Polyglycerol Hyperbranched Polyesters: Synthesis, Properties and Pharmaceutical and Biomedical Applications

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Poly(Glycerol Succinate), PGSuc
2.1. Synthesis and Properties
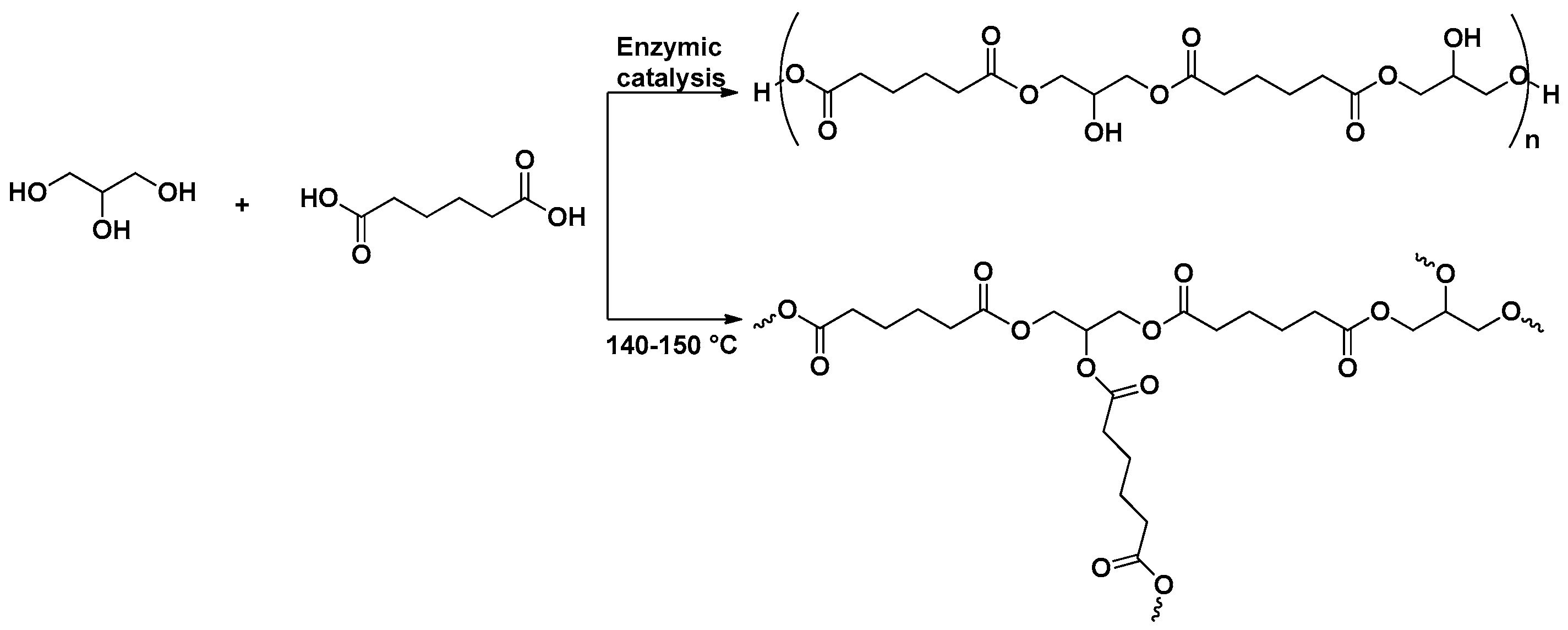
2.1.1. PGSuc
2.1.2. Synthesis of PGSuc Polyesters with Side Groups and PGSuc Copolymers
2.2. Applications
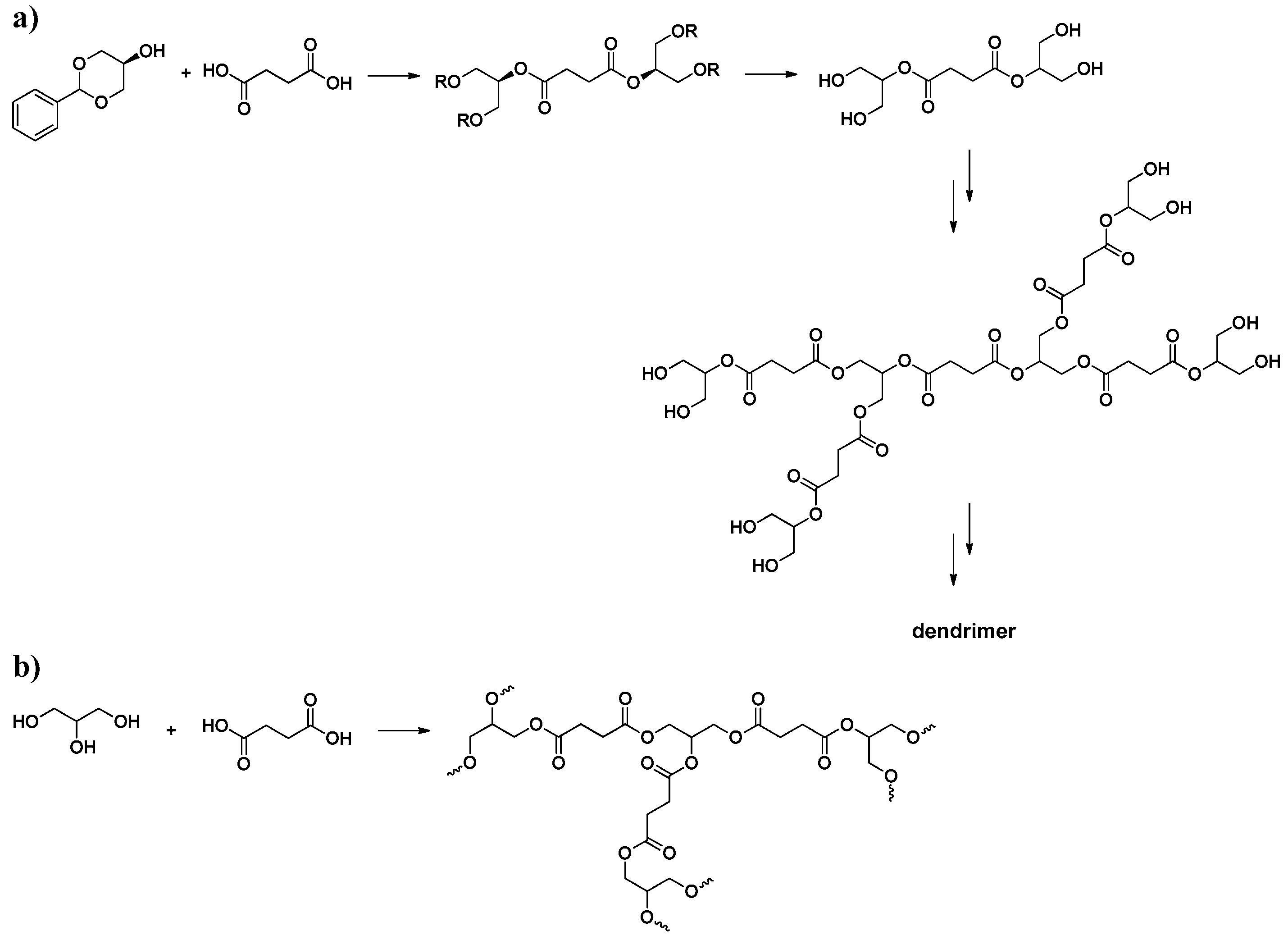
3. Poly(Glycerol Glutarate), PGGlu
4. Poly(Glycerol Adipate), PGAd
4.1. Synthesis and Properties
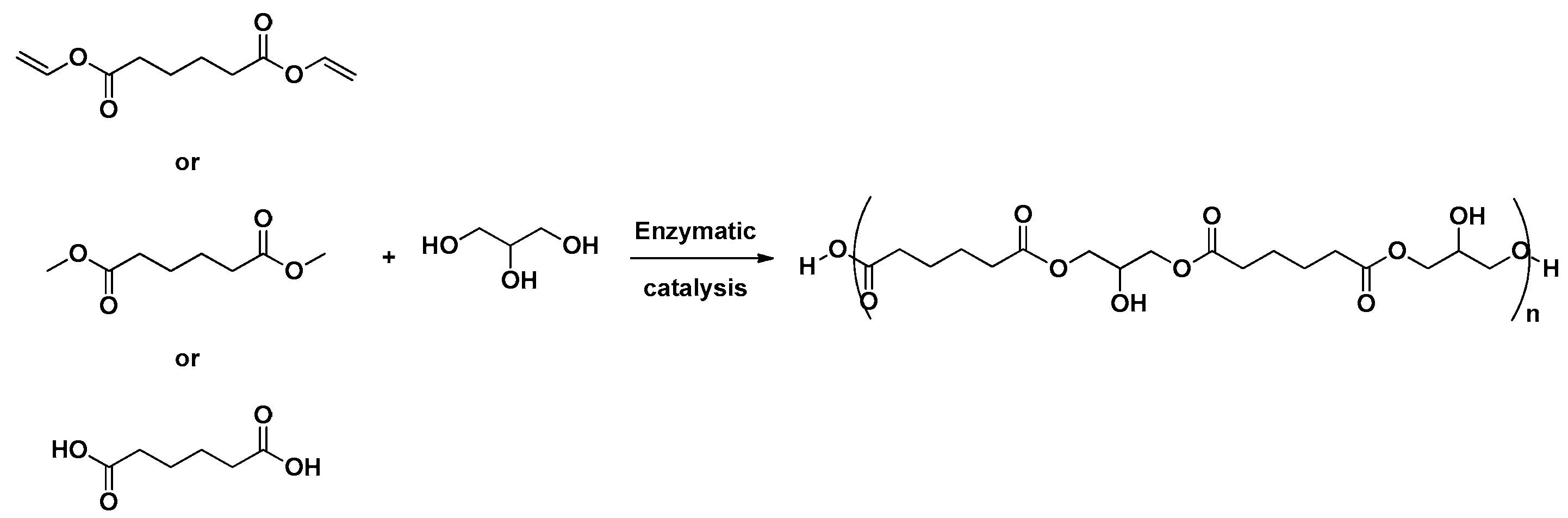
4.1.1. Linear PGAd
4.1.2. Hyperbranched PGAd
4.2. Applications
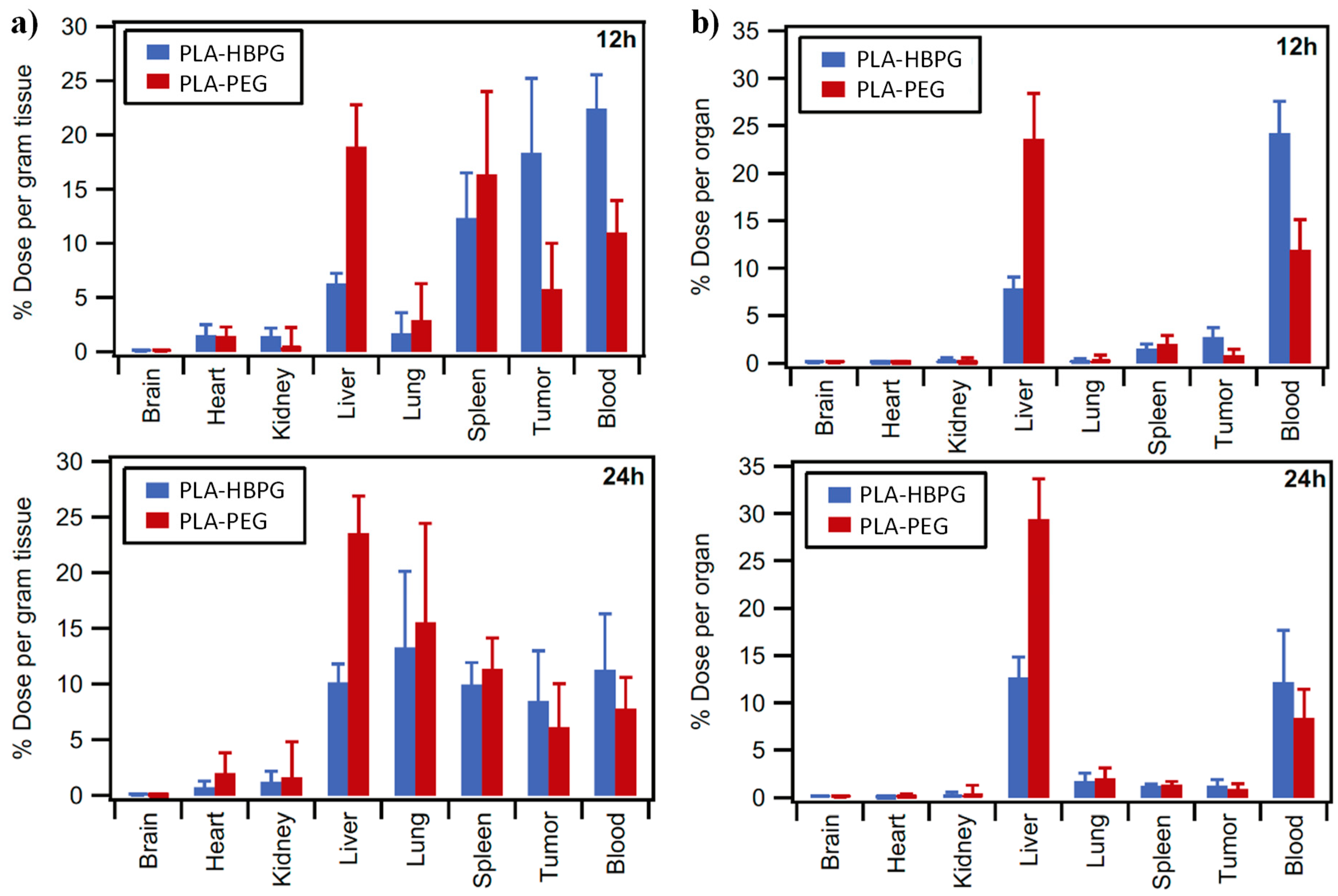
4.2.1. Linear PGAd and Acylated PGAd Nanoparticles
4.2.2. PGAd Conjugates
4.2.3. PGAd Copolymers
5. Poly(Glycerol Suberate), PGSub
6. Poly(glycerol Azelate), PGAz
7. Poly(Glycerol Sebacate), PGSeb
7.1. Synthesis and Properties
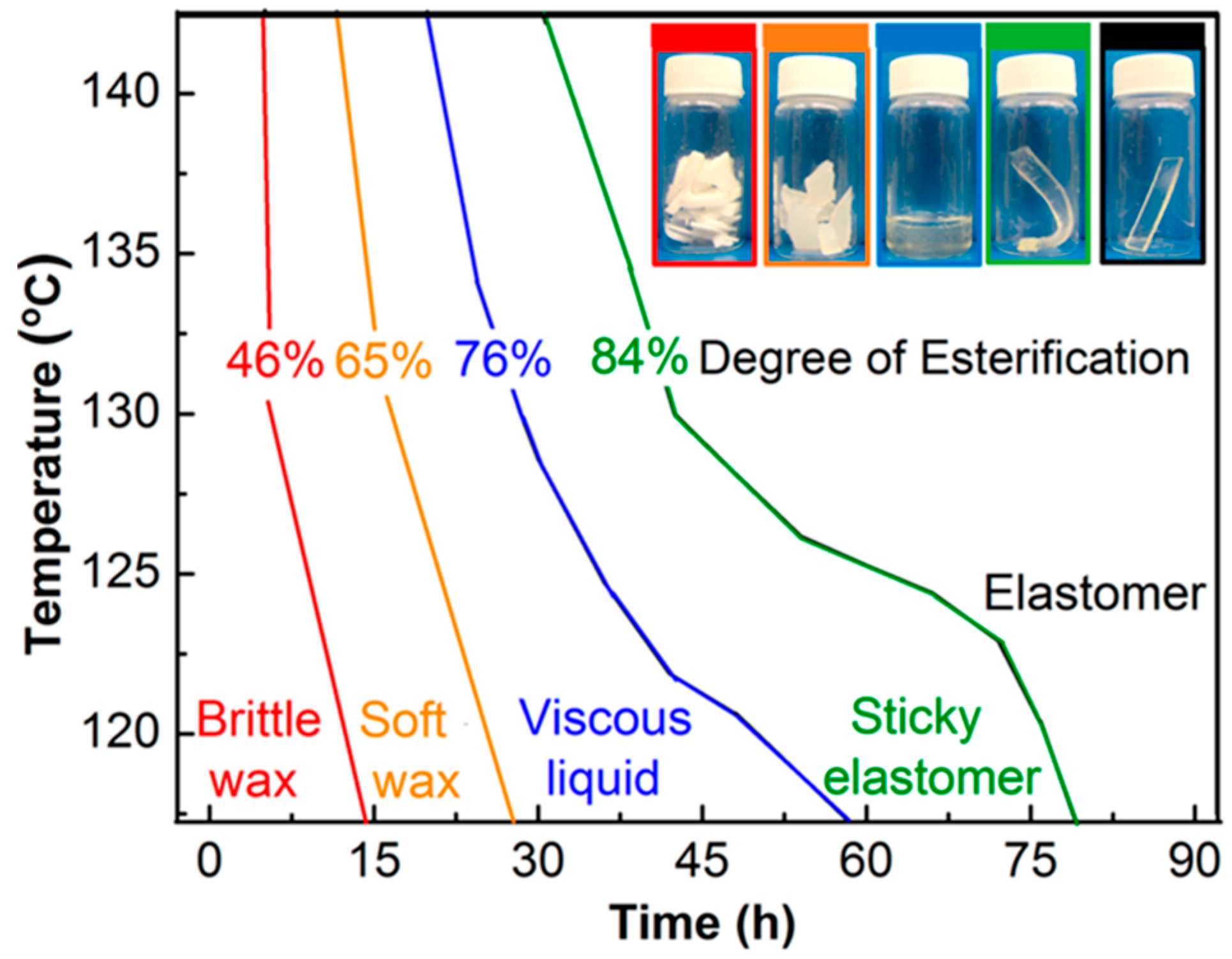
7.1.1. Synthesis
7.1.2. Mechanical Properties
7.1.3. Degradation
7.1.4. Compatibility
7.1.5. Processing
7.2. Applications
7.2.1. Tissue Engineering
Myocardial Tissue Engineering
Vascular Tissue Engineering
Bone and Cartilage Tissue Engineering
Other Tissues
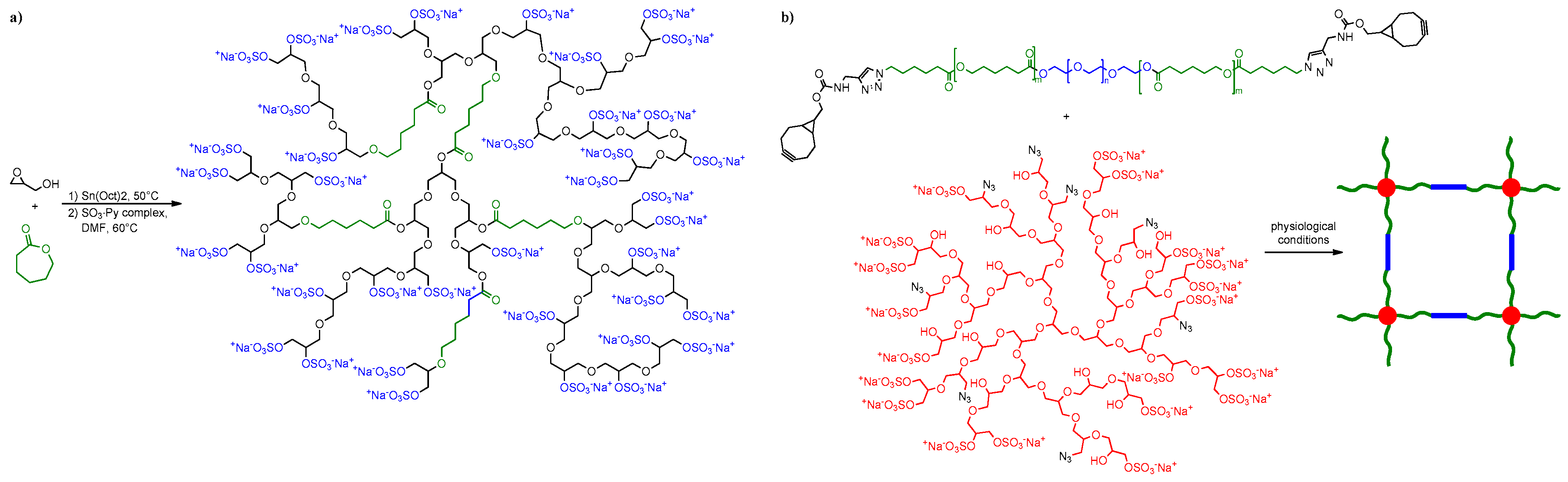
7.2.2. Drug Delivery
7.2.3. Other Applications
8. Poly(Glycerol Lactide), PGL
8.1. Synthesis
8.2. Applications
9. Poly(Glycerol ε-Caprolactone), PGCL
9.1. Synthesis and Properties
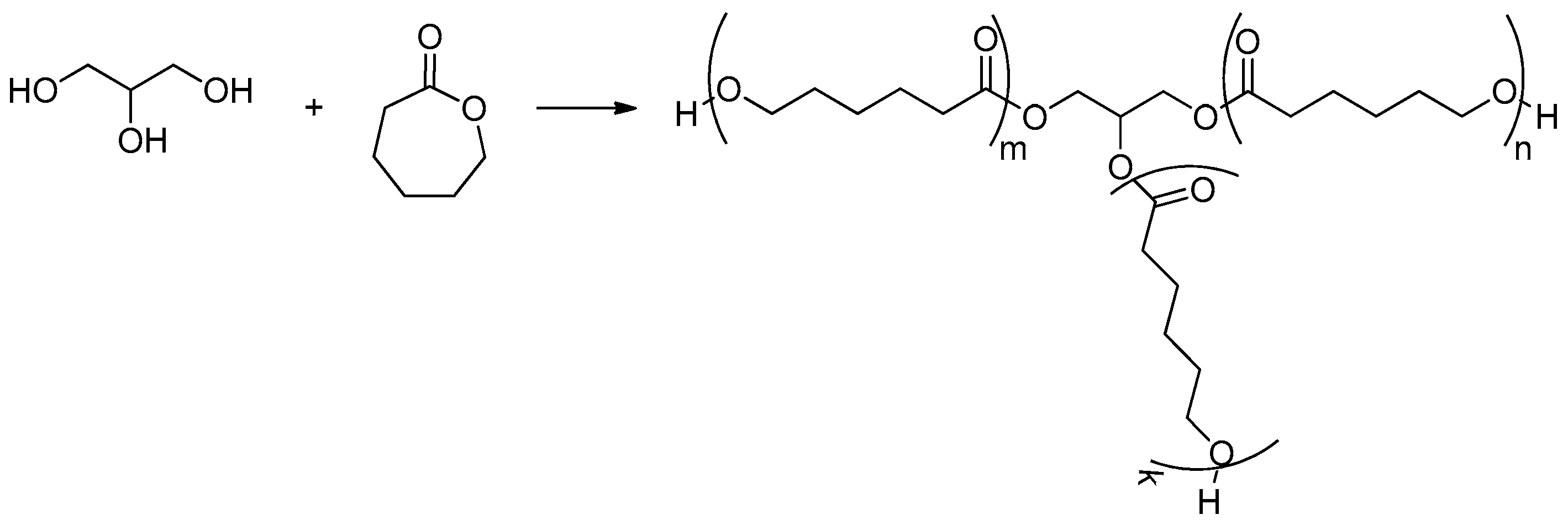
9.1.1. Three-Arm Poly(Glycerol ε-Caprolactone)
9.1.2. Synthesis of Block Polyglycerol/Poly(ε-Caprolactone) Copolymers
9.1.3. Functionalized Polyglycerol/Poly(ε-Caprolactone) Copolymers
9.1.4. Functionalized Polyglycerol/Poly(ε-Caprolactone) Copolymers with Additional Co-Monomers
9.2. Applications
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| AdA | adipic acid |
| AzA | azelaic acid |
| BSA | bovine serum albumin |
| CAL | lipase B from Candida Antarctica |
| CIP | ciprofloxacin-HCl |
| CL | ε-caprolactone |
| Cyt-ara | cytosine arabinoside |
| DB | degree of branching |
| D-lact | D-lactide |
| DMF | N,N-dimethyl formamide |
| DMSO | dimethyl sulfoxide |
| DP | degree of polymerization |
| dPGS | dendritic polyglycerol sulfate |
| DSC | differential scanning calorimetry |
| DXM | dexamethasone |
| DXMP | dexamethasone phosphate |
| ECM | extracellular matrix |
| EDC | 1-ethyl-(3,3-dimethylaminopropyl) carbodiimide |
| FA | folic acid |
| FDA | US Food and Drug Administration |
| FTIR | Fourier-transform infrared spectroscopy |
| Gld | glycidol |
| GA | glutaric acid |
| Gly | glycerol |
| GPC | gel permeation chromatography |
| h | hour |
| HA | hydroxyapatite |
| HBPG | hyperbranched polyglycerol |
| HBPG-NH2 | amino hyperbranched polyglycerol |
| HPMA | N-(2-hydroxypropyl) methacrylamide |
| IBU-Na | ibuprofen sodium salt |
| Ind | indomethacin |
| IV | intrinsic viscosity |
| LaA | lactic acid |
| Lact | lactide |
| L-lact | L-lactide |
| L-leu | L-leucine |
| MG | monoglyceride |
| Mn | number average molecular weight |
| MTX | methotrexate |
| Mw | weight average molecular weight |
| MWD | molecular weight distributions |
| NHS | N-hydroxysuccinimide |
| NMR | nuclear magnetic resonance |
| NP | nanoparticle |
| OX26 | transferrin antibody |
| PAMAM | polyamidoamine |
| PBS | phosphate buffer saline |
| PCL | poly(ε-caprolactone) |
| PDI | polydispersity index |
| PDL | pentadecalactone |
| PDLA | poly(D-lactic acid), poly(D-lactide) |
| PEG | polyethylene glycol |
| PEGDA | poly(ethylene glycol) diacrylate |
| PEI | polyethylenimine |
| PEO | poly(ethylene oxide) |
| PG | polyglycidol |
| PGA | poly(glycolic acid), poly(glycolide) |
| PGAd | poly(glycerol adipate) |
| PGAdS | stearic acid-modified PGAd |
| PGAz | poly(glycerol azelate) |
| PGCL | poly(glycerol ε-caprolactone) |
| PGGlu | poly(glycerol glutarate) |
| PGL | poly(glycerol lactide) |
| PGly | poly(glycerol) |
| PGSeb | poly(glycerol sebacate) |
| PGSMA | poly(glycerol succinate-co-maleate) |
| PGSub | poly(glycerol suberate) |
| PGSuc | poly(glycerol succinate) |
| PHA | poly(hydroxyalkanoates) |
| PLA | poly(lactic acid), poly(lactide) |
| PLGA | poly(lactic-co-glycolic acid), poly(lactide-co-glycolie) |
| PLLA | poly(L-lactic acid), poly(L-lactide) |
| PNIPAAm | poly(N-isopropylacrylamide) |
| PTX | paclitaxel |
| PVA | poly(vinyl alcohol) |
| RBITC | Rhodamine B Isothiocyanate |
| ROP | ring-opening-polymerization |
| RPC | retinal progenitor cells |
| SeA | sebacic acid |
| SEC | size exclusion chromatography |
| SEM | scanning electron microscopy |
| Sn(Oct)2 | tin(II) 2-ethylhexanoate, stannous octoate |
| SuA | succinic acid |
| TAC | tacrolimus |
| Tg | glass transition temperature |
| THF | tetrahydrofurane |
| Tm | melting temperature |
| TPT | tetraphenyl tin |
| TSA | p-toluenesulfonic acid |
References
- Albertsson, A.-C.; Varma, I.K. Aliphatic Polyesters: Synthesis, Properties and Applications BT—Degradable Aliphatic Polyesters; Springer: Berlin/Heidelberg, Germany, 2002; pp. 1–40. ISBN 978-3-540-45734-3. [Google Scholar]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, S.; Weng, Z.; Gao, C. Hyperbranched polymers: Advances from synthesis to applications. Chem. Soc. Rev. 2015, 44, 4091–4130. [Google Scholar] [CrossRef] [PubMed]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass: Volume I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas; National Renewable Energy Lab: Golden, CO, USA, 2004. [Google Scholar]
- Valerio, O.; Misra, M.; Mohanty, A.K. Poly(glycerol-co-diacids) Polyesters: From Glycerol Biorefinery to Sustainable Engineering Applications, A Review. ACS Sustain. Chem. Eng. 2018, 6, 5681–5693. [Google Scholar] [CrossRef]
- Kafouris, D.; Kossivas, F.; Constantinides, C.; Nguyen, N.Q.; Wesdemiotis, C.; Patrickios, C.S. Biosourced Amphiphilic Degradable Elastomers of Poly(glycerol sebacate): Synthesis and Network and Oligomer Characterization. Macromolecules 2013, 46, 622–630. [Google Scholar] [CrossRef]
- Frazza, E.J.; Schmitt, E.E. A new absorbable suture. J. Biomed. Mater. Res. 1971, 5, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Vert, M.; Li, S.M.; Spenlehauer, G.; Guerin, P. Bioresorbability and biocompatibility of aliphatic polyesters. J. Mater. Sci. Mater. Med. 1992, 3, 432–446. [Google Scholar] [CrossRef]
- Shalaby, S.W.; Johnson, R.A. Synthetic absorbable polyesters. In Biomedical Polymers: Designed-to-Degrade Systems; Shalaby, S.W., Ed.; Carl Hanser Verlag: Munchen, Germany, 1994; pp. 2–34. [Google Scholar]
- Puri, S.; Kallinteri, P.; Higgins, S.; Hutcheon, G.A.; Garnett, M.C. Drug incorporation and release of water soluble drugs from novel functionalised poly(glycerol adipate) nanoparticles. J. Control. Release 2008, 125, 59–67. [Google Scholar] [CrossRef]
- Daniel, W.; Stiriba, S.E.; Holger, F. Hyperbranched polyglycerols: From the controlled synthesis of biocompatible polyether polyols to multipurpose applications. Acc. Chem. Res. 2010, 43, 129–141. [Google Scholar]
- Abbina, S.; Vappala, S.; Kumar, P.; Siren, E.M.J.; La, C.C.; Abbasi, U.; Brooks, D.E.; Kizhakkedathu, J.N. Hyperbranched polyglycerols: Recent advances in synthesis, biocompatibility and biomedical applications. J. Mater. Chem. B 2017, 5, 9249–9277. [Google Scholar] [CrossRef]
- Carnahan, M.A.; Grinstaff, M.W. Synthesis and characterization of poly(glycerol-succinic acid) dendrimers. Macromolecules 2001, 34, 7648–7655. [Google Scholar] [CrossRef]
- Agach, M.; Delbaere, S.; Marinkovic, S.; Estrine, B.; Nardello-Rataj, V. Characterization, stability and ecotoxic properties of readily biodegradable branched oligoesters based on bio-sourced succinic acid and glycerol. Polym. Degrad. Stab. 2012, 97, 1956–1963. [Google Scholar] [CrossRef]
- Cheng, K.-C.; Chuang, T.-H.; Tsai, T.-H.; Guo, W.; Su, W.-F. Model of hyperbranched polymers formed by monomers A2 and Bg with end-capping molecules. Eur. Polym. J. 2008, 44, 2998–3004. [Google Scholar] [CrossRef]
- Valerio, O.; Pin, J.M.; Misra, M.; Mohanty, A.K. Synthesis of Glycerol-Based Biopolyesters as Toughness Enhancers for Polylactic Acid Bioplastic through Reactive Extrusion. ACS Omega 2016, 1, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Valerio, O.; Horvath, T.; Pond, C.; Manjusri, M.; Mohanty, A. Improved utilization of crude glycerol from biodiesel industries: Synthesis and characterization of sustainable biobased polyesters. Ind. Crops Prod. 2015, 78, 141–147. [Google Scholar] [CrossRef]
- Wyatt, V.T. Lewis acid-catalyzed synthesis of hyperbranched polymers based on glycerol and diacids in toluene. JAOCS J. Am. Oil Chem. Soc. 2012, 89, 313–319. [Google Scholar] [CrossRef]
- Wyatt, V.T.; Nuñez, A.; Foglia, T.A.; Marmer, W.N. Synthesis of hyperbranched poly(glycerol-diacid) oligomers. JAOCS J. Am. Oil Chem. Soc. 2006, 83, 1033–1039. [Google Scholar] [CrossRef]
