Synaptic Plasticity Shapes Brain Connectivity: Implications for Network Topology



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Brain Network Organization
3. Synaptic Plasticity
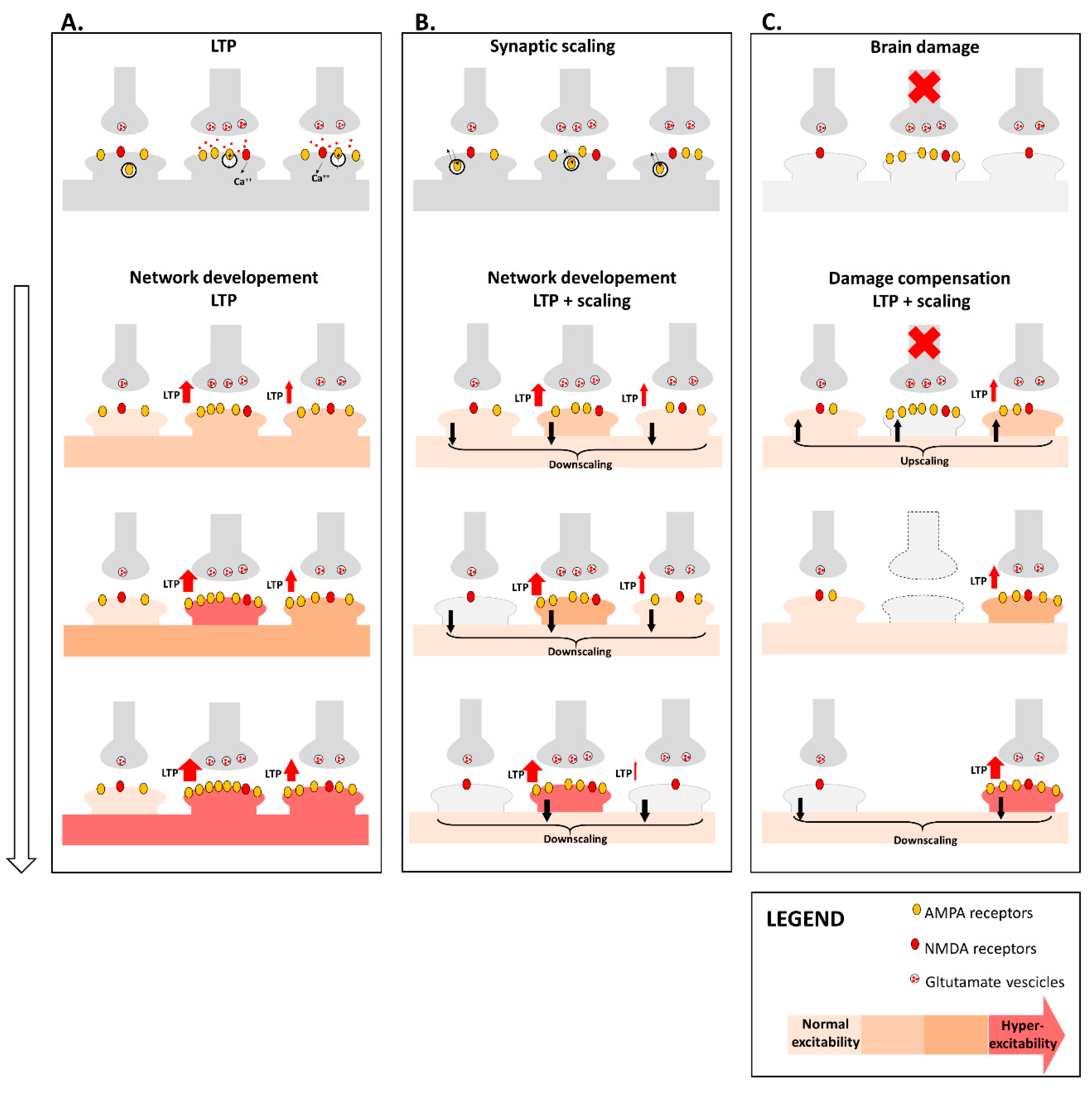
4. Synaptic Plasticity and Brain Network Organization
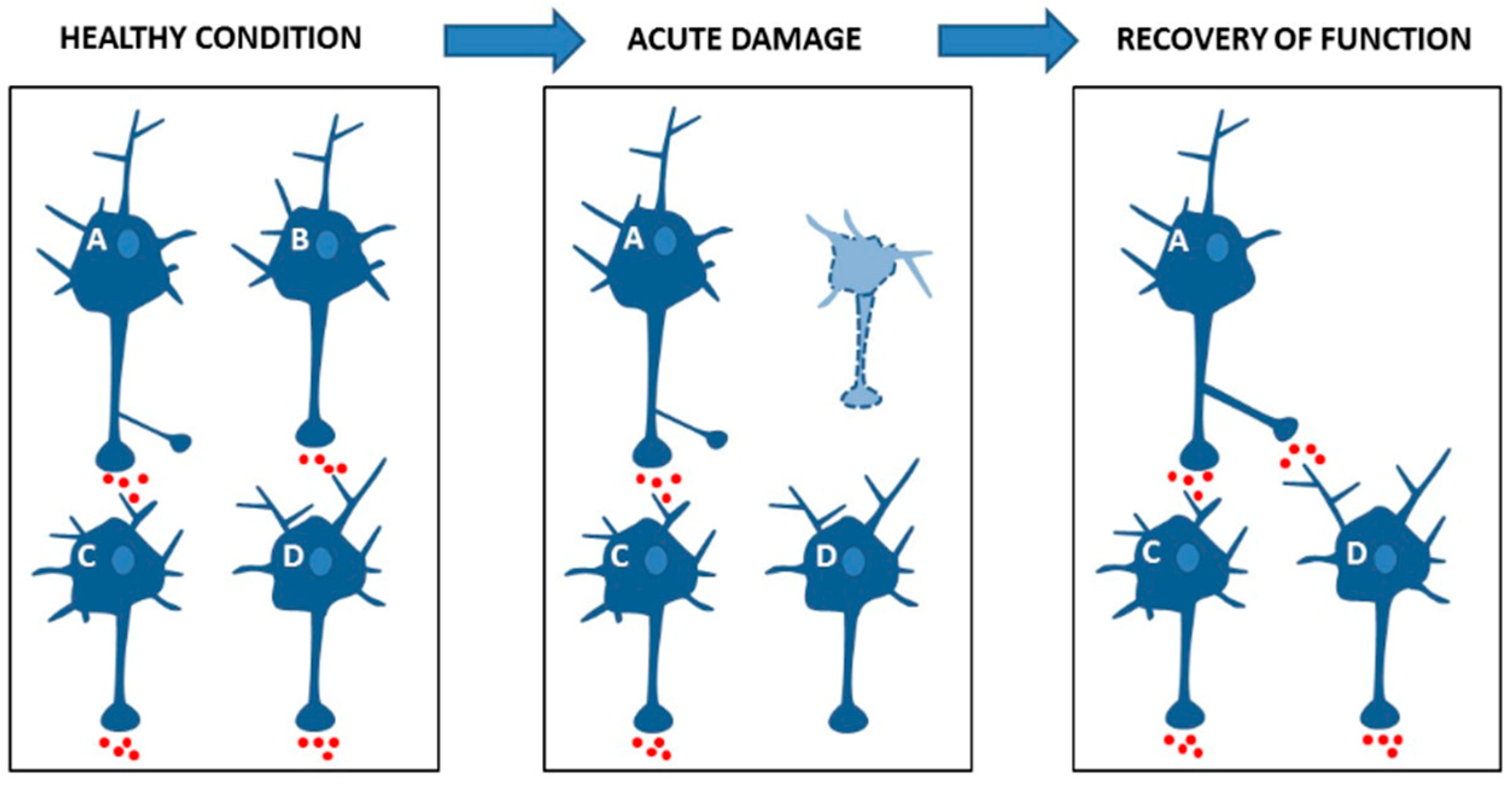
4.1. Synaptic Plasticity Promotes Brain Network Reorganization after Damage
4.2. Synaptic Plasticity Dysfunction May Drive Brain Network Disruption
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| EEG | Electroencephalography |
| MEG | Magnetoencephalography |
| FC | Functional Connectivity |
| fMRI | Functional MRI |
| rs-fMRI | Resting state-fMRI |
| BOLD | Blood-Oxygen-Level-Dependent |
| SC | Structural Connectivity |
| DTI | Diffusion Tensor Imaging |
| P(k) | Degree Distribution of a Graph |
| AD | Alzheimer’s disease |
| LTP | Long term potentiation |
| NMDARs | N-methyl-D-aspartate receptors |
| LTD | Long-Term Depression |
| AMPARs | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors |
| STDP | Spike Timing-Dependent Plasticity |
| TMS | Transcranial Magnetic Stimulation |
| MCI | Mild Cognitive Impairment |
| Aβ | Amyloid-β |
References
- Sporns, O.; Tononi, G.; Kötter, R. The human connectome: A structural description of the human brain. PLoS Comput. Biol. 2005, 1, e42. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.D.; Raichle, M.E. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat. Rev. Neurosci. 2007, 8, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Watts, D.J.; Strogatz, S.H. Collective dynamics of ‘small-world’ networks. Nature 1998, 393, 440–442. [Google Scholar] [CrossRef]
- Bullmore, E.; Sporns, O. Complex brain networks: Graph theoretical analysis of structural and functional systems. Nat. Rev. Neurosci. 2009, 10, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Achard, S.; Bullmore, E. Efficiency and cost of economical brain functional networks. PLoS Comput. Biol. 2007, 3, e17. [Google Scholar] [CrossRef] [PubMed]
- Rubinov, M.; Knock, S.A.; Stam, C.J.; Micheloyannis, S.; Harris, A.W.; Williams, L.M.; Breakspear, M. Small-world properties of nonlinear brain activity in schizophrenia. Hum. Brain Mapp. 2009, 30, 403–416. [Google Scholar] [CrossRef]
- Stam, C.J.; de Haan, W.; Daffertshofer, A.; Jones, B.F.; Manshanden, I.; Van Cappellen van Walsum, A.M.; Montez, T.; Verbunt, J.P.; de Munck, J.C.; van Dijk, B.W.; et al. Graph theoretical analysis of magnetoencephalographic functional connectivity in Alzheimer’s disease. Brain 2009, 132, 213–224. [Google Scholar] [CrossRef]
- Hillary, F.G.; Grafman, J.H. Injured Brains and Adaptive Networks: The Benefits and Costs of Hyperconnectivity. Trends. Cogn. Sci. 2017, 21, 385–401. [Google Scholar] [CrossRef]
- Turrigiano, G.G.; Nelson, S.B. Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 2004, 5, 97–107. [Google Scholar] [CrossRef]
- Varshney, L.R.; Chen, B.L.; Paniagua, E.; Hall, D.H.; Chklovskii, D.B. Structural properties of the Caenorhabditis elegans neuronal network. PLoS Comput. Biol. 2011, 7, e1001066. [Google Scholar] [CrossRef]
- Shih, C.T.; Sporns, O.; Yuan, S.L.; Su, T.S.; Lin, Y.J.; Chuang, C.C.; Wang, T.Y.; Lo, C.C.; Greenspan, R.J.; Chiang, A.S. Connectomics-based analysis of information flow in the Drosophila brain. Curr. Biol. 2015, 25, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Jensen, O.; Spaak, E.; Zumer, J.M. Human brain oscillations: From physiological mechanisms to analysis and cognition. In Magnetoencephalography: From Signals to Dynamic Cortical Networks; Supek, S., Aine, C.J., Eds.; Springer: Berlin, Germany, 2014; pp. 359–403. [Google Scholar]
- Rossini, P.M.; Di Iorio, R.; Bentivoglio, M.; Bertini, G.; Ferreri, F.; Gerloff, C.; Ilmoniemi, R.J.; Miraglia, F.; Nitsche, M.A.; Pestilli, F.; et al. Methods for analysis of brain connectivity: An IFCN-sponsored review. Clin. Neurophysiol. 2019, 130, 1833–1858. [Google Scholar] [CrossRef] [PubMed]
- Bassett, D.S.; Bullmore, E.T.; Verchinski, B.A.; Mattay, V.S.; Weinberger, D.R.; Meyer-Lindenberg, A. Hierarchical organization of human cortical networks in health and schizophrenia. J. Neurosci. 2008, 28, 9239–9248. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, Z.; Evans, A. Structural insights into aberrant topological patterns of large-scale cortical networks in Alzheimer’s disease. J. Neurosci. 2008, 28, 4756–4766. [Google Scholar] [CrossRef]
- Biswal, B.B.; Yetkin, F.Z.; Haughton, V.M.; Hyde, J.S. Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn. Reson. Med. 1995, 34, 537–541. [Google Scholar] [CrossRef]
- Honey, C.J.; Kötter, R.; Breakspear, M.; Sporns, O. Network structure of cerebral cortex shapes functional connectivity on multiple time scales. Proc. Natl. Acad. Sci. USA 2007, 104, 10240–10245. [Google Scholar] [CrossRef]
