Modeling the Effects of Severe Metabolic Disease by Genome Editing of hPSC-Derived Endothelial Cells Reveals an Inflammatory Phenotype

 , ,
, ,
Abstract
1. Introduction
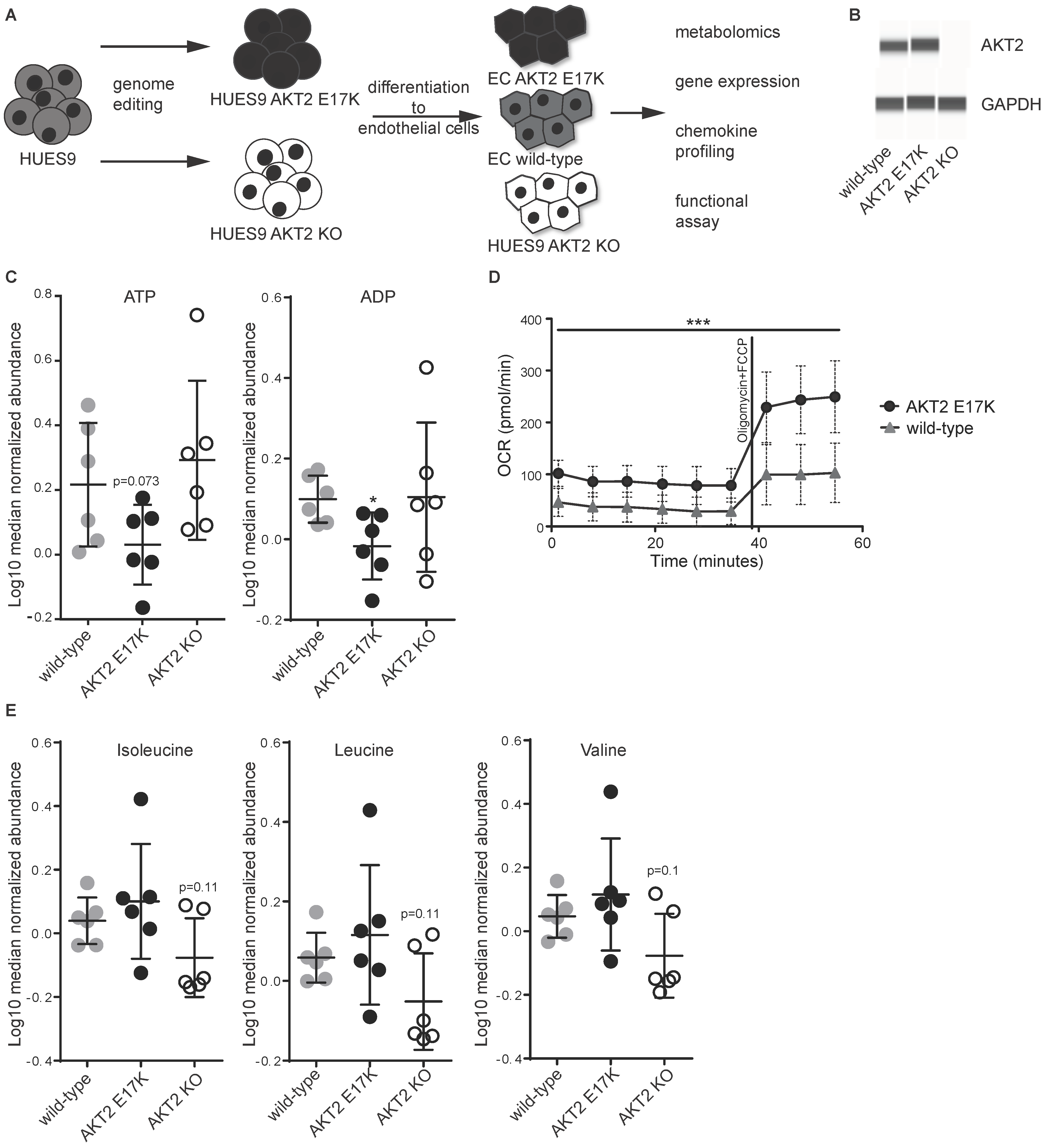
2. Results
3. Discussion
4. Material and Methods
4.1. Human PSC Cell Culture and Endothelial Cell Differentiation
4.2. Western Blotting
4.3. Measurement of Cellular Oxygen Consumption Rate
4.4. Metabolic Profiling
4.5. Multiplexed Sandwich Immunoassay
4.6. Leukocyte Adhesion Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- George, S.; Rochford, J.J.; Wolfrum, C.; Gray, S.L.; Schinner, S.; Wilson, J.C.; Soos, M.A.; Murgatroyd, P.R.; Williams, R.M.; Acerini, C.L.; et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 2004, 304, 1325–1328. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.; Highland, H.M.; Gasser, J.; Sim, X.; Tukiainen, T.; Fontanillas, P.; Grarup, N.; Rivas, M.A.; Mahajan, A.; Locke, A.E.; et al. A Low-Frequency Inactivating AKT2 Variant Enriched in the Finnish Population Is Associated With Fasting Insulin Levels and Type 2 Diabetes Risk. Diabetes 2017, 66, 2019–2032. [Google Scholar] [CrossRef] [PubMed]
- Hussain, K.; Challis, B.; Rocha, N.; Payne, F.; Minic, M.; Thompson, A.; Daly, A.; Scott, C.; Harris, J.; Smillie, B.J.; et al. An activating mutation of AKT2 and human hypoglycemia. Science 2011, 334, 474. [Google Scholar]
- Ding, Q.; Lee, Y.K.; Schaefer, E.A.; Peters, D.T.; Veres, A.; Kim, K.; Kuperwasser, N.; Motola, D.L.; Meissner, T.B.; Hendriks, W.T.; et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 2013, 12, 238–251. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; Kahn, C.R. Tissue-specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2052–2059. [Google Scholar] [CrossRef]
- Barrett, E.J.; Liu, Z. The endothelial cell: An “early responder” in the development of insulin resistance. Rev. Endocr. Metab. Disord. 2013, 14, 21–27. [Google Scholar] [CrossRef]
- Roudnicky, F.; Dieterich, L.C.; Poyet, C.; Buser, L.; Wild, P.; Tang, D.; Camenzind, P.; Ho, C.H.; Otto, V.I.; Detmar, M. High expression of insulin receptor on tumour-associated blood vessels in invasive bladder cancer predicts poor overall and progression-free survival. J. Pathol. 2017, 242, 193–205. [Google Scholar] [CrossRef]
- Lee, M.Y.; Luciano, A.K.; Ackah, E.; Rodriguez-Vita, J.; Bancroft, T.A.; Eichmann, A.; Simons, M.; Kyriakides, T.R.; Morales-Ruiz, M.; Sessa, W.C. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc. Natl. Acad. Sci. USA 2014, 111, 12865–12870. [Google Scholar] [CrossRef]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef]