- Agach, M.; Delbaere, S.; Marinkovic, S.; Estrine, B.; Nardello-Rataj, V. Synthesis, characterization, biodegradability and surfactant properties of bio-sourced lauroyl poly(glycerol-succinate) oligoesters. Colloids Surfaces A Physicochem. Eng. Asp. 2013, 419, 263–273. [Google Scholar] [CrossRef]
- Agach, M.; Marinkovic, S.; Estrine, B.; Nardello-Rataj, V. Acyl Poly(Glycerol-Succinic Acid) Oligoesters: Synthesis, Physicochemical and Functional Properties, and Biodegradability. J. Surfactants Deterg. 2016, 19, 933–941. [Google Scholar] [CrossRef]
- Agach, M.; Marinkovic, S.; Estrine, B.; Nardello-Rataj, V. Biosourced lauroyl poly(glycerol-succinate) oligoesters modified by copolymerizable solvents: A wasteless and eco-friendly surfactants properties enhancement. Colloids Surfaces A Physicochem. Eng. Asp. 2018, 536, 88–95. [Google Scholar] [CrossRef]
- Khongphow, C.; Theerakul, J.; Puttamat, S.; Singkhonrat, J. Characterisation of poly(glycerol-succinate) oligomers as bio-based non-ionic surfactants by nuclear magnetic resonance and mass spectrometry. Colloids Surfaces A Physicochem. Eng. Asp. 2015, 468, 301–308. [Google Scholar] [CrossRef]
- Pin, J.M.; Valerio, O.; Misra, M.; Mohanty, A. Impact of Butyl Glycidyl Ether Comonomer on Poly(glycerol-succinate) Architecture and Dynamics for Multifunctional Hyperbranched Polymer Design. Macromolecules 2017, 50, 732–745. [Google Scholar] [CrossRef]
- Baharu, M.N.; Kadhum, A.A.H.; Al-Amiery, A.A.; Mohamad, A.B. Synthesis and characterization of polyesters derived from glycerol, azelaic acid, and succinic acid. Green Chem. Lett. Rev. 2015, 8, 31–38. [Google Scholar] [CrossRef]
- Medeiros, E.S.; Offeman, R.D.; Klamczynski, A.P.; Glenn, G.M.; Mattoso, L.H.C.; Orts, W.J. Synthesis, Characterization and Nanocomposite Formation of Poly(glycerol succinate-co-maleate) with Nanocrystalline Cellulose. J. Polym. Environ. 2014, 22, 219–226. [Google Scholar] [CrossRef]
- Valerio, O.; Misra, M.; Mohanty, A.K. Sustainable biobased blends of poly(lactic acid) (PLA) and poly(glycerol succinate-: Co-maleate) (PGSMA) with balanced performance prepared by dynamic vulcanization. RSC Adv. 2017, 7, 38594–38603. [Google Scholar] [CrossRef]
- Valerio, O.; Misra, M.; Mohanty, A.K. Statistical design of sustainable thermoplastic blends of poly(glycerol succinate-co-maleate) (PGSMA), poly(lactic acid) (PLA) and poly(butylene succinate) (PBS). Polym. Test. 2018, 65, 420–428. [Google Scholar] [CrossRef]
- Valerio, O.; Misra, M.; Mohanty, A.K. Improvement of Impact Toughness of Biodegradable Poly(butylene succinate) by Melt Blending with Sustainable Biobased Glycerol Elastomers. J. Polym. Environ. 2018, 26, 1078–1087. [Google Scholar] [CrossRef]
- Luman, N.R.; Kim, T.; Grinstaff, M.W. Dendritic polymers composed of glycerol and succinic acid: Synthetic methodologies and medical applications. Pure Appl. Chem. 2007, 76, 1375–1385. [Google Scholar] [CrossRef]
- Immoos, C.E.; Ribeiro, A.A.; Morgan, M.T.; Lee, S.J.; Grinstaff, M.W.; Carnahan, M.A.; Finkelstein, S. Dendritic Molecular Capsules for Hydrophobic Compounds. J. Am. Chem. Soc. 2003, 125, 15485–15489. [Google Scholar]
- Carnahan, M.A.; Middleton, C.; Kim, J.; Kim, T.; Grinstaff, M.W. Hybrid dendritic-linear polyester-ethers for in situ photopolymerization. J. Am. Chem. Soc. 2002, 124, 5291–5293. [Google Scholar] [CrossRef]
- Wyatt, V.T.; Strahan, G.D.; Nuñez, A. The lewis acid-catalyzed synthesis of hyperbranched oligo(glycerol-diacid)s in aprotic polar media. JAOCS J. Am. Oil Chem. Soc. 2010, 87, 1359–1369. [Google Scholar] [CrossRef]
- Wyatt, V.T.; Strahan, G.D.; Nunez, A.; Haas, M.J. Characterization of Thermal and Mechanical Properties of Hyperbranched Oligo(glycerol-glutaric acid)s. J. Biobased Mater. Bioenergy 2011, 5, 92–101. [Google Scholar] [CrossRef]
- Wyatt, V.T.; Strahan, G.D. Degree of Branching in Hyperbranched Poly(glycerol-co-diacid)s Synthesized in Toluene. Polymers 2012, 4, 396–407. [Google Scholar] [CrossRef]
- Wyatt, V.T.; Yadav, M.P.; Latona, N.; Liu, C.-K. Thermal and Mechanical Properties of Glycerol-Based Polymer Films Infused with Plant Cell Wall Polysaccharides. J. Biobased Mater. Bioenergy 2013, 7, 348–356. [Google Scholar] [CrossRef]
- Wyatt, V.; Jones, K. Quantification of Monomers in Poly(glycerol-co-diacid) Gels Using Gas Chromatography. J. Biobased Mater. Bioenergy 2012, 6, 119–124. [Google Scholar] [CrossRef]
- Wyatt, V.T. The effects of solvent polarity and pKa on the absorption of solvents into poly(glutaric acid-glycerol) films. J. Appl. Polym. Sci. 2014, 131, 1–7. [Google Scholar] [CrossRef]
- Wyatt, V.T.; Yadav, M. A multivariant study of the absorption properties of poly(glutaric acid-glycerol) films. J. Appl. Polym. Sci. 2013, 130, 70–77. [Google Scholar] [CrossRef]
- Boakye, P.G.; Jones, K.C.; Latona, N.P.; Liu, C.K.; Besong, S.A.; Lumor, S.E.; Wyatt, V.T. Modification of absorbent poly(glycerol-glutaric acid) films by the addition of monoglycerides. J. Appl. Polym. Sci. 2017, 134, 45381. [Google Scholar] [CrossRef]
- Bilal, M.H.; Prehm, M.; Njau, A.E.; Samiullah, M.H.; Meister, A.; Kressler, J. Enzymatic synthesis and characterization of hydrophilic sugar based polyesters and their modification with stearic acid. Polymers (Basel) 2016, 8, 80. [Google Scholar] [CrossRef]
- Abo-zeid, Y.; Mantovani, G.; Irving, W.L.; Garnett, M.C. Synthesis of nucleoside-boronic esters hydrophobic pro-drugs: A possible route to improve hydrophilic nucleoside drug loading into polymer nanoparticles. J. Drug Deliv. Sci. Technol. 2018, 46, 354–364. [Google Scholar] [CrossRef]
- Iglesias, L.E.; Fukuyama, Y.; Hiroshi, N.; Rosa, E.-B.; Alicia, B. A simple enzymatic procedure for the synthesis of a hydroxylated polyester from glycerol and adipic acid. Biotechnol. Tech. 1999, 13, 923–926. [Google Scholar] [CrossRef]
- Kline, B.J.; Beckman, E.J.; Russell, A.J. One-Step Biocatalytic Synthesis of Linear Polyesters with Pendant Hydroxyl Groups. J. Am. Chem. Soc. 1998, 120, 9475–9480. [Google Scholar] [CrossRef]
- Taresco, V.; Creasey, R.G.; Kennon, J.; Mantovani, G.; Alexander, C.; Burley, J.C.; Garnett, M.C. Variation in structure and properties of poly(glycerol adipate) via control of chain branching during enzymatic synthesis. Polymer (Guildf) 2016, 89, 41–49. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Uyama, H.; Kobayashi, S. Enzymatic synthesis and curing of biodegradable crosslinkable polyesters. Macromol. Biosci. 2002, 2, 329–335. [Google Scholar] [CrossRef]
- Weiss, V.M.; Naolou, T.; Hause, G.; Kuntsche, J.; Kressler, J.; Mäder, K. Poly(glycerol adipate)-fatty acid esters as versatile nanocarriers: From nanocubes over ellipsoids to nanospheres. J. Control. Release 2012, 158, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Naolou, T.; Jbeily, M.; Scholtysek, P.; Kressler, J. Synthesis and Characterization of Stearoyl Modified Poly (Glycerol Adipate) Containing ATRP Initiator on its Backbone. In Progress in Polymer and Rubber Technology; Trans Tech Publications: Zurich, Switzerland, 2013; Volume 812, pp. 1–11. [Google Scholar]
- Naolou, T.; Hussain, H.; Baleed, S.; Busse, K.; Lechner, B.-D.; Kressler, J. The behavior of fatty acid modified poly(glycerol adipate) at the air/water interface. Colloids Surfaces A Physicochem. Eng. Asp. 2015, 468, 22–30. [Google Scholar] [CrossRef]
- Naolou, T.; Weiss, V.M.; Conrad, D.; Busse, K.; Mäder, K.; Kressler, J. Fatty acid modified poly(glycerol adipate)-Polymeric analogues of glycerides. In Tailored Polymer Architectures for Pharmaceutical and Biomedical Applications; American Chemical Society: Washington, DC, USA, 2013; Volume 1135, pp. 39–52. [Google Scholar]
- Villeneuve, P.; Foglia, T.A.; Mangos, T.J.; Nuñez, A. Synthesis of polyfunctional glycerol esters: Lipase-catalyzed esterification of glycerol with diesters. J. Am. Oil Chem. Soc. 1998, 75, 1545. [Google Scholar] [CrossRef]
- Kumar, A.; Kulshrestha, A.S.; Gao, W.; Gross, R.A. Versatile route to polyol polyesters by lipase catalysis. Macromolecules 2003, 36, 8219. [Google Scholar] [CrossRef]
- Navarro, L.; Ceaglio, N.; Rintoul, I. Structure and properties of biocompatible poly(glycerol adipate) elastomers modified with ethylene glycol. Polym. J. 2017, 49, 625. [Google Scholar] [CrossRef]
- Zhang, T.; Howell, B.A.; Dumitrascu, A.; Martin, S.J.; Smith, P.B. Synthesis and characterization of glycerol-adipic acid hyperbranched polyesters. Polymer (Guildf) 2014, 55, 5065–5072. [Google Scholar] [CrossRef]
- Zhang, T.; Howell, B.A.; Smith, P.B. Thermal degradation of glycerol/adipic acid hyperbranched poly(ester)s containing either hydroxyl or carboxyl end-groups. J. Therm. Anal. Calorim. 2015, 122, 1221–1229. [Google Scholar] [CrossRef]