- Rubinov, M.; Sporns, O.; van Leeuwen, C.; Breakspear, M. Symbiotic relationship between brain structure and dynamics. BMC Neurosci. 2009, 10, 55. [Google Scholar] [CrossRef]
- Honey, C.J.; Sporns, O.; Cammoun, L.; Gigandet, X.; Thiran, J.P.; Meuli, R.; Hagmann, P. Predicting human resting-state functional connectivity from structural connectivity. Proc. Natl. Acad. Sci. USA 2009, 106, 2035–2040. [Google Scholar] [CrossRef]
- Rubinov, M.; Sporns, O. Complex network measures of brain connectivity: Uses and interpretations. Neuroimage 2010, 52, 1059–1069. [Google Scholar] [CrossRef]
- Smith, S.M.; Miller, K.L.; Salimi-Khorshidi, G.; Webster, M.; Beckmann, C.F.; Nichols, T.E.; Ramsey, J.D.; Woolrich, M.W. Network modeling methods for FMRI. Neuroimage 2011, 54, 875–891. [Google Scholar] [CrossRef]
- Friston, K.J. Functional and effective connectivity: A review. Brain Connect. 2011, 1, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Stam, C.J.; Reijneveld, J.C. Graph theoretical analysis of complex networks in the brain. Nonlinear Biomed. Phys. 2007, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Bullmore, E.; Sporns, O. The economy of brain network organization. Nat. Rev. Neurosci. 2012, 13, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Sporns, O.; Honey, C.J.; Kötter, R. Identification and classification of hubs in brain networks. PLoS ONE 2007, 2, e1049. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel, M.P.; Sporns, O. An anatomical substrate for integration among functional networks in human cortex. J. Neurosci. 2013, 33, 14489–14500. [Google Scholar] [CrossRef] [PubMed]
- Sporns, O. Network attributes for segregation and integration in the human brain. Curr. Opin. Neurobiol. 2013, 23, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Albert, R. Emergence of scaling in random networks. Science 1999, 286, 509–512. [Google Scholar] [CrossRef]
- Achard, S.; Salvador, R.; Whitcher, B.; Suckling, J.; Bullmore, E. A resilient, low-frequency, small-world human brain functional network with highly connected association cortical hubs. J. Neurosci. 2006, 26, 63–72. [Google Scholar] [CrossRef]
- Crossley, N.A.; Mechelli, A.; Scott, J.; Carletti, F.; Fox, P.T.; McGuire, P.; Bullmore, E.T. The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain 2014, 137, 2382–2395. [Google Scholar] [CrossRef]
- Yu, M.; Engels, M.M.A.; Hillebrand, A.; van Straaten, E.C.W.; Gouw, A.A.; Teunissen, C.; van der Flier, W.M.; Scheltens, P.; Stam, C.J. Selective impairment of hippocampus and posterior hub areas in Alzheimer’s disease: An MEG-based multiplex network study. Brain 2017, 140, 1466–1485. [Google Scholar] [CrossRef]
- Rubinov, M.; Bullmore, E. Fledgling pathoconnectomics of psychiatric disorders. Trends Cogn. Sci. 2013, 17, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Klauser, P.; Baker, S.T.; Cropley, V.L.; Bousman, C.; Fornito, A.; Cocchi, L.; Fullerton, J.M.; Rasser, P.; Schall, U.; Henskens, F.; et al. White matter disruptions in schizophrenia are spatially widespread and topologically converge on brain network hubs. Schizophr. Bull. 2016, 43, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Lømo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356. [Google Scholar] [CrossRef]
- Morris, R.G.; Anderson, E.; Lynch, G.S.; Baudry, M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature 1986, 319, 774–776. [Google Scholar] [CrossRef]
- Centonze, D.; Rossi, S.; Tortiglione, A.; Picconi, B.; Prosperetti, C.; De Chiara, V.; Bernardi, G.; Calabresi, P. Synaptic plasticity during recovery from permanent occlusion of the middle cerebral artery. Neurobiol. Dis. 2007, 27, 44–53. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Kehl, S.J.; McLennan, H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J. Physiol. 1983, 334, 33–46. [Google Scholar] [CrossRef]
- Malenka, R.C.; Kauer, J.A.; Zucker, R.S.; Nicoll, R.A. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science 1988, 242, 81–84. [Google Scholar] [CrossRef]
- Engert, F.; Bonhoeffer, T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 1999, 399, 66–70. [Google Scholar] [CrossRef]
- De Roo, M.; Klauser, P.; Mendez, P.; Poglia, L.; Muller, D. Activity-dependent PSD formation and stabilization of newly formed spines in hippocampal slice cultures. Cereb. Cortex 2008, 18, 151–161. [Google Scholar] [CrossRef]
- Hill, T.C.; Zito, K. LTP-induced long-term stabilization of individual nascent dendritic spines. J. Neurosci. 2013, 33, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.W.; Ganong, A.H.; Cotman, C.W. Long-term potentiation in the hippocampus involves activation of N-methyl-D-aspartate receptors. Brain Res. 1984, 323, 132–137. [Google Scholar] [CrossRef]
