More Than One HMG-CoA Lyase: The Classical Mitochondrial Enzyme Plus the Peroxisomal and the Cytosolic Ones


 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
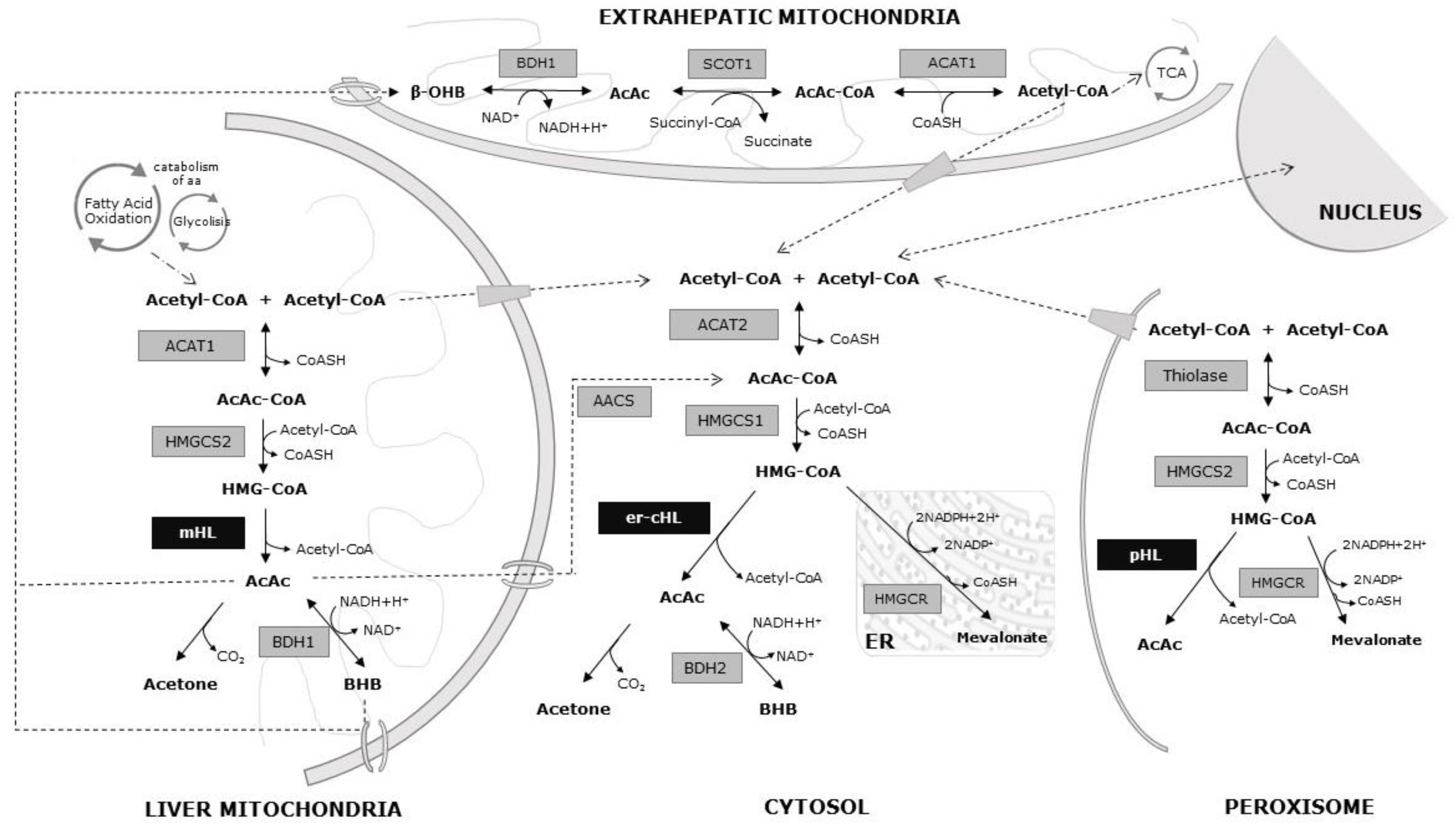
2. Ketone Body Metabolism
3. Roles of Ketone Bodies: More Than Energy Fuel
3.1. Metabolic Remodeling
3.2. Physiological Homeostasis
3.3. Signaling
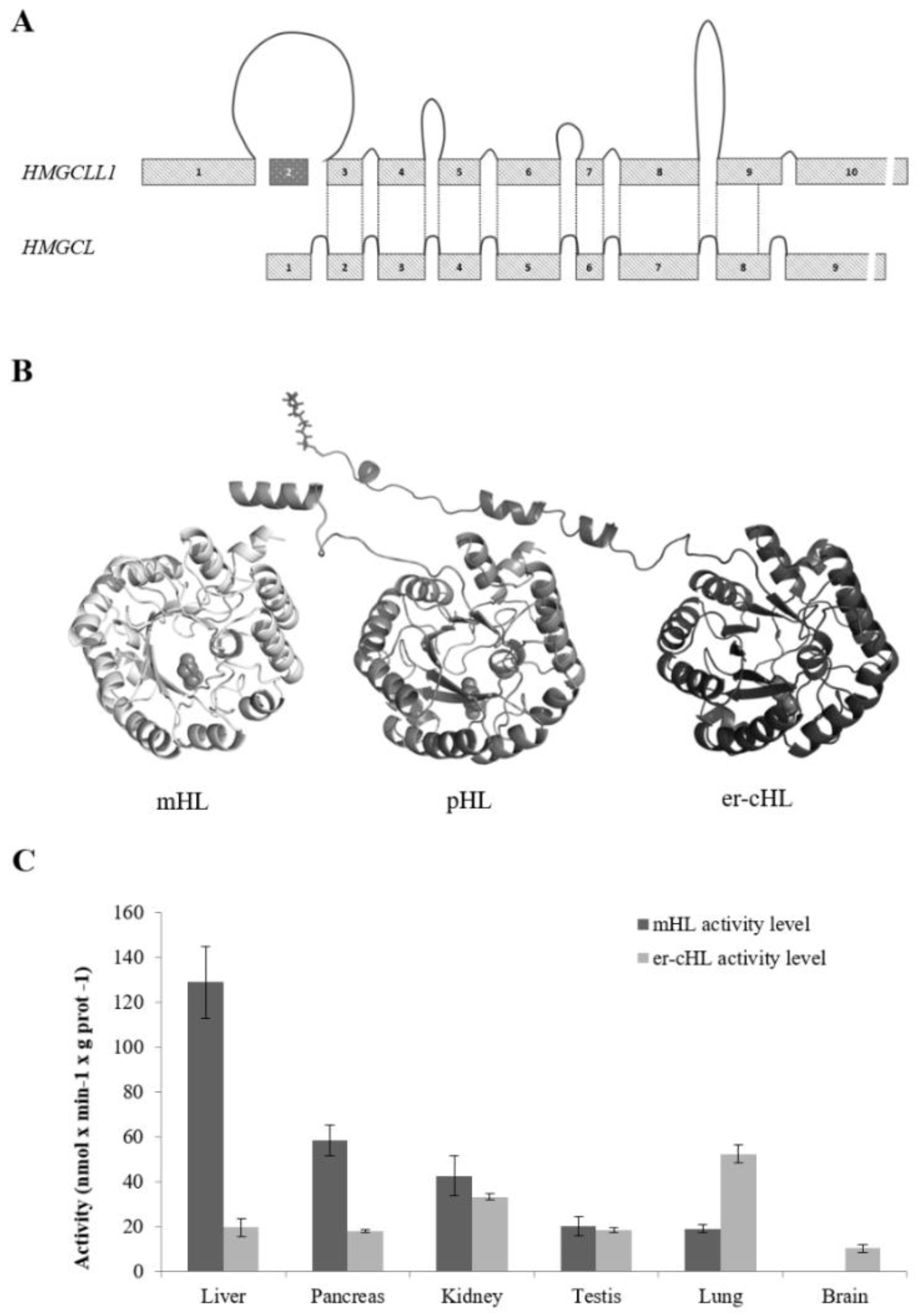
4. Gene Structure and Protein Features of HMG-CoA Lyases
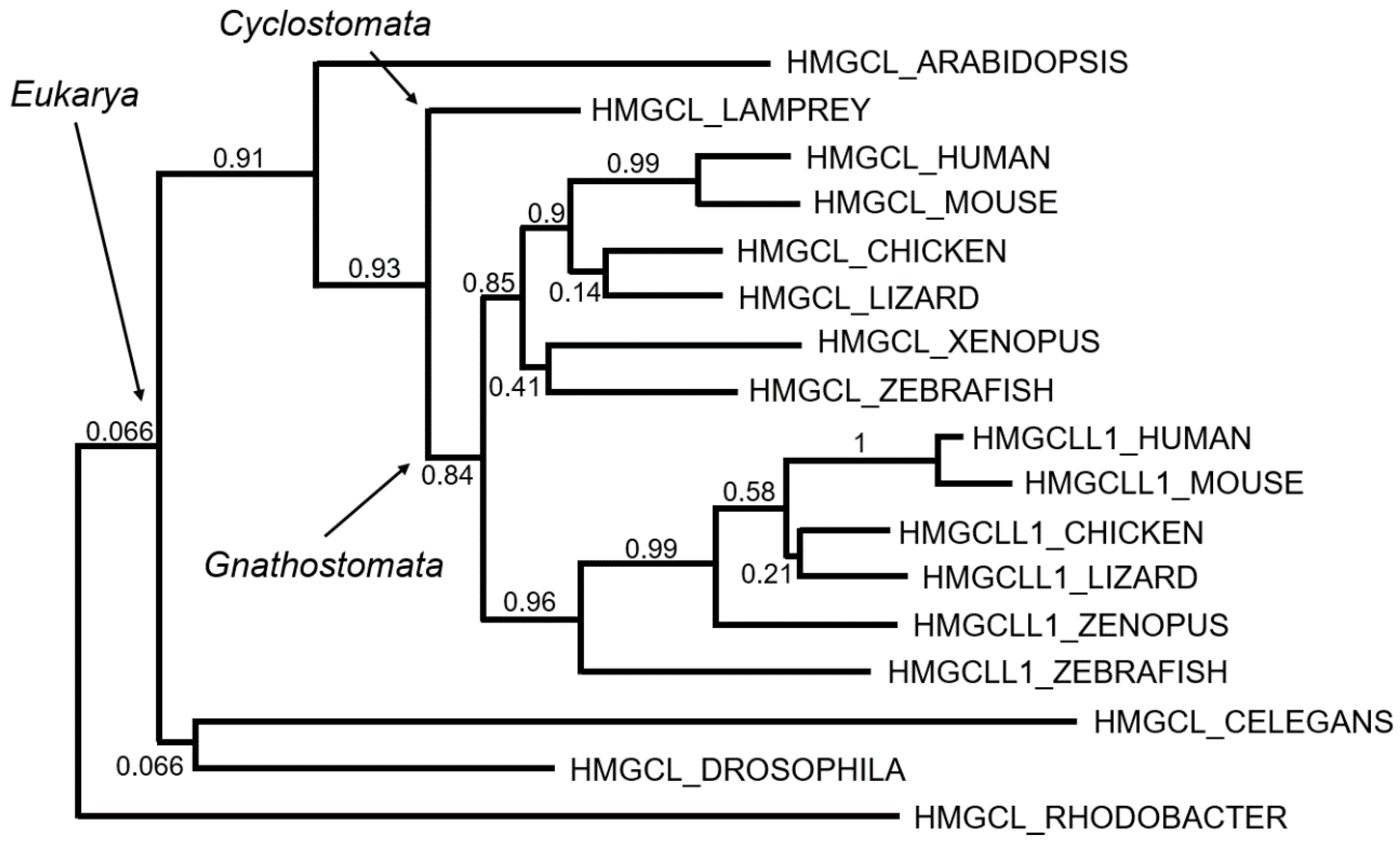
5. Phylogenetic Evolution of the HMG-CoA Lyases
6. Tissue-Specific Expression of HMG-CoA Lyases
7. HMG-CoA Lyase Related Diseases
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HMG-CoA | 3-Hydroxy-3-methylglutaryl coenzyme A |