- Holser, R.A. Degradation rates of glycerol polyesters at acidic and basic conditions. Mater. Chem. Phys. 2011, 128, 10–11. [Google Scholar] [CrossRef]
- Suksiriworapong, J.; Taresco, V.; Ivanov, D.P.; Styliari, I.D.; Sakchaisri, K.; Junyaprasert, V.B.; Garnett, M.C. Synthesis and properties of a biodegradable polymer-drug conjugate: Methotrexate-poly(glycerol adipate). Colloids Surfaces B Biointerfaces 2018, 167, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Weiss, V.M.; Naolou, T.; Groth, T.; Kressler, J.; Mäder, K. In Vitro Toxicity of Stearoyl-poly(glycerol adipate) Nanoparticles. J. Appl. Biomater. Funct. Mater. 2012, 10, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Parker, T.L.; Kallinteri, P.; Walker, D.A.; Higgins, S.; Hutcheon, G.A.; Garnett, M.C. Uptake and metabolism of novel biodegradable poly (glycerol-adipate) nanoparticles in DAOY monolayer. J. Control. Release 2006, 116, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Kallinteri, P.; Walker, D.A.; Parker, T.L.; Garnett, M.C. Evaluation of Poly (Glycerol-Adipate) Nanoparticle Uptake in an In Vitro 3-D Brain Tumor Co-Culture Model. Exp. Biol. Med. 2007, 232, 1100–1108. [Google Scholar] [CrossRef]
- Meng, W.; Garnett, M.C.; Walker, D.A.; Parker, T.L. Penetration and intracellular uptake of poly(glycerol-adipate) nanoparticles into three-dimensional brain tumour cell culture models. Exp. Biol. Med. 2016, 241, 466–477. [Google Scholar] [CrossRef]
- Abo-zeid, Y.; Urbanowicz, R.A.; Thomson, B.J.; Irving, W.L.; Tarr, A.W.; Garnett, M.C. Enhanced nanoparticle uptake into virus infected cells: Could nanoparticles be useful in antiviral therapy? Int. J. Pharm. 2018, 547, 572–581. [Google Scholar] [CrossRef]
- Orafai, H.; Kallinteri, P.; Garnett, M.; Huggins, S.; Hutcheon, G.; Pourcain, C. Novel poly(glycerol-adipate) polymers used for nanoparticle making: A study of surface free energy. Iran. J. Pharm. Res. 2008, 7, 11–19. [Google Scholar]
- Taresco, V.; Suksiriworapong, J.; Creasey, R.; Burley, J.C.; Mantovani, G.; Alexander, C.; Treacher, K.; Booth, J.; Garnett, M.C. Properties of acyl modified poly(glycerol-adipate) comb-like polymers and their self-assembly into nanoparticles. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 3267–3278. [Google Scholar] [CrossRef]
- Mackenzie, R.; Booth, J.; Alexander, C.; Garnett, M.C.; Laughton, C.A. Multiscale Modeling of Drug–Polymer Nanoparticle Assembly Identifies Parameters Influencing Drug Encapsulation Efficiency. J. Chem. Theory Comput. 2015, 11, 2705–2713. [Google Scholar] [CrossRef]
- Kallinteri, P.; Higgins, S.; Hutcheon, G.A.; St. Pourçain, C.B.; Garnett, M.C. Novel functionalized biodegradable polymers for nanoparticle drug delivery systems. Biomacromolecules 2005, 6, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Wahab, A.; Favretto, M.E.; Onyeagor, N.D.; Khan, G.M.; Douroumis, D.; Casely-Hayford, M.A.; Kallinteri, P. Development of poly(glycerol adipate) nanoparticles loaded with non-steroidal anti-inflammatory drugs. J. Microencapsul. 2012, 29, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Wersig, T.; Hacker, M.C.; Kressler, J.; Mäder, K. Poly(glycerol adipate)—Indomethacin drug conjugates—Synthesis and in vitro characterization. Int. J. Pharm. 2017, 531, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Wersig, T.; Krombholz, R.; Janich, C.; Meister, A.; Kressler, J.; Mäder, K. Indomethacin functionalised poly(glycerol adipate) nanospheres as promising candidates for modified drug release. Eur. J. Pharm. Sci. 2018, 123, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Howell, B.A.; Zhang, D.; Zhu, B.; Smith, P.B. Hyperbranched poly (ester) s for delivery of small molecule therapeutics. Polym. Adv. Technol. 2018, 29, 2352–2363. [Google Scholar] [CrossRef]
- Kunda, N.K.; Alfagih, I.M.; Dennison, S.R.; Tawfeek, H.M.; Somavarapu, S.; Hutcheon, G.A.; Saleem, I.Y. Bovine serum albumin adsorbed PGA-CO-PDL nanocarriers for vaccine delivery via dry powder inhalation. Pharm. Res. 2015, 32, 1341–1353. [Google Scholar] [CrossRef]
- Saleem, I.Y.; Mohammed, A.R.; Abdellatif, A.A.H.; Dennison, T.J.; Tawfeek, H.M.; Sadiq, Y. Colonic delivery of indometacin loaded PGA-co-PDL microparticles coated with Eudragit L100-55 from fast disintegrating tablets. Int. J. Pharm. 2017, 531, 80–89. [Google Scholar]
- Weiss, V.M.; Lucas, H.; Mueller, T.; Chytil, P.; Etrych, T.; Naolou, T.; Kressler, J.; Mäder, K. Intended and Unintended Targeting of Polymeric Nanocarriers: The Case of Modified Poly(glycerol adipate) Nanoparticles. Macromol. Biosci. 2018, 18, 1700240. [Google Scholar] [CrossRef]
- Nusrath Unnisa, C.; Chitra, S.; Selvasekarapandian, S.; Monisha, S.; Nirmala Devi, G.; Moniha, V.; Hema, M. Development of poly(glycerol suberate) polyester (PGS)–PVA blend polymer electrolytes with NH4SCN and its application. Ionics (Kiel) 2018, 24, 1979–1993. [Google Scholar] [CrossRef]
- Chongcharoenchaikul, T.; Thamyongkit, P.; Poompradub, S. Synthesis, characterization and properties of a bio-based poly(glycerol azelate) polyester. Mater. Chem. Phys. 2016, 177, 485–495. [Google Scholar] [CrossRef]
- Kadhum, A.A.H.; Baharu, M.N.; Mahmood, M.H. Elastic polyesters from glycerol and azelaic acid. Adv. Mater. Res. 2011, 233–235, 2571–2575. [Google Scholar] [CrossRef]
- Wang, Y.; Ameer, G.A.; Sheppard, B.J.; Langer, R. A tough biodegradable elastomer. Nat. Biotechnol. 2002, 20, 602. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Thouas, G.A.; Chen, Q.-Z. Biodegradable soft elastomers: Synthesis/properties of materials and fabrication of scaffolds. RSC Adv. 2012, 2, 8229–8242. [Google Scholar] [CrossRef]
- Rai, R.; Tallawi, M.; Grigore, A.; Boccaccini, A.R. Synthesis, properties and biomedical applications of poly(glycerol sebacate) (PGS): A review. Prog. Polym. Sci. 2012, 37, 1051–1078. [Google Scholar] [CrossRef]
- Liu, Q.; Jiang, L.; Shi, R.; Zhang, L. Synthesis, preparation, in vitro degradation, and application of novel degradable bioelastomers—A review. Prog. Polym. Sci. 2012, 37, 715–765. [Google Scholar] [CrossRef]
- Loh, X.J.; Abdul Karim, A.; Owh, C. Poly(glycerol sebacate) biomaterial: Synthesis and biomedical applications. J. Mater. Chem. B 2015, 3, 7641–7652. [Google Scholar] [CrossRef]
- Gao, J.; Crapo, P.M.; Wang, Y. Macroporous Elastomeric Scaffolds with Extensive Micropores for Soft Tissue Engineering. Tissue Eng. 2006, 12, 917–925. [Google Scholar] [CrossRef]
- Li, Y.; Cook, W.D.; Moorhoff, C.; Huang, W.-C.; Chen, Q.-Z. Synthesis, characterization and properties of biocompatible poly(glycerol sebacate) pre-polymer and gel. Polym. Int. 2013, 62, 534–547. [Google Scholar] [CrossRef]
- Uyama, H.; Inada, K.; Kobayashi, S. Regioselective polymerization of divinyl sebacate and triols using lipase catalyst. Macromol. Rapid Commun. 1999, 20, 171–174. [Google Scholar] [CrossRef]
- Uyama, H.; Inada, K.; Kobayashi, S. Regioselectivity Control in Lipase-Catalyzed Polymerization of Divinyl Sebacate and Triols. Macromol. Biosci. 2001, 1, 40–44. [Google Scholar] [CrossRef]
- Moorhoff, C.; Li, Y.; Cook, W.D.; Braybrook, C.; Chen, Q.-Z. Characterization of the prepolymer and gel of biocompatible poly(xylitol sebacate) in comparison with poly(glycerol sebacate) using a combination of mass spectrometry and nuclear magnetic resonance. Polym. Int. 2015, 64, 668–688. [Google Scholar] [CrossRef]
- Kiyotsukuri, T.; Kanaboshi, M.; Tsutsumi, N. Network polyester films from glycerol and dicarboxylic acids. Polym. Int. 1994, 33, 1–8. [Google Scholar] [CrossRef]
- Nagata, M.; Kiyotsukuri, T.; Ibuki, H.; Tsutsumi, N.; Sakai, W. Synthesis and enzymatic degradation of regular network aliphatic polyesters. React. Funct. Polym. 1996, 30, 165–171. [Google Scholar] [CrossRef]
- Maliger, R.; Halley, P.J.; Cooper-White, J.J. Poly(glycerol–sebacate) bioelastomers—Kinetics of step-growth reactions using Fourier Transform (FT)-Raman spectroscopy. J. Appl. Polym. Sci. 2013, 127, 3980–3986. [Google Scholar] [CrossRef]
- Liu, Q.; Tian, M.; Ding, T.; Shi, R.; Feng, Y.; Zhang, L.; Chen, D.; Tian, W. Preparation and characterization of a thermoplastic poly(glycerol sebacate) elastomer by two-step method. J. Appl. Polym. Sci. 2007, 103, 1412–1419. [Google Scholar] [CrossRef]
- Liu, Q.; Tian, M.; Shi, R.; Zhang, L.; Chen, D.; Tian, W. Structure and properties of thermoplastic poly(glycerol sebacate) elastomers originating from prepolymers with different molecular weights. J. Appl. Polym. Sci. 2007, 104, 1131–1137. [Google Scholar] [CrossRef]
- Maliger, R.B.; Halley, P.J.; Cooper-White, J.J. Poly (glycerol-sebacate) bioelastomers: 2. Synthesis using Brabender Plasticoder® as a batch reactor. J. Appl. Polym. Sci. 2016, 133, 42852. [Google Scholar] [CrossRef]