- Malinow, R.; Schulman, H.; Tsien, R.W. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 1989, 245, 862–866. [Google Scholar] [CrossRef]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef]
- Tada, T.; Sheng, M. Molecular mechanisms of dendritic spine morphogenesis. Curr. Opin. Neurobiol. 2006, 16, 95–101. [Google Scholar] [CrossRef]
- Zhou, Q.; Homma, K.J.; Poo, M.M. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 2004, 44, 749–757. [Google Scholar] [CrossRef]
- Turrigiano, G.G.; Leslie, K.R.; Desai, N.S.; Rutherford, L.C.; Nelson, S.B. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 1998, 391, 892–896. [Google Scholar] [CrossRef]
- Lissin, D.V.; Gomperts, S.N.; Carroll, R.C.; Christine, C.W.; Kalman, D.; Kitamura, M.; Hardy, S.; Nicoll, R.A.; Malenka, R.C.; von Zastrow, M. Activity differentially regulates the surface expression of synaptic AMPA and NMDA glutamate receptors. Proc. Natl. Acad. Sci. USA 1998, 95, 7097–7102. [Google Scholar] [CrossRef]
- Tzingounis, V.; Nicoll, R.A. Arc/Arg3.1: Linking gene expression to synaptic plasticity and memory. Neuron 2006, 52, 403–407. [Google Scholar] [CrossRef]
- Shepherd, J.D.; Rumbaugh, G.; Wu, J.; Chowdhury, S.; Plath, N.; Kuhl, D.; Huganir, R.L.; Worley, P.F. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 2006, 52, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Donoghue, J.P. Reshaping the cortical motor map by unmasking latent intracortical connections. Science 1991, 251, 944–947. [Google Scholar] [CrossRef] [PubMed]
- Ziemann, U.; Rothwell, J.C.; Ridding, M.C. Interaction between intracortical inhibition and facilitation in human motor cortex. J. Physiol. 1996, 496, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Baroncelli, L.; Braschi, C.; Spolidoro, M.; Begenisic, T.; Maffei, L.; Sale, A. Brain plasticity and disease: A matter of inhibition. Neural. Plast. 2011, 2011. [Google Scholar] [CrossRef]
- Imbrosci, B.; Mittmann, T. Functional consequences of the disturbances in the GABA-mediated inhibition induced by injuries in the cerebral cortex. Neural. Plast. 2011, 2011. [Google Scholar] [CrossRef]
- Zhao, L.; Beverlin, B.I.; Netoff, T.; Nykamp, D.Q. Synchronization from second order network connectivity statistics. Front. Comput. Neurosci. 2011, 5, 28. [Google Scholar] [CrossRef]
- Roxin, A. The role of degree distribution in shaping the dynamics in networks of sparsely connected spiking neurons. Front. Comput. Neurosci. 2011, 5, 8. [Google Scholar] [CrossRef]
- Pernice, V.; Staude, B.; Cardanobile, S.; Rotter, S. How structure determines correlations in neuronal networks. PLoS Comput. Biol. 2011, 7, e1002059. [Google Scholar] [CrossRef]
- Pernice, V.; Deger, M.; Cardanobile, S.; Rotter, S. The relevance of network micro-structure for neural dynamics. Front. Comput. Neurosci. 2013, 7, 72. [Google Scholar] [CrossRef]
- Trousdale, J.; Hu, Y.; Shea-Brown, E.; Josić, K. Impact of network structure and cellular response on spike time correlations. PLoS Comput. Biol. 2012, 8, e1002408. [Google Scholar] [CrossRef]
- Hu, Y.; Trousdale, J.; Josić, K.; Shea-Brown, E. Motif statistics and spike correlations in neuronal networks. BMC Neurosci. 2013, 2013, P03012. [Google Scholar] [CrossRef]
- Hu, Y.; Trousdale, J.; Josić, K.; Shea-Brown, E. Local paths to global coherence: Cutting networks down to size. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2014, 89, 032802. [Google Scholar] [CrossRef]
- Helias, M.; Tetzlaff, T.; Diesmann, M. The correlation structure of local neuronal networks intrinsically results from recurrent dynamics. PLoS Comput. Biol. 2014, 10, e1003428. [Google Scholar] [CrossRef]
- Buzsáki, G. Two-stage model of memory trace formation: A role for “noisy” brain states. Neuroscience 1989, 31, 551–570. [Google Scholar] [CrossRef]
- Wilson, M.A.; McNaughton, B.L. Reactivation of hippocampal ensemble memories during sleep. Science 1994, 29, 676–679. [Google Scholar] [CrossRef]
- Babiloni, C.; Vecchio, F.; Lizio, R.; Ferri, R.; Rodriguez, G.; Marzano, N.; Frisoni, G.B.; Rossini, P.M. Resting state cortical rhythms in mild cognitive impairment and Alzheimer’s disease: Electroencephalographic evidence. J. Alzheimers Dis. 2011, 26, 201–214. [Google Scholar] [CrossRef]
- Buzsáki, G.; Watson, B.O. Brain rhythms and neural syntax: Implications for efficient coding of cognitive content and neuropsychiatric disease. Dialogues Clin. Neurosci. 2012, 14, 345–367. [Google Scholar] [PubMed]
- Uhlhaas, P.J.; Singer, W. Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci. 2010, 11, 100–113. [Google Scholar] [CrossRef]