- Boye, K.; Grotterod, I.; Aasheim, H.C.; Hovig, G.M. Maelandsmo, Activation of NF-kappaB by extracellular S100A4: Analysis of signal transduction mechanisms and identification of target genes. Int. J. Cancer 2008, 123, 1301–1310. [Google Scholar] [CrossRef]
- Geback, T.; Schulz, M.M.; Koumoutsakos, P.; Detmar, M. TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays. BioTechniques 2009, 46, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.R.; O’Sullivan, J.F.; Friesen, M.; Becker, C.E.; Zhang, X.; Liu, P.; Wakabayashi, Y.; Morningstar, J.E.; Shi, X.; Choi, J.; et al. Cowan, Induced Pluripotent Stem Cell Differentiation Enables Functional Validation of GWAS Variants in Metabolic Disease. Cell Stem Cell 2017, 20, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Balhara, B.; Burkart, A.; Topcu, V.; Lee, Y.K.; Cowan, C.; Kahn, C.R.; Patti, M.E. Severe insulin resistance alters metabolism in mesenchymal progenitor cells. Endocrinology 2015, 156, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Drawnel, F.M.; Boccardo, S.; Prummer, M.; Delobel, F.; Graff, A.; Weber, M.; Gerard, R.; Badi, L.; Kam-Thong, T.; Bu, L.; et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 2014, 9, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Carcamo-Orive, I.; Huang, N.F.; Quertermous, T.; Knowles, J.W. Induced Pluripotent Stem Cell-Derived Endothelial Cells in Insulin Resistance and Metabolic Syndrome. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2038–2042. [Google Scholar] [CrossRef]
- Feener, E.P.; King, G.L. Vascular dysfunction in diabetes mellitus. Lancet 1997, 350, S9–S13. [Google Scholar] [CrossRef]
- Christensen, K.; Roudnicky, F.; Burcin, M.; Patsch, C. Monolayer Generation of Vascular Endothelial Cells from Human Pluripotent Stem Cells. Methods Mol. Biol. 2019, 1994, 17–29. [Google Scholar]
- O’Connell, T.M. The complex role of branched chain amino acids in diabetes and cancer. Metabolites 2013, 3, 931–945. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Gogg, S.; Lotta, L.A.; Stančáková, A.; Nerstedt, A.; Boren, J.; Blüher, M.; Ferrannini, E.; Langenberg, C.; Wareham, N.J.; et al. Elevated Plasma Levels of 3-Hydroxyisobutyric Acid Are Associated With Incident Type 2 Diabetes. EBioMedicine 2018, 27, 151–155. [Google Scholar] [CrossRef]