- Li, Y.; Huang, W.; Cook, W.D.; Chen, Q. A comparative study on poly(xylitol sebacate) and poly(glycerol sebacate): Mechanical properties,biodegradation and cytocompatibility. Biomed. Mater. 2013, 8, 035006. [Google Scholar] [CrossRef]
- Li, X.; Hong, A.T.L.; Naskar, N.; Chung, H.J. Criteria for quick and consistent synthesis of poly(glycerol sebacate) for tailored mechanical properties. Biomacromolecules 2015, 16, 1525–1533. [Google Scholar] [CrossRef]
- Lau, C.C.; Bayazit, M.K.; Knowles, J.C.; Tang, J. Tailoring degree of esterification and branching of poly(glycerol sebacate) by energy efficient microwave irradiation. Polym. Chem. 2017, 8, 3937–3947. [Google Scholar] [CrossRef]
- Gadomska-Gajadhur, A.; Wrzecionek, M.; Matyszczak, G.; Piętowski, P.; Więcław, M.; Ruśkowski, P. Optimization of Poly(glycerol sebacate) Synthesis for Biomedical Purposes with the Design of Experiments. Org. Process Res. Dev. 2018, 22, 1793–1800. [Google Scholar] [CrossRef]
- Liu, Q.; Tian, M.; Ding, T.; Shi, R.; Zhang, L. Preparation and characterization of a biodegradable polyester elastomer with thermal processing abilities. J. Appl. Polym. Sci. 2005, 98, 2033–2041. [Google Scholar] [CrossRef]
- Conejero-García, Á.; Gimeno, H.R.; Sáez, Y.M.; Vilariño-Feltrer, G.; Ortuño-Lizarán, I.; Vallés-Lluch, A. Correlating synthesis parameters with physicochemical properties of poly(glycerol sebacate). Eur. Polym. J. 2017, 87, 406–419. [Google Scholar] [CrossRef]
- Aydin, H.M.; Salimi, K.; Rzayev, Z.M.O.; Pişkin, E. Microwave-assisted rapid synthesis of poly(glycerol-sebacate) elastomers. Biomater. Sci. 2013, 1, 503–509. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, K.-W.; Gade, P.S.; Robertson, A.M.; Wang, Y. Microwave-assisted facile fabrication of porous poly (glycerol sebacate) scaffolds. J. Biomater. Sci. Polym. Ed. 2018, 29, 907–916. [Google Scholar] [CrossRef]
- Chen, Q.-Z.; Bismarck, A.; Hansen, U.; Junaid, S.; Tran, M.Q.; Harding, S.E.; Ali, N.N.; Boccaccini, A.R. Characterisation of a soft elastomer poly(glycerol sebacate) designed to match the mechanical properties of myocardial tissue. Biomaterials 2008, 29, 47–57. [Google Scholar] [CrossRef]
- Jaafar, I.H.; Ammar, M.M.; Jedlicka, S.S.; Pearson, R.A.; Coulter, J.P. Spectroscopic evaluation, thermal, and thermomechanical characterization of poly(glycerol-sebacate) with variations in curing temperatures and durations. J. Mater. Sci. 2010, 45, 2525–2529. [Google Scholar] [CrossRef]
- Kemppainen, J.M.; Hollister, S.J. Tailoring the mechanical properties of 3D-designed poly(glycerol sebacate) scaffolds for cartilage applications. J. Biomed. Mater. Res. Part A 2010, 94, 9–18. [Google Scholar] [CrossRef]
- Chen, Q.; Yang, X.; Li, Y. A comparative study on in vitro enzymatic degradation of poly(glycerol sebacate) and poly(xylitol sebacate). RSC Adv. 2012, 2, 4125–4134. [Google Scholar] [CrossRef]
- Sundback, C.A.; McFadden, J.; Hart, A.; Kulig, K.M.; Wieland, A.M.; Pereira, M.J.N.; Pomerantseva, I.; Hartnick, C.J.; Masiakos, P.T. Behavior of poly(glycerol sebacate) plugs in chronic tympanic membrane perforations. J. Biomed. Mater. Res. Part B Appl. Biomater. 2012, 100, 1943–1954. [Google Scholar] [CrossRef]
- Mitsak, A.G.; Dunn, A.M.; Hollister, S.J. Mechanical characterization and non-linear elastic modeling of poly(glycerol sebacate) for soft tissue engineering. J. Mech. Behav. Biomed. Mater. 2012, 11, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Thouas, G.A.; Shi, H.; Chen, Q. Enzymatic and oxidative degradation of poly(polyol sebacate). J. Biomater. Appl. 2013, 28, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-L.; Lu, X.-L.; Dong, D.-L.; Sun, Z.-J. Characterization and optimization of glycerol/sebacate ratio in poly(glycerol-sebacate) elastomer for cell culture application. J. Biomed. Mater. Res. Part A 2014, 102, 3903–3907. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Tallawi, M.; Roether, J.A.; Detsch, R.; Barbani, N.; Rosellini, E.; Kaschta, J.; Schubert, D.W.; Boccaccini, A.R. Sterilization effects on the physical properties and cytotoxicity of poly(glycerol sebacate). Mater. Lett. 2013, 105, 32–35. [Google Scholar] [CrossRef]
- Sundback, C.A.; Shyu, J.Y.; Wang, Y.; Faquin, W.C.; Langer, R.S.; Vacanti, J.P.; Hadlock, T.A. Biocompatibility analysis of poly(glycerol sebacate) as a nerve guide material. Biomaterials 2005, 26, 5454–5464. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, D.J.; Ishii, H.; Chen, Q.-Z.; Boccaccini, A.R.; Hansen, U.; Carr, C.A.; Roether, J.A.; Jawad, H.; Tyler, D.J.; Ali, N.N.; et al. Magnetic Resonance Imaging Evaluation of Remodeling by Cardiac Elastomeric Tissue Scaffold Biomaterials in a Rat Model of Myocardial Infarction. Tissue Eng. Part A 2010, 16, 3395–3402. [Google Scholar] [CrossRef]
- Liang, S.L.; Yang, X.Y.; Fang, X.Y.; Cook, W.D.; Thouas, G.A.; Chen, Q.Z. In Vitro enzymatic degradation of poly (glycerol sebacate)-based materials. Biomaterials 2011, 32, 8486–8496. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, Y.M.; Langer, R. In vivo degradation characteristics of poly(glycerol sebacate). J. Biomed. Mater. Res. Part A 2003, 66, 192–197. [Google Scholar] [CrossRef]
- LeBlon, C.E.; Pai, R.; Fodor, C.R.; Golding, A.S.; Coulter, J.P.; Jedlicka, S.S. In vitro comparative biodegradation analysis of salt-leached porous polymer scaffolds. J. Appl. Polym. Sci. 2013, 128, 2701–2712. [Google Scholar] [CrossRef]
- Gade, P.S.; Lee, K.; Pfaff, B.N.; Wang, Y.; Robertson, A.M. Degradation and erosion mechanisms of bioresorbable porous acellular vascular grafts: An in vitro investigation. J. R. Soc. Interface 2017, 14, 20170102. [Google Scholar] [CrossRef]
- Motlagh, D.; Yang, J.; Lui, K.Y.; Webb, A.R.; Ameer, G.A. Hemocompatibility evaluation of poly(glycerol-sebacate) in vitro for vascular tissue engineering. Biomaterials 2006, 27, 4315–4324. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Ensley, A.E.; Nerem, R.M.; Wang, Y. Poly(glycerol sebacate) supports the proliferation and phenotypic protein expression of primary baboon vascular cells. J. Biomed. Mater. Res. Part A 2007, 83, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-Z.; Ishii, H.; Thouas, G.A.; Lyon, A.R.; Wright, J.S.; Blaker, J.J.; Chrzanowski, W.; Boccaccini, A.R.; Ali, N.N.; Knowles, J.C.; et al. An elastomeric patch derived from poly(glycerol sebacate) for delivery of embryonic stem cells to the heart. Biomaterials 2010, 31, 3885–3893. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Jin, L.; Cook, W.D.; Mohn, D.; Lagerqvist, E.L.; Elliott, D.A.; Haynes, J.M.; Boyd, N.; Stark, W.J.; Pouton, C.W.; et al. Elastomeric nanocomposites as cell delivery vehicles and cardiac support devices. Soft Matter 2010, 6, 4715–4726. [Google Scholar] [CrossRef]
- Tallawi, M.; Rai, R.; R-Gleixner, M.; Roerick, O.; Weyand, M.; Roether, J.A.; Schubert, D.W.; Kozlowska, A.; El Fray, M.; Merle, B.; et al. Poly(glycerol sebacate)\poly(butylene succinate-dilinoleate) blends as candidate materials for cardiac tissue engineering. Macromol. Symp. 2013, 334, 57–67. [Google Scholar] [CrossRef]
- Ye, X.; Lu, L.; Kolewe, M.E.; Park, H.; Larson, B.L.; Kim, E.S.; Freed, L.E. A biodegradable microvessel scaffold as a framework to enable vascular support of engineered tissues. Biomaterials 2013, 34, 10007–10015. [Google Scholar] [CrossRef]
- Bettinger, C.J.; Orrick, B.; Misra, A.; Langer, R.; Borenstein, J.T. Microfabrication of poly (glycerol–sebacate) for contact guidance applications. Biomaterials 2006, 27, 2558–2565. [Google Scholar] [CrossRef]
- Neeley, W.L.; Redenti, S.; Klassen, H.; Tao, S.; Desai, T.; Young, M.J.; Langer, R. A microfabricated scaffold for retinal progenitor cell grafting. Biomaterials 2008, 29, 418–426. [Google Scholar] [CrossRef]
- Neal, R.A.; Jean, A.; Park, H.; Wu, P.B.; Hsiao, J.; Engelmayr, G.C.; Langer, R.; Freed, L.E. Three-Dimensional Elastomeric Scaffolds Designed with Cardiac-Mimetic Structural and Mechanical Features. Tissue Eng. Part A 2012, 19, 793–807. [Google Scholar] [CrossRef]
- Hsieh, Y.-K.; Chen, S.-C.; Huang, W.-L.; Hsu, K.-P.; Gorday, K.A.V.; Wang, T.; Wang, J. Direct Micromachining of Microfluidic Channels on Biodegradable Materials Using Laser Ablation. Polymers (Basel) 2017, 9, 242. [Google Scholar] [CrossRef]
- Engelmayr, G.C., Jr.; Cheng, M.; Bettinger, C.J.; Borenstein, J.T.; Langer, R.; Freed, L.E. Accordion-like honeycombs for tissue engineering of cardiac anisotropy. Nat. Mater. 2008, 7, 1003. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Larson, B.L.; Guillemette, M.D.; Jain, S.R.; Hua, C.; Engelmayr, G.C.; Freed, L.E. The significance of pore microarchitecture in a multi-layered elastomeric scaffold for contractile cardiac muscle constructs. Biomaterials 2011, 32, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Bettinger, C.J.; Weinberg, E.J.; Kulig, K.M.; Vacanti, J.P.; Wang, Y.; Borenstein, J.T.; Langer, R. Three-dimensional microfluidic tissue-engineering scaffolds using a flexible biodegradable polymer. Adv. Mater. 2006, 18, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Kolewe, M.E.; Park, H.; Gray, C.; Ye, X.; Langer, R.; Freed, L.E. 3D Structural Patterns in Scalable, Elastomeric Scaffolds Guide Engineered Tissue Architecture. Adv. Mater. 2013, 25, 4459–4465. [Google Scholar] [CrossRef] [PubMed]