- Wang, X.J.; Buzsáki, G. Gamma oscillation by synaptic inhibition in a hippocampal interneuronal network model. J. Neurosci. 1996, 16, 6402–6413. [Google Scholar] [CrossRef]
- Whittington, M.A.; Traub, R.D.; Jefferys, J.G. Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature 1995, 373, 612–615. [Google Scholar] [CrossRef]
- Lynch, G.; Larson, J.; Kelso, S.; Barrionuevo, G.; Schottler, F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature 1983, 305, 719–721. [Google Scholar] [CrossRef]
- Stanton, P.K.; Sejnowski, T.J. Associative long-term depression in the hippocampus induced by Hebbian covariance. Nature 1989, 339, 215–218. [Google Scholar] [CrossRef]
- Dudek, S.M.; Bear, M.F. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc. Natl. Acad. Sci. USA 1992, 89, 4363–4367. [Google Scholar] [CrossRef]
- Markram, H.; Lübke, J.; Frotscher, M.; Sakmann, B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 1997, 275, 213–215. [Google Scholar] [CrossRef]
- Markram, H.; Gerstner, W.; Sjostrom, P.J. A history of spike-timing-dependent plasticity. Front. Synaptic Neurosci. 2011, 3, 4. [Google Scholar] [CrossRef]
- Harris, K.D. Neural signatures of cell assembly organization. Nat. Rev. Neurosci. 2005, 6, 399–407. [Google Scholar] [CrossRef]
- Singer, W. Neuronal synchrony: A versatile code for the definition of relations? Neuron 1999, 24, 49–65. [Google Scholar] [CrossRef]
- Zanos, S.; Rembado, I.; Chen, D.; Fetz, E.E. Phase-Locked Stimulation during Cortical Beta Oscillations Produces Bidirectional Synaptic Plasticity in Awake Monkeys. Curr. Biol. 2018, 28, R879–R882. [Google Scholar] [CrossRef]
- Nevian, T.; Sakmann, B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J. Neurosci. 2006, 26, 11001–11013. [Google Scholar] [CrossRef]
- Egelman, D.M.; Montague, P.R. Calcium dynamics in the extracellular space of mammalian neural tissue. Biophys. J. 1999, 76, 1856–1867. [Google Scholar] [CrossRef]
- Abbott, L.F.; Nelson, S.B. Synaptic plasticity: Taming the beast. Nat. Neurosci. 2000, 3, 1178–1183. [Google Scholar] [CrossRef]
- Canals, S.; Beyerlein, M.; Merkle, H.; Logothetis, N.K. Functional MRI evidence for LTP-induced neural network reorganization. Curr. Biol. 2009, 19, 398–403. [Google Scholar] [CrossRef]
- Alvarez-Salvado, E.; Pallarés, V.; Moreno, A.; Canals, S. Functional MRI of long-term potentiation: Imaging network plasticity. Philos. Trans. R. Soc. B Biol. Sci. 2013, 369, 20130152. [Google Scholar] [CrossRef]
- Miller, K.D. Synaptic economics: Competition and cooperation in synaptic plasticity. Neuron 1996, 17, 371–374. [Google Scholar] [CrossRef]
- Koch, G.; Di Lorenzo, F.; Bonnì, S.; Ponzo, V.; Caltagirone, C.; Martorana, A. Impaired LTP- but not LTD-like cortical plasticity in Alzheimer’s disease patients. J. Alzheimers Dis. 2012, 31, 593–599. [Google Scholar] [CrossRef]
- Ribolsi, M.; Lisi, G.; Ponzo, V.; Siracusano, A.; Caltagirone, C.; Niolu, C.; Koch, G. Left hemispheric breakdown of LTP-like cortico-cortical plasticity in schizophrenic patients. Clin. Neurophysiol. 2017, 128, 2037–2042. [Google Scholar] [CrossRef]
- Guerriero, R.M.; Giza, C.C.; Rotenberg, A. Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2015, 15, 27. [Google Scholar] [CrossRef]
- Khazipov, R.; Valeeva, G.; Khalilov, I. Depolarizing GABA and developmental epilepsies. CNS Neurosci. Ther. 2015, 21, 83–91. [Google Scholar] [CrossRef]
- Bonansco, C.; Fuenzalida, M. Plasticity of hippocampal excitatory-inhibitory balance: Missing the synaptic control in the epileptic brain. Neural Plast. 2016, 2016, 8607038. [Google Scholar] [CrossRef]
- Desai, N.S.; Cudmore, R.H.; Nelson, S.B.; Turrigiano, G.G. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat. Neurosci. 2002, 5, 783–789. [Google Scholar] [CrossRef]
- Turrigiano, G. Homeostatic synaptic plasticity: Local and global mechanisms for stabilizing neuronal function. Cold. Spring Harb. Perspect. Biol. 2012, 4, a005736. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Profice, P.; Pilato, F.; Capone, F.; Ranieri, F.; Pasqualetti, P.; Colosimo, C.; Pravatà, E.; Cianfoni, A.; Dileone, M. Motor cortex plasticity predicts recovery in acute stroke. Cereb. Cortex 2010, 20, 1523–1528. [Google Scholar] [CrossRef]
- Mori, F.; Kusayanagi, H.; Nicoletti, C.G.; Weiss, S.; Marciani, M.G.; Centonze, D. Cortical plasticity predicts recovery from relapse in multiple sclerosis. Mult. Scler. 2014, 20, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Hillary, F.G.; Rajtmajer, S.M.; Roman, C.A.; Medaglia, J.D.; Slocomb-Dluzen, J.E.; Calhoun, V.D.; Good, D.C.; Wylie, G.R. The rich get richer: Brain injury elicits hyperconnectivity in core subnetworks. PLoS ONE 2014, 9, e113545. [Google Scholar] [CrossRef] [PubMed]