- Jang, C.; Oh, S.F.; Wada, S.; Rowe, G.C.; Liu, L.; Chan, M.C.; Rhee, J.; Hoshino, A.; Kim, B.; Ibrahim, A.; et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med. 2016, 22, 421–426. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J.; Rowlinson, S.W.; Goodwin, D.C.; Kalgutkar, A.S.; Lanzo, C.A. Arachidonic acid oxygenation by COX-1 and COX-2. Mechanisms of catalysis and inhibition. J. Biol. Chem. 1999, 274, 22903–22906. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Yoshida, M.; Nakano, Y.; Suganami, T.; Satoh, N.; Mita, T.; Azuma, K.; Itoh, M.; Yamamoto, Y.; Kamei, Y.; et al. In vivo and in vitro inhibition of monocyte adhesion to endothelial cells and endothelial adhesion molecules by eicosapentaenoic acid. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2173–2179. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Weber, C. Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc. Res. 2010, 86, 192–201. [Google Scholar] [CrossRef]
- Calles-Escandon, J.; Cipolla, M. Diabetes and endothelial dysfunction: A clinical perspective. Endocr. Rev. 2001, 22, 36–52. [Google Scholar] [CrossRef]
- Zhu, Z.; Huangfu, D. Human pluripotent stem cells: An emerging model in developmental biology. Development 2013, 140, 705–717. [Google Scholar] [CrossRef]
- Chen, Y.L.; Qiao, Y.C.; Pan, Y.H.; Xu, Y.; Huang, Y.C.; Wang, Y.H.; Geng, L.J.; Zhao, H.L.; Zhang, X.X. Correlation between serum interleukin-6 level and type 1 diabetes mellitus: A systematic review and meta-analysis. Cytokine 2017, 94, 14–20. [Google Scholar] [CrossRef]
- Van Ravenzwaay, B.; Cunha, G.C.; Leibold, E.; Looser, R.; Mellert, W.; Prokoudine, A.; Walk, T.; Wiemer, J. The use of metabolomics for the discovery of new biomarkers of effect. Toxicol. Lett. 2007, 172, 21–28. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate-a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Analytes Analyzed in: | Supernatant | Cell Lysate | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Comparison between experimental groups | E17K | KO | E17K | KO | WT | E17K | E17K | KO | E17K |
| WT | WT | KO | fresh media | fresh media | fresh media | WT | WT | KO | |
| Significance Thresholds | |||||||||
| p < 0.1 | 22 | 23 | 34 | 39 | 38 | 35 | 51 | 39 | 63 |
| p < 0.05 | 15 | 12 | 15 | 31 | 28 | 32 | 39 | 29 | 39 |
| p < 0.01 | 4 | 3 | 7 | 22 | 20 | 21 | 21 | 12 | 15 |
| Analytes Analyzed in: | Supernatant | Cell Lysate | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Comparison between experimental groups | E17K | KO | E17K | KO | WT | E17K | E17K | KO | E17K |
| WT | WT | KO | Fresh media | Fresh media | Fresh media | WT | WT | KO | |