- Fidkowski, C.; Kaazempur-Mofrad, M.R.; Borenstein, J.; Vacanti, J.P.; Langer, R.; Wang, Y. Endothelialized Microvasculature Based on a Biodegradable Elastomer. Tissue Eng. 2005, 11, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Radisic, M.; Park, H.; Martens, T.P.; Salazar-Lazaro, J.E.; Geng, W.; Wang, Y.; Langer, R.; Freed, L.E.; Vunjak-Novakovic, G. Pre-treatment of synthetic elastomeric scaffolds by cardiac fibroblasts improves engineered heart tissue. J. Biomed. Mater. Res.Part A 2008, 86, 713–724. [Google Scholar] [CrossRef]
- Yi, F.; LaVan, D.A. Poly(glycerol sebacate) Nanofiber Scaffolds by Core/Shell Electrospinning. Macromol. Biosci. 2008, 8, 803–806. [Google Scholar] [CrossRef]
- Hsu, C.-N.; Lee, P.-Y.; Tuan-Mu, H.-Y.; Li, C.-Y.; Hu, J.-J. Fabrication of a mechanically anisotropic poly(glycerol sebacate) membrane for tissue engineering. J. Biomed. Mater. Res. Part B Appl. Biomater. 2018, 106, 760–770. [Google Scholar] [CrossRef]
- Crapo, P.M.; Gao, J.; Wang, Y. Seamless tubular poly(glycerol sebacate) scaffolds: High-yield fabrication and potential applications. J. Biomed. Mater. Res. Part A 2008, 86, 354–363. [Google Scholar] [CrossRef]
- Crapo, P.M.; Wang, Y. Physiologic compliance in engineered small-diameter arterial constructs based on an elastomeric substrate. Biomaterials 2010, 31, 1626–1635. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, C.; Qiao, X.; Liu, T.; Sun, K. Porous poly(glycerol sebacate) (PGS) elastomer scaffolds for skin tissue engineering. Polym. Test. 2016, 54, 118–125. [Google Scholar] [CrossRef]
- Jeong, C.G.; Hollister, S.J. A comparison of the influence of material on in vitro cartilage tissue engineering with PCL, PGS, and POC 3D scaffold architecture seeded with chondrocytes. Biomaterials 2010, 31, 4304–4312. [Google Scholar] [CrossRef] [PubMed]
- Frydrych, M.; Chen, B. Large three-dimensional poly(glycerol sebacate)-based scaffolds—A freeze-drying preparation approach. J. Mater. Chem. B 2013, 1, 6650–6661. [Google Scholar] [CrossRef]
- You, Z.R.; Hu, M.H.; Tuan-Mu, H.Y.; Hu, J.J. Fabrication of poly(glycerol sebacate) fibrous membranes by coaxial electrospinning: Influence of shell and core solutions. J. Mech. Behav. Biomed. Mater. 2016, 63, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Li, Y.; Zhu, C.; Cook, W.D.; Forsythe, J.; Chen, Q. Fabrication, mechanical properties and cytocompatibility of elastomeric nanofibrous mats of poly(glycerol sebacate). Eur. Polym. J. 2015, 64, 79–92. [Google Scholar] [CrossRef]
- Khosravi, R.; Best, C.A.; Allen, R.A.; Stowell, C.E.T.; Onwuka, E.; Zhuang, J.J.; Lee, Y.U.; Yi, T.; Bersi, M.R.; Shinoka, T.; et al. Long-Term Functional Efficacy of a Novel Electrospun Poly(Glycerol Sebacate)-Based Arterial Graft in Mice. Ann. Biomed. Eng. 2016, 44, 2402–2416. [Google Scholar] [CrossRef]
- Jeffries, E.M.; Allen, R.A.; Gao, J.; Pesce, M.; Wang, Y. Highly elastic and suturable electrospun poly(glycerol sebacate) fibrous scaffolds. Acta Biomater. 2015, 18, 30–39. [Google Scholar] [CrossRef]
- Vogt, L.; Rivera, L.R.; Liverani, L.; Piegat, A.; El Fray, M.; Boccaccini, A.R. Poly(ε-caprolactone)/poly(glycerol sebacate) electrospun scaffolds for cardiac tissue engineering using benign solvents. Mater. Sci. Eng. C 2019, 103, 109712. [Google Scholar] [CrossRef]
- Louage, B.; Tack, L.; Wang, Y.; De Geest, B.G. Poly(glycerol sebacate) nanoparticles for encapsulation of hydrophobic anti-cancer drugs. Polym. Chem. 2017, 8, 5033–5038. [Google Scholar] [CrossRef]
- Rai, R.; Tallawi, M.; Barbani, N.; Frati, C.; Madeddu, D.; Cavalli, S.; Graiani, G.; Quaini, F.; Roether, J.A.; Schubert, D.W.; et al. Biomimetic poly(glycerol sebacate) (PGS) membranes for cardiac patch application. Mater. Sci. Eng. C 2013, 33, 3677–3687. [Google Scholar] [CrossRef]
- Marsano, A.; Maidhof, R.; Wan, L.Q.; Wang, Y.; Gao, J.; Tandon, N.; Vunjak-Novakovic, G. Scaffold stiffness affects the contractile function of three-dimensional engineered cardiac constructs. Biotechnol. Prog. 2010, 26, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Rodda, A.E.; Truong, V.X.; Shi, Y.; Zhou, K.; Haynes, J.M.; Wang, B.; Cook, W.D.; Forsythe, J.S. Increased Cardiomyocyte Alignment and Intracellular Calcium Transients Using Micropatterned and Drug-Releasing Poly(Glycerol Sebacate) Elastomers. ACS Biomater. Sci. Eng. 2018, 4, 2494–2504. [Google Scholar] [CrossRef]
- Masoumi, N.; Johnson, K.L.; Howell, M.C.; Engelmayr, G.C. Valvular interstitial cell seeded poly(glycerol sebacate) scaffolds: Toward a biomimetic in vitro model for heart valve tissue engineering. Acta Biomater. 2013, 9, 5974–5988. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, R.; Venugopal, J.R.; Sundarrajan, S.; Mukherjee, S.; Sridhar, R.; Ramakrishna, S. Minimally invasive injectable short nanofibers of poly(glycerol sebacate) for cardiac tissue engineering. Nanotechnology 2012, 23, 385102. [Google Scholar] [CrossRef]
- Rai, R.; Tallawi, M.; Frati, C.; Falco, A.; Gervasi, A.; Quaini, F.; Roether, J.A.; Hochburger, T.; Schubert, D.W.; Seik, L.; et al. Bioactive Electrospun Fibers of Poly(glycerol sebacate) and Poly(ε-caprolactone) for Cardiac Patch Application. Adv. Healthc. Mater. 2015, 4, 2012–2025. [Google Scholar] [CrossRef]
- Tallawi, M.; Rai, R.; Boccaccini, A.R.; Zebrowski, D.C.; Engel, F.B.; Roether, J.A.; Schubert, D.W.; El Fray, M.; Aifantis, K.E. Poly(Glycerol Sebacate)/Poly(Butylene Succinate-Butylene Dilinoleate) Fibrous Scaffolds for Cardiac Tissue Engineering. Tissue Eng. Part C Methods 2015, 21, 585–596. [Google Scholar] [CrossRef]
- Lee, K.-W.; Stolz, D.B.; Wang, Y. Substantial expression of mature elastin in arterial constructs. Proc. Natl. Acad. Sci. USA 2011, 108, 2705. [Google Scholar] [CrossRef]
- Crapo, P.M.; Wang, Y. Hydrostatic pressure independently increases elastin and collagen co-expression in small-diameter engineered arterial constructs. J. Biomed. Mater. Res. Part A 2011, 96, 673–681. [Google Scholar] [CrossRef]
- Wu, W.; Allen, R.A.; Wang, Y. Fast-degrading elastomer enables rapid remodeling of a cell-free synthetic graft into a neoartery. Nat. Med. 2012, 18, 1148. [Google Scholar] [CrossRef]
- Zaky, S.H.; Lee, K.W.; Gao, J.; Jensen, A.; Verdelis, K.; Wang, Y.; Almarza, A.J.; Sfeir, C. Poly (glycerol sebacate) elastomer supports bone regeneration by its mechanical properties being closer to osteoid tissue rather than to mature bone. Acta Biomater. 2017, 54, 95–106. [Google Scholar] [CrossRef]
- Zaky, S.H.; Lee, K.-W.; Gao, J.; Jensen, A.; Close, J.; Wang, Y.; Almarza, A.J.; Sfeir, C. Poly(Glycerol Sebacate) Elastomer: A Novel Material for Mechanically Loaded Bone Regeneration. Tissue Eng. Part A 2013, 20, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Feins, E.N.; Lee, Y.; O’Cearbhaill, E.D.; Vasilyev, N.V.; Shimada, S.; Friehs, I.; Perrin, D.; Hammer, P.E.; Yamauchi, H.; Marx, G.; et al. A growth-accommodating implant for paediatric applications. Nat. Biomed. Eng. 2017, 1, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Redenti, S.; Neeley, W.L.; Rompani, S.; Saigal, S.; Yang, J.; Klassen, H.; Langer, R.; Young, M.J. Engineering retinal progenitor cell and scrollable poly(glycerol-sebacate) composites for expansion and subretinal transplantation. Biomaterials 2009, 30, 3405–3414. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, F.; Neeley, W.L.; Arnér, K.; Langer, R. Selective Removal of Photoreceptor Cells In Vivo Using the Biodegradable Elastomer Poly(Glycerol Sebacate). Tissue Eng. Part A 2009, 17, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Hagandora, C.K.; Gao, J.; Wang, Y.; Almarza, A.J. Poly (Glycerol Sebacate): A Novel Scaffold Material for Temporomandibular Joint Disc Engineering. Tissue Eng. Part A 2012, 19, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Wieland, A.M.; Sundback, C.A.; Hart, A.; Kulig, K.; Masiakos, P.T.; Hartnick, C.J. Poly(glycerol sebacate)-engineered plugs to repair chronic tympanic membrane perforations in a chinchilla model. Otolaryngol. Neck Surg. 2010, 143, 127–133. [Google Scholar] [CrossRef]
- Yeh, C.W.; Wang, L.W.; Wu, H.C.; Hsieh, Y.K.; Wang, J.; Chen, M.H.; Wang, T.W. Development of biomimetic micro-patterned device incorporated with neurotrophic gradient and supportive Schwann cells for the applications in neural tissue engineering. Biofabrication 2017, 9, 015024. [Google Scholar] [CrossRef]