- Iraji, A.; Benson, R.R.; Welch, R.D.; O’Neil, B.J.; Woodard, J.L.; Ayaz, S.I.; Kulek, A.; Mika, V.; Medado, P.; Soltanian-Zadeh, H.; et al. Resting state functional connectivity in mild traumatic brain injury at the acute stage: Independent component and seed-based analyses. J. Neurotrauma 2016, 32, 1031–1045. [Google Scholar] [CrossRef]
- Bharath, R.D.; Munivenkatappa, A.; Gohel, S.; Panda, R.; Saini, J.; Rajeswaran, J.; Shukla, D.; Bhagavatula, I.D.; Biswal, B.B. Recovery of resting brain connectivity ensuing mild traumatic brain injury. Front. Hum. Neurosci. 2015, 9, 513. [Google Scholar] [CrossRef] [PubMed]
- Nicolo, P.; Rizk, S.; Magnin, C.; Pietro, M.D.; Schnider, A.; Guggisberg, A.G. Coherent neural oscillations predict future motor and language improvement after stroke. Brain 2015, 138, 3048–3060. [Google Scholar] [CrossRef]
- Gorges, M.; Müller, H.P.; Lulé, D.; LANDSCAPE Consortium; Pinkhardt, E.H.; Ludolph, A.C.; Kassubek, J. To rise and to fall: Functional connectivity in cognitively normal and cognitively impaired patients with Parkinson’s disease. Neurobiol. Aging 2014, 36, 1727–1735. [Google Scholar] [CrossRef]
- Fernández-Seara, M.A.; Mengual, E.; Vidorreta, M.; Castellanos, G.; Irigoyen, J.; Erro, E.; Pastor, M.A. Resting state functional connectivity of the subthalamic nucleus in Parkinson’s disease assessed using arterial spin-labeled perfusion fMRI. Hum. Brain Mapp. 2015, 36, 1937–1950. [Google Scholar] [CrossRef]
- Cohen, A.D.; Price, J.C.; Weissfeld, L.A.; James, J.; Rosario, B.L.; Bi, W.; Nebes, R.D.; Saxton, J.A.; Snitz, B.E.; Aizenstein, H.A.; et al. Basal Cerebral Metabolism May Modulate the Cognitive Effects of Aβ in Mild Cognitive Impairment: An Example of Brain Reserve. J. Neurosci. 2009, 29, 14770–14778. [Google Scholar] [CrossRef]
- Gour, N.; Ranjeva, J.P.; Ceccaldi, M.; Confort-Gouny, S.; Barbeau, E.; Soulier, E.; Guye, M.; Didic, M.; Felician, O. Basal functional connectivity within the anterior temporal network is associated with performance on declarative memory tasks. Neuroimage 2011, 58, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef]
- Valenzuela, M.J.; Sachdev, P. Brain reserve and dementia: A systematic review. Psychol. Med. 2006, 36, 441–454. [Google Scholar] [CrossRef]
- Okonkwo, O.C.; Schultz, S.A.; Oh, J.M.; Larson, J.; Edwards, D.; Cook, D.; Koscik, R.; Gallagher, C.L.; Dowling, N.M.; Carlsson, C.M.; et al. Physical activity attenuates age-related biomarker alterations in preclinical AD. Neurology 2014, 83, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Tolppanen, A.M.; Solomon, A.; Kulmala, J.; Kåreholt, I.; Ngandu, T.; Rusanen, M.; Laatikainen, T.; Soininen, H.; Kivipelto, M. Leisure-time physical activity from mid- to late life, body mass index, and risk of dementia. Alzheimers Dement. 2015, 11, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Duzel, E.; van Praag, H.; Sendtner, M. Can physical exercise in old age improve memory and hippocampal function? Brain 2016, 139, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Frick, K.M.; Stearns, N.A.; Pan, J.Y.; Berger-Sweeney, J. Effects of environmental enrichment on spatial memory and neurochemistry in middle-aged mice. Learn. Mem. 2003, 10, 187–198. [Google Scholar] [CrossRef]
- Leggio, M.G.; Mandolesi, L.; Federico, F.; Spirito, F.; Ricci, B.; Gelfo, F.; Petrosini, L. Environmental enrichment promotes improved spatial abilities and enhanced dendritic growth in the rat. Behav. Brain Res. 2005, 163, 78–90. [Google Scholar] [CrossRef]
- Malik, R.; Chattarji, S. Enhanced intrinsic excitability and EPSP-spike coupling accompany enriched environment-induced facilitation of LTP in hippocampal CA1 pyramidal neurons. J. Neurophysiol. 2012, 107, 1366–1378. [Google Scholar] [CrossRef]
- Hullinger, R.; O’Riordan, K.; Burger, C. Environmental enrichment improves learning and memory and long-term potentiation in young adult rats through a mechanism requiring mGluR5 signaling and sustained activation of p70s6k. Neurobiol. Learn. Mem. 2015, 125, 126–134. [Google Scholar] [CrossRef]
- Stein, L.R.; O’Dell, K.A.; Funatsu, M.; Zorumski, C.F.; Izumi, Y. Short-term environmental enrichment enhances synaptic plasticity in hippocampal slices from aged rats. Neuroscience 2016, 329, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Cortese, G.P.; Olin, A.; O’Riordan, K.; Hullinger, R.; Burger, C. Environmental enrichment improves hippocampal function in aged rats by enhancing learning and memory, LTP, and mGluR5-Homer1c activity. Neurobiol. Aging 2018, 63, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Arenaza-Urquijo, E.M.; Landeau, B.; La Joie, R.; Mevel, K.; Mézenge, F.; Perrotin, A.; Desgranges, B.; Bartrés-Faz, D.; Eustache, F.; Chételat, G. Relationships between years of education and gray matter volume, metabolism and functional connectivity in healthy elders. Neuroimage 2013, 83, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Franzmeier, N.; Duering, M.; Weiner, M.; Dichgans, M.; Ewers, M.; Alzheimer’s Disease Neuroimaging Initiative (ADNI). Left frontal cortex connectivity underlies cognitive reserve in prodromal Alzheimer disease. Neurology 2017, 88, 1054–1061. [Google Scholar] [CrossRef]
