et al. Kit regulates HSC engraftment across the human-mouse species barrier. Cell Stem Cell 2014, 15, 227–238. [Google Scholar] [CrossRef]
- Aumann, K.; Lassmann, S.; Schöpflin, A.; May, A.M.; Wöhrle, F.U.; Zeiser, R.; Waller, C.F.; Hauschke, D.; Werner, M.; Brummer, T. The immunohistochemical staining pattern of Gab2 correlates with distinct stages of chronic myeloid leukemia. Hum. Pathol. 2011, 42, 719–726. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1. Take 20–40 mL of fresh peripheral blood (PB) in EDTA tubes from CLL patients (>20.000 WBC/µL) |
| 2. Isolate PBMCs (CLL cells, T- and B cells, monocytes) by Ficoll density centrifugation |
| 3. Count isolated PBMCs, quantify CLL cells, T cells and B cells by flow cytometry (CD19, CD5) Step 1–3 within 3 h |
4.
|
| 5. Dilute 70 x 10 E6 CLL-PBMCs in 400 µL of HBSS |
| 6. Inject cell suspension half i.p. and half retro-orbital into 8–11 weeks old NOG mice |
| 7. For treatment studies: Start at day 7 post transplantation, 14–21 days of treatment |
8. Final analysis:
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Decker, S.; Zwick, A.; Khaja Saleem, S.; Kissel, S.; Rettig, A.; Aumann, K.; Dierks, C. Optimized Xenograft Protocol for Chronic Lymphocytic Leukemia Results in High Engraftment Efficiency for All CLL Subgroups. Int. J. Mol. Sci. 2019, 20, 6277. https://doi.org/10.3390/ijms20246277
Decker S, Zwick A, Khaja Saleem S, Kissel S, Rettig A, Aumann K, Dierks C. Optimized Xenograft Protocol for Chronic Lymphocytic Leukemia Results in High Engraftment Efficiency for All CLL Subgroups. International Journal of Molecular Sciences. 2019; 20(24):6277. https://doi.org/10.3390/ijms20246277
Chicago/Turabian StyleDecker, Sarah, Anabel Zwick, Shifa Khaja Saleem, Sandra Kissel, Andres Rettig, Konrad Aumann, and Christine Dierks. 2019. "Optimized Xenograft Protocol for Chronic Lymphocytic Leukemia Results in High Engraftment Efficiency for All CLL Subgroups" International Journal of Molecular Sciences 20, no. 24: 6277. https://doi.org/10.3390/ijms20246277
APA StyleDecker, S., Zwick, A., Khaja Saleem, S., Kissel, S., Rettig, A., Aumann, K., & Dierks, C. (2019). Optimized Xenograft Protocol for Chronic Lymphocytic Leukemia Results in High Engraftment Efficiency for All CLL Subgroups. International Journal of Molecular Sciences, 20(24), 6277. https://doi.org/10.3390/ijms20246277