- Pan, Q.; Guo, Y.; Kong, F. Poly(glycerol sebacate) combined with chondroitinase ABC promotes spinal cord repair in rats. J. Biomed. Mater. Res. Part B Appl. Biomater. 2018, 106, 1770–1777. [Google Scholar] [CrossRef]
- Sun, Z.-J.; Chen, C.; Sun, M.-Z.; Ai, C.-H.; Lu, X.-L.; Zheng, Y.-F.; Yang, B.-F.; Dong, D.-L. The application of poly (glycerol–sebacate) as biodegradable drug carrier. Biomaterials 2009, 30, 5209–5214. [Google Scholar] [CrossRef]
- Sun, Z.J.; Sun, C.W.; Sun, B.; Lu, X.L.; Dong, D.L. The polycondensing temperature rather than time determines the degradation and drug release of poly(glycerol-sebacate) doped with 5-fluorouracil. J. Biomater. Sci. Polym. Ed. 2012, 23, 833–841. [Google Scholar] [CrossRef]
- Sun, Z.-J.; Sun, B.; Sun, C.-W.; Wang, L.-B.; Xie, X.; Ma, W.-C.; Lu, X.-L.; Dong, D.-L. A poly(glycerol-sebacate-(5-fluorouracil-1-acetic acid)) polymer with potential use for cancer therapy. J. Bioact. Compat. Polym. 2012, 27, 18–30. [Google Scholar] [CrossRef]
- Sun, Z.-J.; Sun, B.; Tao, R.-B.; Xie, X.; Lu, X.-L.; Dong, D.-L. A poly(glycerol–sebacate–curcumin) polymer with potential use for brain gliomas. J. Biomed. Mater. Res. Part A 2013, 101, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Lv, W.; Deng, Y. Drug loaded poly(glycerol sebacate) as a local drug delivery system for the treatment of periodontal disease. RSC Adv. 2017, 7, 37426–37435. [Google Scholar] [CrossRef]
- Tobias, I.S.; Lee, H.; Engelmayr, G.C.; Macaya, D.; Bettinger, C.J.; Cima, M.J. Zero-order controlled release of ciprofloxacin-HCl from a reservoir-based, bioresorbable and elastomeric device. J. Control. Release 2010, 146, 356–362. [Google Scholar] [CrossRef]
- Jen, M.C.; Baler, K.; Hood, A.R.; Shin, S.; Shea, L.D.; Ameer, G.A. Sustained, localized transgene expression mediated from lentivirus-loaded biodegradable polyester elastomers. J. Biomed. Mater. Res. Part A 2013, 101, 1328–1335. [Google Scholar] [CrossRef]
- Lee, K.W.; Johnson, N.R.; Gao, J.; Wang, Y. Human progenitor cell recruitment via SDF-1α coacervate-laden PGS vascular grafts. Biomaterials 2013, 34, 9877–9885. [Google Scholar] [CrossRef]
- Pryor, H.I., II; O’Doherty, E.; Hart, A.; Owens, G.; Hoganson, D.; Vacanti, J.P.; Masiakos, P.T.; Sundback, C.A. Poly(glycerol sebacate) films prevent postoperative adhesions and allow laparoscopic placement. Surgery 2009, 146, 490–497. [Google Scholar] [CrossRef]
- Boutry, C.M.; Nguyen, A.; Lawal, Q.O.; Chortos, A.; Rondeau-Gagné, S.; Bao, Z. A Sensitive and Biodegradable Pressure Sensor Array for Cardiovascular Monitoring. Adv. Mater. 2015, 27, 6954–6961. [Google Scholar] [CrossRef]
- Boutry, C.M.; Kaizawa, Y.; Schroeder, B.C.; Chortos, A.; Legrand, A.; Wang, Z.; Chang, J.; Fox, P.; Bao, Z. A stretchable and biodegradable strain and pressure sensor for orthopaedic application. Nat. Electron. 2018, 1, 314–321. [Google Scholar] [CrossRef]
- Lin, D.; Yang, K.; Tang, W.; Liu, Y.; Yuan, Y.; Liu, C. A poly(glycerol sebacate)-coated mesoporous bioactive glass scaffold with adjustable mechanical strength, degradation rate, controlled-release and cell behavior for bone tissue engineering. Colloids Surfaces B Biointerfaces 2015, 131, 1–11. [Google Scholar] [CrossRef]
- Kim, M.J.; Hwang, M.Y.; Kim, J.; Chung, D.J. Biodegradable and elastomeric poly(glycerol sebacate) as a coating material for nitinol bare stent. BioMed Res. Int. 2014, 2014, 956952. [Google Scholar] [CrossRef] [PubMed]
- Arvanitoyannis, I.; Nakayama, A.; Kawasaki, N.; Yamamoto, N. Novel star-shaped polylactide with glycerol using stannous octoate or tetraphenyl tin as catalyst: 1. Synthesis, characterization and study of their biodegradability. Polymer (Guildf) 1995, 36, 2947–2956. [Google Scholar] [CrossRef]
- Luo, S.H.; Wang, Z.Y.; Mao, C.X.; Huo, J.P. Synthesis of biodegradable material poly(lactic acid-co-glycerol) via direct melt polycondensation and its reaction mechanism. J. Polym. Res. 2011, 18, 2093–2102. [Google Scholar] [CrossRef]
- Basko, M.; Bednarek, M.; Kubisa, P. Cationic copolymerization of L,L-lactide with hydroxyl substituted cyclic ethers. Polym. Adv. Technol. 2015, 26, 804–813. [Google Scholar] [CrossRef]
- Pitet, L.M.; Hait, S.B.; Lanyk, T.J.; Knauss, D.M. Linear and branched architectures from the polymerization of lactide with glycidol. Macromolecules 2007, 40, 2327–2334. [Google Scholar] [CrossRef]
- Petchsuk, A.; Nakayama, A.; Aiba, S. Synthesis and biodegradability of L-lactide/glycidol copolymers. Polym. Degrad. Stab. 2009, 94, 1700–1706. [Google Scholar] [CrossRef]
- Gottschalk, C.; Wolf, F.; Frey, H. Multi-arm star poly(L-lactide) with hyperbranched polyglycerol core. Macromol. Chem. Phys. 2007, 208, 1657–1665. [Google Scholar] [CrossRef]
- Ouchi, T.; Ichimura, S.; Ohya, Y. Synthesis of branched poly(lactide) using polyglycidol and thermal, mechanical properties of its solution-cast film. Polymer (Guildf) 2006, 47, 429–434. [Google Scholar] [CrossRef]
- Adeli, M.; Haag, R.; Zarnegar, Z. Effect of the shell on the transport properties of poly(glycerol) and Poly(ethylene imine) nanoparticles. J. Nanoparticle Res. 2007, 9, 1057–1065. [Google Scholar] [CrossRef]
- Adeli, M.; Namazi, H.; Du, F.; Hönzke, S.; Hedtrich, S.; Keilitz, J.; Haag, R. Synthesis of multiarm star copolymers based on polyglycerol cores with polylactide arms and their application as nanocarriers. RSC Adv. 2015, 5, 14958–14966. [Google Scholar] [CrossRef]
- Kundys, A.; Plichta, A.; Florjańczyk, Z.; Zychewicz, A.; Lisowska, P.; Parzuchowski, P.; Wawrzyńska, E. Multi-arm star polymers of lactide obtained in melt in the presence of hyperbranched oligoglycerols. Polym. Int. 2016, 65, 927–937. [Google Scholar] [CrossRef]
- Gardella, L.; Forouharshad, M.; Pastorino, L.; Monticelli, O. Hyperbranched PDLA-polyglicerol: A novel additive for tuning PLLA electrospun fiber degradation and properties. Eur. Polym. J. 2017, 91, 21–30. [Google Scholar] [CrossRef]
- Petchsuk, A.; Buchatip, S.; Supmak, W.; Opaprakasit, M.; Opaprakasit, P. Preparation and properties of multi-branched poly(D-lactide) derived from polyglycidol and its stereocomplex blends. Express Polym. Lett. 2014, 8, 779–789. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, X.; Zhang, X.; Wang, Y.; Sun, L.; Li, C. Amphiphilic polylactic acid-hyperbranched polyglycerol nanoparticles as a controlled release system for poorly water-soluble drugs: Physicochemical characterization. J. Pharm. Pharmacol. 2011, 63, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.K.; Fischer, A.M.; Frey, H. Poly(glycolide) multi-arm star polymers: Improved solubility via limited arm length. Beilstein J. Org. Chem. 2010, 6, 67. [Google Scholar] [CrossRef]
- Bao, H.; Jin, X.; Li, L.; Lv, F.; Liu, T. OX26 modified hyperbranched polyglycerol-conjugated poly(lactic-co-glycolic acid) nanoparticles: Synthesis, characterization and evaluation of its brain delivery ability. J. Mater. Sci. Mater. Med. 2012, 23, 1891–1901. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, X.; Wu, Z.; Zhang, X.; Wang, Z.; Li, C. Synthesis and physicochemical characterization of a novel amphiphilic polylactic acid-hyperbranched polyglycerol conjugate for protein delivery. J. Control. Release 2009, 140, 141–147. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, C.H.; Cheng, S.X.; Zhuo, R.X. Hyperbranched amphiphilic polymer with folate mediated targeting property. Colloids Surfaces B Biointerfaces 2010, 79, 427–433. [Google Scholar] [CrossRef]
- Deng, Y.; Saucier-Sawyer, J.K.; Hoimes, C.J.; Zhang, J.; Seo, Y.E.; Andrejecsk, J.W.; Saltzman, W.M. The effect of hyperbranched polyglycerol coatings on drug delivery using degradable polymer nanoparticles. Biomaterials 2014, 35, 6595–6602. [Google Scholar] [CrossRef]
- Saucier-Sawyer, J.K.; Deng, Y.; Seo, Y.-E.; Cheng, C.J.; Zhang, J.; Quijano, E.; Saltzman, W.M. Systemic delivery of blood–brain barrier-targeted polymeric nanoparticles enhances delivery to brain tissue. J. Drug Target. 2015, 23, 736–749. [Google Scholar] [CrossRef]
- Hedtrich, S.; Graff, P.; Haag, R.; Zabihi, F.; Schumacher, F.; Kleuser, B. Synthesis of poly(lactide-co-glycerol) as a biodegradable and biocompatible polymer with high loading capacity for dermal drug delivery. Nanoscale 2018, 10, 16848–16856. [Google Scholar]
- Istratov, V.V.; Krupina, T.V.; Gomzyak, V.I.; Vasnev, V.A. Development and characterization of bioresorbable polyglycerol esters and drug-loaded microparticles. High Perform. Polym. 2017, 29, 708–715. [Google Scholar] [CrossRef]
- Mohammadifar, E.; Adeli, M.; Kharat, A.N.; Namazi, H.; Haag, R. Stimuli-Responsive Core Multishell Dendritic Nanocarriers. Macromol. Chem. Phys. 2017, 218, 1600525. [Google Scholar] [CrossRef]