| ONTOLOGY | number of significantly changed analytes | ||||||||
| Amino acids | 4 | 1 | 1 | 6 | 6 | 6 | 1 | 2 | 7 |
| Amino acids related | 0 | 1 | 1 | 4 | 3 | 4 | 0 | 0 | 0 |
| Carbohydrates and related | 3 | 4 | 2 | 4 | 4 | 6 | 0 | 0 | 0 |
| Complex lipids, fatty acids and related | 3 | 1 | 0 | 0 | 0 | 2 | 13 | 8 | 11 |
| Energy metabolism and related | 1 | 1 | 1 | 2 | 3 | 2 | 0 | 0 | 2 |
| Hormones, signal substances and related | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 |
| Miscellaneous | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 2 |
| Nucleobases and related | 0 | 0 | 0 | 3 | 3 | 3 | 0 | 0 | 0 |
| Unknown | 4 | 4 | 9 | 10 | 8 | 7 | 24 | 18 | 15 |
| Vitamins, cofactors and related | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 2 |
| Total number of analytes | 15 | 12 | 15 | 31 | 28 | 32 | 39 | 29 | 39 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roudnicky, F.; Lan, Y.; Friesen, M.; Dernick, G.; Zhang, J.D.; Staempfli, A.; Bordag, N.; Wagner-Golbs, A.; Christensen, K.; Ebeling, M.; et al. Modeling the Effects of Severe Metabolic Disease by Genome Editing of hPSC-Derived Endothelial Cells Reveals an Inflammatory Phenotype. Int. J. Mol. Sci. 2019, 20, 6201. https://doi.org/10.3390/ijms20246201
Roudnicky F, Lan Y, Friesen M, Dernick G, Zhang JD, Staempfli A, Bordag N, Wagner-Golbs A, Christensen K, Ebeling M, et al. Modeling the Effects of Severe Metabolic Disease by Genome Editing of hPSC-Derived Endothelial Cells Reveals an Inflammatory Phenotype. International Journal of Molecular Sciences. 2019; 20(24):6201. https://doi.org/10.3390/ijms20246201
Chicago/Turabian StyleRoudnicky, Filip, Yanjun Lan, Max Friesen, Gregor Dernick, Jitao David Zhang, Andreas Staempfli, Natalie Bordag, Antje Wagner-Golbs, Klaus Christensen, Martin Ebeling, and et al. 2019. "Modeling the Effects of Severe Metabolic Disease by Genome Editing of hPSC-Derived Endothelial Cells Reveals an Inflammatory Phenotype" International Journal of Molecular Sciences 20, no. 24: 6201. https://doi.org/10.3390/ijms20246201
APA StyleRoudnicky, F., Lan, Y., Friesen, M., Dernick, G., Zhang, J. D., Staempfli, A., Bordag, N., Wagner-Golbs, A., Christensen, K., Ebeling, M., Graf, M., Burcin, M., Meyer, C. A., Cowan, C. A., & Patsch, C. (2019). Modeling the Effects of Severe Metabolic Disease by Genome Editing of hPSC-Derived Endothelial Cells Reveals an Inflammatory Phenotype. International Journal of Molecular Sciences, 20(24), 6201. https://doi.org/10.3390/ijms20246201