- Franzmeier, N.; Düzel, E.; Jessen, F.; Buerger, K.; Levin, J.; Duering, M.; Dichgans, M.; Haass, C.; Suárez-Calvet, M.; Fagan, A.M.; et al. Left frontal hub connectivity delays cognitive impairment in autosomal-dominant and sporadic Alzheimer’s disease. Brain 2018, 141, 1186–1200. [Google Scholar] [CrossRef]
- Sumowski, J.F.; Wylie, G.R.; Deluca, J.; Chiaravalloti, N. Intellectual enrichment is linked to cerebral efficiency in multiple sclerosis: Functional magnetic resonance imaging evidence for cognitive reserve. Brain 2010, 133, 362–374. [Google Scholar] [CrossRef]
- Santarnecchi, E.; Rossi, S.; Rossi, A. The smarter, the stronger: Intelligence level correlates with brain resilience to systematic insults. Cortex 2015, 293–309. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Ponzo, V.; Bonnì, S.; Motta, C.; Negrão Serra, P.C.; Bozzali, M.; Caltagirone, C.; Martorana, A.; Koch, G. Long-term potentiation-like cortical plasticity is disrupted in Alzheimer’s disease patients independently from age of onset. Ann. Neurol. 2016, 80, 202–210. [Google Scholar] [CrossRef]
- Buckner, R.L.; Sepulcre, J.; Talukdar, T.; Krienen, F.M.; Liu, H.; Hedden, T.; Andrews-Hanna, J.R.; Sperling, R.A.; Johnson, K.A. Cortical hubs revealed by intrinsic functional connectivity: Mapping, assessment of stability, and relation to Alzheimer’s disease. J. Neurosci. 2009, 29, 1860–1873. [Google Scholar] [CrossRef]
- Uhlhaas, P.J.; Singer, W. Neural synchrony in brain disorders: Relevance for cognitive dysfunctions and pathophysiology. Neuron 2006, 52, 155–168. [Google Scholar] [CrossRef]
- Yener, G.G.; Başar, E. Brain oscillations as biomarkers in neuropsychiatric disorders: Following an interactive panel discussion and synopsis. Suppl. Clin. Neurophysiol. 2013, 62, 343–363. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Holtzman, D.M. Biomarker modeling of Alzheimer’s disease. Neuron 2013, 80, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, S.; Wicklund, A.H.; Salmon, D.P. The neuropsychological profile of Alzheimer disease. Cold. Spring Harb. Perspect. Med. 2012, 2, a006171. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W., Jr.; Morris, J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001, 56, 127–129. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Dekosky, S.T.; Mufson, E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef]
- Chapman, P.F.; White, G.L.; Jones, M.W.; Cooper-Blacketer, D.; Marshall, V.J.; Irizarry, M.; Younkin, L.; Good, M.A.; Bliss, T.V.; Hyman, B.T.; et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci. 1999, 2, 271–276. [Google Scholar] [CrossRef]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef]
- Jacobsen, J.S.; Wu, C.C.; Redwine, J.M.; Comery, T.A.; Arias, R.; Bowlby, M.; Martone, R.; Morrison, J.H.; Pangalos, M.N.; Reinhart, P.H.; et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5161–5166. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural Oligomers of the Alzheimer Amyloid-β Protein Induce Reversible Synapse Loss by Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s disease: Synaptic dysfunction and Aβ. Mol. Neurodegen. 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Brewer, G.J. Amyloid-β as a modulator of synaptic plasticity. J. Alzheimers Dis. 2010, 22, 741–763. [Google Scholar] [CrossRef] [PubMed]
- Small, D.H. Network dysfunction in Alzheimer’s disease: Does synaptic scaling drive disease progression? Trends Mol. Med. 2008, 14, 103–108. [Google Scholar] [CrossRef]
- Jang, S.S.; Chung, H.J. Emerging Link between Alzheimer’s Disease and Homeostatic Synaptic Plasticity. Neural. Plast. 2016, 2016. [Google Scholar] [CrossRef]
- Stam, C.J.; Jones, B.F.; Nolte, G.; Breakspear, M.; Scheltens, P. Small-world networks and functional connectivity in Alzheimer’s disease. Cereb. Cortex 2007, 17, 92–99. [Google Scholar] [CrossRef]
- De Haan, W.; Pijnenburg, Y.A.; Strijers, R.L.; Van der Made, Y.; Van der Flier, W.M.; Scheltens, P.; Stam, C.J. Functional neural network analysis in frontotemporal dementia and Alzheimer’s disease using EEG and graph theory. BMC Neurosci. 2009, 10, 101. [Google Scholar] [CrossRef]