- Yang, X.; Liu, L. Synthesis and characterization of novel polyglycerol hydrogels containing L-lactic acid groups as pendant acidic substituents: pH-Responsive polyglycerol-based hydrogels. J. Appl. Polym. Sci. 2009, 112, 3209–3216. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, C.; Fan, Y.; Wu, Y. Binary gene vectors based on hyperbranched poly(l-lactide-co-polyglycerol) and polyethylenimine for prolonged transgene expression via co-assembly with DNA into fiber core-shell triplexes. J. Mater. Chem. B 2013, 1, 6271–6282. [Google Scholar] [CrossRef]
- Cameron, D.J.A.; Shaver, M.P. Aliphatic polyester polymer stars: Synthesis, properties and applications in biomedicine and nanotechnology. Chem. Soc. Rev. 2011, 40, 1761–1776. [Google Scholar] [CrossRef]
- Zhang, H.; Grinstaff, M.W. Recent advances in glycerol polymers: Chemistry and biomedical applications. Macromol. Rapid Commun. 2014, 35, 1906–1924. [Google Scholar] [CrossRef]
- Lang, M.; Wong, R.P.; Chu, C.-C. Synthesis and structural analysis of functionalized poly (ϵ-caprolactone)-based three-arm star polymers. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 1127–1141. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, X.; Chu, C.C. Synthesis, characterization and drug release from three-arm poly(ε-caprolactone) maleic acid/poly(ethylene glycol) diacrylate hydrogels. J. Biomater. Sci. Polym. Ed. 2003, 14, 777–802. [Google Scholar] [CrossRef]
- Zhang, X.-Z.; Jo Lewis, P.; Chu, C.-C. Fabrication and characterization of a smart drug delivery system: Microsphere in hydrogel. Biomaterials 2005, 26, 3299–3309. [Google Scholar] [CrossRef]
- Xie, W.; Jiang, N.; Gan, Z. Effects of Multi-Arm Structure on Crystallization and Biodegradation of Star-Shaped Poly(ε-caprolactone). Macromol. Biosci. 2008, 8, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, M.; Witkowska, E.; Oledzka, E.; Kolodziejski, W. Synthesis and structural analysis of polyester prodrugs of norfloxacin. Molecules 2008, 13, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Gan, Z. Thermal degradation of star-shaped poly(ɛ-caprolactone). Polym. Degrad. Stab. 2009, 94, 1040–1046. [Google Scholar] [CrossRef]
- Sobczak, M. Synthesis and characterization of polyester conjugates of ciprofloxacin. Eur. J. Med. Chem. 2010, 45, 3844–3849. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, M.; Nałȩcz-Jawecki, G.; Kołodziejski, W.L.; Goś, P.; Żółtowska, K. Synthesis and study of controlled release of ofloxacin from polyester conjugates. Int. J. Pharm. 2010, 402, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Sugane, K.; Takahashi, H.; Shimasaki, T.; Teramoto, N.; Shibata, M. Stereocomplexation, thermal and mechanical properties of conetworks composed of star-shaped L-lactide, D-lactide and ε-caprolactone oligomers utilizing sugar alcohols as core molecules. Polymers (Basel) 2017, 9, 582. [Google Scholar] [CrossRef]
- Christian, P.; Jones, I.A. Polymerisation and stabilisation of polycaprolactone using a borontrifluoride–glycerol catalyst system. Polymer (Guildf) 2001, 42, 3989–3994. [Google Scholar] [CrossRef]
- Jiang, G.; Walker, G.S.; Jones, I.A.; Rudd, C.D. Mechanistic study of boron trifluoride catalyzed ε-caprolactone polymerization in the presence of glycerol. J. Appl. Polym. Sci. 2006, 102, 3900–3906. [Google Scholar] [CrossRef]
- Xue, L.; Dai, S.; Li, Z. Synthesis and Characterization of Three-Arm Poly(ε-caprolactone)-Based Poly(ester−urethanes) with Shape-Memory Effect at Body Temperature. Macromolecules 2009, 42, 964–972. [Google Scholar] [CrossRef]
- Chakraborty, S.; Pagaduan, J.N.M.; Melgar, Z.K.A.; Seitz, S.; Kan, K.; Ajiro, H. Glycerol-modified poly(ε-caprolactone): An biocatalytic approach to improve the hydrophilicity of poly(ε-caprolactone). Polym. Bull. 2018, 76, 1915–1928. [Google Scholar] [CrossRef]
- Jun, H.; Le Kim, T.H.; Han, S.W.; Seo, M.; Kim, J.W.; Nam, Y.S. Polyglycerol-poly(ε-caprolactone) block copolymer as a new semi-solid polymeric emulsifier to stabilize O/W nanoemulsions. Colloid Polym. Sci. 2015, 293, 2949–2956. [Google Scholar] [CrossRef]
- Le Kim, T.H.; Jun, H.; Kim, J.H.; Park, K.; Kim, J.S.; Nam, Y.S. Lipiodol nanoemulsions stabilized with polyglycerol-polycaprolactone block copolymers for theranostic applications. Biomater. Res. 2017, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Le Kim, T.H.; Yu, J.H.; Jun, H.; Yang, M.Y.; Yang, M.J.; Cho, J.W.; Kim, J.W.; Kim, J.S.; Nam, Y.S. Polyglycerolated nanocarriers with increased ligand multivalency for enhanced in vivo therapeutic efficacy of paclitaxel. Biomaterials 2017, 145, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.P.K.; Laurila, T.; Kivilahti, J.K. Reactive blending approach to modify spin-coated epoxy film: Part I. Synthesis and characterization of star-shaped poly(e-caprolactone). J. Appl. Polym. Sci. 2006, 101, 3677–3688. [Google Scholar] [CrossRef]
- Adeli, M.; Kakanejadifard, A.; Khani, M.; Bani, F.; Kabiri, R.; Sadeghizad, M. A polyglycerol-polycaprolactone-polycitric acid copolymer and its self-assembly to produce medium-responsive nanoparticles. J. Mater. Chem. B 2014, 2, 3589–3596. [Google Scholar] [CrossRef]
- Du, F.; Hönzke, S.; Neumann, F.; Keilitz, J.; Chen, W.; Ma, N.; Hedtrich, S.; Haag, R. Development of biodegradable hyperbranched core-multishell nanocarriers for efficient topical drug delivery. J. Control. Release 2016, 242, 42–49. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, L.; Xu, J.; Wu, J.; Kirk, T.B.; Ma, D.; Xue, W. Construction of a High-Efficiency Drug and Gene Co-Delivery System for Cancer Therapy from a pH-Sensitive Supramolecular Inclusion between Oligoethylenimine-graft-β-cyclodextrin and Hyperbranched Polyglycerol Derivative. ACS Appl. Mater. Interfaces 2018, 10, 35812–35829. [Google Scholar] [CrossRef]
- Cai, T.; Li, M.; Neoh, K.G.; Kang, E.T. Surface-functionalizable membranes of polycaprolactone-click-hyperbranched polyglycerol copolymers from combined atom transfer radical polymerization, ring-opening polymerization and click chemistry. J. Mater. Chem. B 2013, 1, 1304–1315. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, B.; Hyun, D.C.; Kim, J.W.; Shim, H.E.; Kang, S.W.; Jeong, U. Photocrosslinkable poly(ε-caprolactone)-b-hyperbranched polyglycerol (PCL-b-hbPG) with improved biocompatibility and stability for drug delivery. Macromol. Chem. Phys. 2015, 216, 1161–1170. [Google Scholar] [CrossRef]
- Ejaz, M.; Alb, A.M.; Grayson, S.M. Amphiphilic hyperbranched polyglycerol-block-polycaprolactone copolymer-grafted nanoparticles with improved encapsulation properties. React. Funct. Polym. 2016, 102, 39–46. [Google Scholar] [CrossRef]
- Ferraro, M.; Silberreis, K.; Mohammadifar, E.; Neumann, F.; Dernedde, J.; Haag, R. Biodegradable Polyglycerol Sulfates Exhibit Promising Features for Anti-inflammatory Applications. Biomacromolecules 2018, 19, 4524–4533. [Google Scholar] [CrossRef] [PubMed]
- Mohammadifar, E.; Zabihi, F.; Tu, Z.; Hedtrich, S.; Nemati Kharat, A.; Adeli, M.; Haag, R. One-pot and gram-scale synthesis of biodegradable polyglycerols under ambient conditions: Nanocarriers for intradermal drug delivery. Polym. Chem. 2017, 8, 7375–7383. [Google Scholar] [CrossRef]
- Dey, P.; Hemmati-Sadeghi, S.; Haag, R. Hydrolytically degradable, dendritic polyglycerol sulfate based injectable hydrogels using strain promoted azide-alkyne cycloaddition reaction. Polym. Chem. 2016, 7, 375–383. [Google Scholar] [CrossRef]
- Zhong, Y.; Dimde, M.; Stöbener, D.; Meng, F.; Deng, C.; Zhong, Z.; Haag, R. Micelles with Sheddable Dendritic Polyglycerol Sulfate Shells Show Extraordinary Tumor Targetability and Chemotherapy in Vivo. ACS Appl. Mater. Interfaces 2016, 8, 27530–27538. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.B.; Yohe, S.T.; Colson, Y.L.; Grinstaff, M.W. Functionalized Hydrophobic Poly(glycerol-co-ε-caprolactone) Depots for Controlled Drug Release. Biomacromolecules 2012, 13, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.B.; Ray, W.C.; Colson, Y.L.; Grinstaff, M.W. Poly(carbonate ester)s Based on Units of 6-Hydroxyhexanoic Acid and Glycerol. Macromolecules 2007, 40, 7065–7068. [Google Scholar] [CrossRef]
- Aydin, H.M.; Salimi, K.; Yilmaz, M.; Turk, M.; Rzayev, Z.M.O.; Pişkin, E. Synthesis and characterization of poly(glycerol-co-sebacate-co-ε-caprolactone) elastomers. J. Tissue Eng. Regen. Med. 2016, 10, E14–E22. [Google Scholar] [CrossRef]
- Tzeng, J.J.; Hsiao, Y.T.; Wu, Y.C.; Chen, H.; Lee, S.Y.; Lin, Y.M. Synthesis, Characterization, and Visible Light Curing Capacity of Polycaprolactone Acrylate. Biomed Res. Int. 2018, 2018, 8719624. [Google Scholar] [CrossRef]
- Carna, M.S.; Chakraborty, S. Glycerol-modified poly-ε-caprolactone nanoparticles for drug delivery application. Kimika 2014, 25, 38–46. [Google Scholar] [CrossRef][Green Version]
- Yohe, S.T.; Herrera, V.L.M.; Colson, Y.L.; Grinstaff, M.W. 3D superhydrophobic electrospun meshes as reinforcement materials for sustained local drug delivery against colorectal cancer cells. J. Control. Release 2012, 162, 92–101. [Google Scholar] [CrossRef]
- Yohe, S.T.; Colson, Y.L.; Grinstaff, M.W. Superhydrophobic materials for tunable drug release: Using displacement of air to control delivery rates. J. Am. Chem. Soc. 2012, 134, 2016–2019. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wolinsky, J.B.; Walpole, J.; Southard, E.; Chirieac, L.R.; Grinstaff, M.W.; Colson, Y.L. Prevention of Local Tumor Recurrence Following Surgery Using Low-Dose Chemotherapeutic Polymer Films. Ann. Surg. Oncol. 2010, 17, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Grinstaff, M.W.; Chirieac, L.R.; Colson, Y.L.; Catalano, P.J.; Wagner, A.J.; Wolinsky, J.B.; Liu, R.; Raut, C.P. Paclitaxel-Eluting Polymer Film Reduces Locoregional Recurrence and Improves Survival in a Recurrent Sarcoma Model: A Novel Investigational Therapy. Ann. Surg. Oncol. 2011, 19, 199–206. [Google Scholar]
































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gly/SuA Molar Ratio | Experimental Conditions | Mn (g/mol) | Tg (°C) | Ref. | |||
|---|---|---|---|---|---|---|---|
| Solvent | Catalyst | T (°C) | Time (h) | ||||
| 1.5 | - | - | 190 | 24 | 844 e, 868 f | - | [14] |
| 1.7 | - | - | 190 | 24 | 673 e, 680 f | - | [14] |
| 1.9 | - | - | 190 | 24 | 606 e, 623 f | - | [14] |
| 0.6 | - | - | 180 | 1.1 | 938 g | −15.2 | [16] |
| 0.9 | - | - | 180 | 2.9 | 1011 g | −14.8 | [16] |
| 1.2 | - | - | 180 | 5.3 | 754 g | −29.9 | [16] |
| 0.5 a,b | - | - | 180 | 1.2 | - | −10.9 | [17] |
| 0.8 a,c | - | - | 180 | 1.8 | - | −2.7 | [17] |
| 1.3, 1.9 a,d | - | - | 180 | 1.5 or 6 | - | 16.6 or 33.4 | [17] |
| 1.0 | Toluene | Dibutyl tin(IV) oxide | 135 | 24 | 22,280 g | - | [18] |
| 0.5 | Toluene | Dibutyl tin(IV) oxide | 135 | 24 | 112,770 g | - | [18] |
| 1.0 h | - | Titanium(IV) butoxide | initial 60 °C, final 150 °C | 16 h in total | 992 e | - | [19] |
| Gly/GA Molar Ratio | Experimental Conditions | Mn (g/mol) | DB (%) | Tg (°C) | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Solvent | Catalyst | T (°C) | Time (h) | |||||
| 1:2 | DMF | Titanium(IV) butoxide | 150 | 24 | 2402 | 54 | - | [33] |
| 1:2 | DMF | Dibutyl tin(IV) oxide | 150 | 10 | 1058 | 41 | −50.1 | [34] |
| 1:2 | Toluene | Dibutyl tin(IV) oxide | 155 | 10 | 23,570 | 70.5 | - | [18] |
| 1:2 | DMF | Dibutyl tin(IV) oxide | 150 | 24 | 1474 | 45 | - | [34] |
| 1:2 | Toluene | Dibutyl tin(IV) oxide | 155 | 24 | 29,380 | 76.7 | - | [18] |
| 1:2 a | Neat | Dibutyl tin(IV) oxide | initial 60 °C, final 150 °C | 24 h in total | 445,000 | 82 | −31.3 | [34] |
| 1:1 | Toluene | Dibutyl tin(IV) oxide | 155 | 24 | 252,060 | 33.3 | - | [18] |
| Gly/AdA Molar Ratio | Experimental Conditions | Mw (g/mol) | Tg (°C) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Catalyst | T (°C) | Time (h) | Pressure | Thermal Curing (°C) | ||||
| 1:1 b | - | a) 150 | 17 | atm | 120 | 4500 a | −15 | [53] |
| b) 150 | 4 | 0.5 mm Hg | ||||||
| 2:3 b | - | a) 150 | 16 | atm | 120 | 12,500 a | −2.5 | [53] |
| b) 150 | 0.5 | 0.5 mm Hg | ||||||
| 4:3 | Dibutyltin oxide | 140 | 11.5 | atm | - | 1250 | - | [54] |
| 1:1 | Dibutyltin oxide | 140 | 11.5 | atm | - | 14,000 | - | [54] |
| Gly/AzA Molar Ratio | Experimental Conditions | Mn (g/mol) | Ref. | |||
|---|---|---|---|---|---|---|
| Solvent | Catalyst | T (°C) | Time (h) | |||
| 1:1 a | - | Titanium(IV) butoxide | initial 60 °C, final 150 °C | 16 h in total | 2316 | [19] |
| 1:1 | - | - | 140 | 14 | 1852 | [75] |
| 1:1 | DMF or DMSO | Titanium(IV) butoxide | 150 | 22 | 3245 | [33] |
| 1:1 | Toluene | Dibutyltin(IV) oxide | 135 | 24 | 4160 | [18] |
| 1:1.5 | - | - | 140 | 14 | 2873 | [75] |
| 1:1.6 | - | - | 140 | 14 | 2574 | [75] |
| 1:2 | Toluene | Dibutyltin(IV) oxide | 135 | 24 | 5410 | [18] |
| Process | Gly/SeA Molar Ratio | Sol Content (%) | Tensile Strength (MPa) | Tensile Stress (MPa) | Elastic Modulus (MPa) | Elongation at Break (%) | Tensile Strain (%) | Tg (°C) | Water Contact Angle (°) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| A | 2:2.5 | 75.13 | 0.210 | - | 0.07 | 114.5 | - | −25.1 | 45.2 | [97] |
| A | 2:3 | 74.81 | 0.141 | - | 0.14 | 101.3 | - | −27.1 | 47.9 | [97] |
| A | 2:3.5 | 69.23 | 0.699 | - | 1.45 | 57.2 | - | −29.9 | 52.3 | [97] |
| A | 2:4 | 84.23 | 0.425 | - | 7.05 | 12.0 | - | −32.2 | 56.6 | [97] |
| B | 2:2.5 | 61.87 | - | 0.61 | 0.55 | - | 236 | −22.5 | 37.8 | [91] |
| B a | 2:2.5 | 66.88 | - | 0.41 | 0.49 | - | 109 | −26.1 | 43.4 | [91] |
| B a | 2:2.5 | 62.75 | - | 0.64 | 0.45 | - | 179 | −26.2 | 42.5 | [91] |
| Gly/SeA Molar Ratio | Conditions | Ultimate Tensile Strength (MPa) | Young’s Modulus (MPa) | Rupture Strain (%) | Crosslinking Density (mol/m3) | Ref. | |
|---|---|---|---|---|---|---|---|
| Solvent | T (°C) | ||||||
| 2:2 | Toluene | 130 | 0.25 | 25.21 ± 8.84 a | [89] | ||
| 2:2.5 | Toluene | 130 | 0.57 | 56.73 ± 5.94 a | [89] | ||
| 2:3.3 | Toluene | 130 | 0.72 | 71.48 ± 8.84 a | [89] | ||
| 1:1 | Neat | 110 | 0.23 | 0.06 | 4.48 | 10 ± 4 b | [101] |
| 1:1 | Neat | 120 | 0.39 | 0.22 | 2.56 | 42 ± 17 b | [101] |
| 1:1 | Neat | 140 | 0.47 | 1.20 | 0.41 | 131 ± 50 b | [101] |
| Monomer/Gly Molar Ratio | Experimental Conditions | Mn (g/mol) | Tg (°C) | Tm (°C) | X (%) | Ref | |||
|---|---|---|---|---|---|---|---|---|---|
| Catalyst | T (°C) | Time | Pressure | ||||||
| 3:1 a | Sn(Oct)2 | 130 | 4 days | 1400 | −10 | - d | [177] | ||
| 3:1 a | TPhT | 130 | 4 days | 4300 | −13 | - d | [177] | ||
| 10:1 a | Sn(Oct)2 | 130 | 4 days | 5400 | 15 | - d | [177] | ||
| 10:1 a | TPhT | 130 | 4 days | 2200 | 14 | - d | [177] | ||
| 20:1 b,c | SnO | a) 140 | 8 h | 4000 Pa | 2600 | 6 | - d | [178] | |
| b) 160 | 8 h | 70 Pa | |||||||
| 40:1 a | Sn(Oct)2 | 130 | 4 days | 9800 | 40 | 138 | 32.0 | [177] | |
| 40:1 a | TPhT | 130 | 4 days | 13,000 | 40 | 128 | 26.5 | [177] | |
| 40:1 b,c | SnO | a) 140 | 8 h | 4000 Pa | 3100 | 25 | - d | [178] | |
| b) 160 | 8 h | 70 Pa | |||||||
| 80:1 b,c | SnO | a) 140 | 8 h | 4000 Pa | 3300 | 37 | 98 | 16.6 | [178] |
| b) 160 | 8 h | 70 Pa | |||||||
| 100:1 a | Sn(Oct)2 | 130 | 4 days | 14,000 | 47 | 158 | 58.9 | [177] | |
| 100:1 a | TPhT | 130 | 4 days | 43,000 | 170 | 55.8 | [177] | ||
| 100:1 b,c | SnO | a) 140 | 8 h | 4000 Pa | 4700 | 41 | 119 | 17.7 | [178] |
| b) 160 | 8 h | 70 Pa | |||||||
| 140:1 b,c | SnO | a) 140 | 8 h | 4000 Pa | 3200 | 41 | 123 | 20.8 | [178] |
| b) 160 | 8 h | 70 Pa | |||||||
| CL/Gly Molar Ratio | Experimental Conditions | Mn (g/mol) | Tm (°C) | Xc a (%) | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Catalyst | Solvent | T (°C) | Time (h) | |||||
| 1.5:1 | Novozym 435 | Toluene | 70 | 4 | 1500 | 46 | 49 | [215] |
| 1.5:1 | Novozym 435 | Toluene | 70 | 6 | 2400 | 48 | 51 | [215] |
| 9:1 | Novozym 435 | Toluene | 70 | 4 | 2500 | 48 | 53 | [215] |
| 10:1 | Novozym 435 | Dioxane | 70 | 4 | 2900 | 46 | 57 | [214] |
| 30:1 | Novozym 435, | Dioxane | 70 | 3 | 4900 | 53 | 68 | [214] |
| 10:1 | Sn(oOct)2 | - | 130 | 48 | 6280 | n.d. | n.d. | [203] |
| 40:1 | Sn(Oct)2 | - | 130 | 48 | 18,900 | n.d. | n.d. | [203] |
| 90:1 | Sn(Oct)2 | - | 110 | 48 | 22,000 | n.d. | n.d. | [206] |
| 270:1 | Sn(Oct)2 | - | 110 | 48 | 58,200 | n.d. | n.d. | [206] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamboulis, A.; Nakiou, E.A.; Christodoulou, E.; Bikiaris, D.N.; Kontonasaki, E.; Liverani, L.; Boccaccini, A.R. Polyglycerol Hyperbranched Polyesters: Synthesis, Properties and Pharmaceutical and Biomedical Applications. Int. J. Mol. Sci. 2019, 20, 6210. https://doi.org/10.3390/ijms20246210
Zamboulis A, Nakiou EA, Christodoulou E, Bikiaris DN, Kontonasaki E, Liverani L, Boccaccini AR. Polyglycerol Hyperbranched Polyesters: Synthesis, Properties and Pharmaceutical and Biomedical Applications. International Journal of Molecular Sciences. 2019; 20(24):6210. https://doi.org/10.3390/ijms20246210
Chicago/Turabian StyleZamboulis, Alexandra, Eirini A. Nakiou, Evi Christodoulou, Dimitrios N. Bikiaris, Eleana Kontonasaki, Liliana Liverani, and Aldo R. Boccaccini. 2019. "Polyglycerol Hyperbranched Polyesters: Synthesis, Properties and Pharmaceutical and Biomedical Applications" International Journal of Molecular Sciences 20, no. 24: 6210. https://doi.org/10.3390/ijms20246210
APA StyleZamboulis, A., Nakiou, E. A., Christodoulou, E., Bikiaris, D. N., Kontonasaki, E., Liverani, L., & Boccaccini, A. R. (2019). Polyglycerol Hyperbranched Polyesters: Synthesis, Properties and Pharmaceutical and Biomedical Applications. International Journal of Molecular Sciences, 20(24), 6210. https://doi.org/10.3390/ijms20246210