- De Haan, W.; van der Flier, W.M.; Wang, H.; Van Mieghem, P.F.; Scheltens, P.; Stam, C.J. Disruption of functional brain networks in Alzheimer’s disease: What can we learn from graph spectral analysis of resting-state magnetoencephalography? Brain Connect. 2012, 2, 45–55. [Google Scholar] [CrossRef]
- Vecchio, F.; Miraglia, F.; Marra, C.; Quaranta, D.; Vita, M.G.; Bramanti, P.; Rossini, P.M. Human brain networks in cognitive decline: A graph theoretical analysis of cortical connectivity from EEG data. J. Alzheimers Dis. 2014, 41, 113–127. [Google Scholar] [CrossRef]
- Canuet, L.; Pusil, S.; Lopez, M.E.; Bajo, R.; Pineda-Pardo, J.A.; Cuesta, P.; Gálvez, G.; Gaztelu, J.M.; Lourido, D.; García-Ribas, G.; et al. Network disruption and cerebrospinal fluid amyloid-beta and phospho-tau levels in mild cognitive impairment. J. Neurosci. 2015, 35, 10325–10330. [Google Scholar] [CrossRef]
- Dai, Z.; Yan, C.; Li, K.; Wang, Z.; Wang, J.; Cao, M.; Lin, Q.; Shu, N.; Xia, M.; Bi, Y.; et al. Identifying and mapping connectivity patterns of brain network hubs in Alzheimer’s disease. Cereb. Cortex 2015, 25, 3723–3742. [Google Scholar] [CrossRef] [PubMed]
- De Haan, W.; Mott, K.; van Straaten, E.C.; Scheltens, P.; Stam, C.J. Activity dependent degeneration explains hub vulnerability in Alzheimer’s disease. PLoS Comput. Biol. 2012, 8, e1002582. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Laviolette, P.S.; O’Keefe, K.; O’Brien, J.; Rentz, D.M.; Pihlajamaki, M.; Marshall, G.; Hyman, B.T.; Selkoe, D.J.; Hedden, T.; et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 2009, 63, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Drzezga, A.; Becker, J.A.; Van Dijk, K.R.; Sreenivasan, A.; Talukdar, T.; Sullivan, C.; Schultz, A.P.; Sepulcre, J.; Putcha, D.; Greve, D.; et al. Neuronal dysfunction and disconnection of cortical hubs in non-demented subjects with elevated amyloid burden. Brain 2011, 134, 1635–1646. [Google Scholar] [CrossRef]
- Buchanan, R.W.; Carpenter, W.T. Schizophrenia: Introduction and overview. In Comprehensive Textbook of Psychiatry; Sadock, B.J., Sadock, V.A., Eds.; Lippincott, Williams, and Wilkins: Philadelphia, PA, USA, 2000; Volume 1, pp. 1096–1110. [Google Scholar]
- Coyle, J.T. The glutamatergic dysfunction hypothesis for schizophrenia. Harv. Rev. Psychiat. 1996, 3, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Levitt, P. Schizophrenia as a disorder of neurodevelopment. Annu. Rev. Neurosci. 2002, 25, 409–432. [Google Scholar] [CrossRef]
- Rubinov, M.; Bullmore, E.T. Schizophrenia and abnormal brain network hubs. Dialogues Clin. Neurosci. 2013, 15, 339–349. [Google Scholar] [PubMed]
- Hasan, A.; Nitsche, M.A.; Rein, B.; Schneider-Axmann, T.; Guse, B.; Gruber, O.; Falkai, P.; Wobrock, T. Dysfunctional long term potentiation-like plasticity in schizophrenia revealed by transcranial direct current stimulation. Behav. Brain Res. 2011, 224, 15–22. [Google Scholar] [CrossRef]
- Hasan, A.; Nitsche, M.A.; Herrmann, M.; Schneider-Axmann, T.; Marshall, L.; Gruber, O. Impaired long-term depression in schizophrenia: A cathodal tDCS pilot study. Brain Stim. 2012, 5, 475–483. [Google Scholar] [CrossRef]
- Glantz, L.A.; Lewis, D.A. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 2000, 57, 65–73. [Google Scholar] [CrossRef]
- Hill, J.J.; Hashimoto, T.; Lewis, D.A. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 2006, 11, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Sweet, R.A.; Henteleff, R.A.; Zhang, W.; Sampson, A.R.; Lewis, D.A. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology 2009, 34, 374–389. [Google Scholar] [CrossRef] [PubMed]
- Glausier, J.R.; Lewis, D.A. Dendritic spine pathology in schizophrenia. Neuroscience 2013, 251, 90–107. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.L.; Ding, Y.; Newman, J.; Hemby, S.; Penzes, P.; Lewis, D.A.; Yates, N.A.; Sweet, R.A. Altered glutamate protein coexpression network topology linked to spine loss in the auditory cortex of schizophrenia. Biol. Psychiatry 2015, 77, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Sucher, N.J.; Bradley, D.; Tafazzoli, A.; Trinh, D.; Hetrick, W.P.; Potkin, S.G.; Sandman, C.A.; Bunney, W.E., Jr.; Jones, E.G. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 1996, 16, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Beneyto, M.; Meador-Woodruff, J.H. Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse 2006, 60, 585–598. [Google Scholar] [CrossRef]
- Emamian, E.S.; Karayiorgou, M.; Gogos, J.A. Decreased phosphorylation of NMDA receptor type 1 at serine 897 in brains of patients with Schizophrenia. J. Neurosci. 2004, 24, 1561–1564. [Google Scholar] [CrossRef][Green Version]
- Funk, A.J.; Rumbaugh, G.; Harotunian, V.; McCullumsmith, R.E.; Meador-Woodruff, J.H. Decreased expression of NMDA receptor-associated proteins in frontal cortex of elderly patients with schizophrenia. Neuroreport 2009, 20, 1019–1022. [Google Scholar] [CrossRef]
- Javitt, D.C. Glutamate and schizophrenia: Phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int. Rev. Neurobiol. 2007, 78, 69–108. [Google Scholar] [CrossRef]
- Frantseva, M.V.; Fitzgerald, P.B.; Chen, R.; Möller, B.; Daigle, M.; Daskalakis, Z.J. Evidence for impaired long-term potentiation in schizophrenia and its relationship to motor skill learning. Cereb. Cortex 2008, 18, 990–996. [Google Scholar] [CrossRef]
- Koch, G.; Ponzo, V.; Di Lorenzo, F.; Caltagirone, C.; Veniero, D. Hebbian and anti-Hebbian spike-timing-dependent plasticity of human cortico-cortical connections. J Neurosci. 2013, 33, 9725–9733. [Google Scholar] [CrossRef] [PubMed]
- Veniero, D.; Ponzo, V.; Koch, G. Paired associative stimulation enforces the communication between interconnected areas. J. Neurosci. 2013, 33, 13773–13783. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, M.; Zhou, Y.; He, Y.; Hao, Y.; Song, M.; Yu, C.; Liu, H.; Liu, Z.; Jiang, T. Disrupted small-world networks in schizophrenia. Brain 2008, 131, 945–961. [Google Scholar] [CrossRef] [PubMed]
- Lynall, M.E.; Bassett, D.S.; Kerwin, R.; McKenna, P.J.; Kitzbichler, M.; Muller, U.; Bullmore, E. Functional connectivity and brain networks in schizophrenia. J Neurosci. 2010, 30, 9477–9487. [Google Scholar] [CrossRef]
- Rubinov, M.; Bassett, D.S. Emerging evidence of connectomic abnormalities in schizophrenia. J. Neurosci. 2011, 31, 6263–6265. [Google Scholar] [CrossRef]
- Skudlarski, P.; Jagannathan, K.; Anderson, K.; Stevens, M.C.; Calhoun, V.D.; Skudlarska, B.A.; Pearlson, G. Brain connectivity is not only lower but different in schizophrenia: A combined anatomical and functional approach. Biol. Psychiatry 2010, 68, 61–69. [Google Scholar] [CrossRef]
- Zalesky, A.; Fornito, A.; Seal, M.L.; Cocchi, L.; Westin, C.F.; Bullmore, E.T.; Egan, G.F.; Pantelis, C. Disrupted axonal fiber connectivity in schizophrenia. Biol. Psychiatry 2011, 69, 80–90. [Google Scholar] [CrossRef]
- Kambeitz, J.; Kambeitz-Ilankovic, L.; Cabral, C.; Dwyer, D.B.; Calhoun, V.D.; van den Heuvel, M.P.; Falkai, P.; Koutsouleris, N.; Malchow, B. Aberrant Functional Whole-Brain Network Architecture in Patients with Schizophrenia: A Meta-analysis. Schizophr. Bull. 2016, 42, 13–21. [Google Scholar] [CrossRef]
- Van den Heuvel, M.P.; Sporns, O.; Collin, G.; Scheewe, T.; Mandl, R.C.; Cahn, W.; Goñi, J.; Hulshoff Pol, H.E.; Kahn, R.S. Abnormal rich club organization and functional brain dynamics in schizophrenia. JAMA Psychiat. 2013, 70, 783–792. [Google Scholar] [CrossRef]
- Zhao, X.; Tian, L.; Yan, J.; Yue, W.; Yan, H.; Zhang, D. Abnormal Rich-Club Organization Associated with Compromised Cognitive Function in Patients with Schizophrenia and Their Unaffected Parents. Neurosci. Bull. 2017, 33, 445–454. [Google Scholar] [CrossRef]
- Van den Heuvel, M.P.; Mandl, R.C.W.; Stam, C.J.; Kahn, R.S.; Hulshoff Pol, H.E. Aberrant frontal and temperal network structure in schizophrenia: A graph theoretical analysis. J. Neurosci. 2010, 30, 15915–15926. [Google Scholar] [CrossRef] [PubMed]
- Gollo, L.L.; Roberts, J.A.; Cropley, V.L.; Di Biase, M.A.; Pantelis, C.; Zalesky, A.; Breakspear, M. Fragility and volatility of structural hubs in the human connectome. Nat. Neurosci. 2018, 21, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Zick, J.L.; Blackman, R.K.; Crowe, D.A.; Amirikian, B.; DeNicola, A.L.; Netoff, T.I.; Chafee, M.V. Blocking NMDAR Disrupts Spike Timing and Decouples Monkey Prefrontal Circuits: Implications for Activity-Dependent Disconnection in Schizophrenia. Neuron 2018, 98, 1243–1255. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stampanoni Bassi, M.; Iezzi, E.; Gilio, L.; Centonze, D.; Buttari, F. Synaptic Plasticity Shapes Brain Connectivity: Implications for Network Topology. Int. J. Mol. Sci. 2019, 20, 6193. https://doi.org/10.3390/ijms20246193
Stampanoni Bassi M, Iezzi E, Gilio L, Centonze D, Buttari F. Synaptic Plasticity Shapes Brain Connectivity: Implications for Network Topology. International Journal of Molecular Sciences. 2019; 20(24):6193. https://doi.org/10.3390/ijms20246193
Chicago/Turabian StyleStampanoni Bassi, Mario, Ennio Iezzi, Luana Gilio, Diego Centonze, and Fabio Buttari. 2019. "Synaptic Plasticity Shapes Brain Connectivity: Implications for Network Topology" International Journal of Molecular Sciences 20, no. 24: 6193. https://doi.org/10.3390/ijms20246193
APA StyleStampanoni Bassi, M., Iezzi, E., Gilio, L., Centonze, D., & Buttari, F. (2019). Synaptic Plasticity Shapes Brain Connectivity: Implications for Network Topology. International Journal of Molecular Sciences, 20(24), 6193. https://doi.org/10.3390/ijms20246193