| HL | HMG-CoA lyase |
| mHL | Mitochondrial HMG-CoA lyase |
| pHL | Peroxisomal HMG-CoA lyase |
| er-cHL | HMG-CoA lyase-like 1 |
| BHB | β-Hydroxybutyrate |
| AcAc | Acetoacetate |
| FAO | β-Oxidation |
| TCA | Tricarboxylic acid cycle |
| ACAT1 | Acetoacetyl-CoA thiolase |
| HMGCS1 | HMG-CoA synthetase 1 |
| HMGCS2 | HMG-CoA synthetase 2 |
| SCOT1 | 3-Oxoacid-CoA 1 |
| BDH1 | β-Hydroxybutyrate dehydrogenase |
| HMGCR | HMG-CoA reductase |
| ROS | Reactive oxygen species |
| HCA2 | Hydroxycarboxylic acid receptor 2 |
| Kbhb | β-Hydroxybutyrylation |
| HDAC | Class I histone deacetylase |
| CML | Chronic myeloid leukemia |
References
- Bachhawat, B.K.; Robinson, W.G.; Coon, M.J. The enzymatic cleavage of beta-hydroxy-beta-methylglutaryl coenzyme A to acetoacetate and acetyl coenzyme A. J. Biol. Chem. 1955, 216, 727–736. [Google Scholar] [PubMed]
- Fu, Z.; Runquist, J.A.; Forouhar, F.; Hussain, M.; Hunt, J.F.; Miziorko, H.M.; Kim, J.J. Crystal structure of human 3-hydroxy-3-methylglutaryl-CoA Lyase: Insights into catalysis and the molecular basis for hydroxymethylglutaric aciduria. J. Biol. Chem. 2006, 281, 7526–7532. [Google Scholar] [CrossRef] [PubMed]
- Forouhar, F.; Hussain, M.; Farid, R.; Benach, J.; Abashidze, M.; Edstrom, W.C.; Vorobiev, S.M.; Xiao, R.; Acton, T.B.; Fu, Z.; et al. Crystal structures of two bacterial 3-hydroxy-3-methylglutaryl-CoA lyases suggest a common catalytic mechanism among a family of TIM barrel metalloenzymes cleaving carbon-carbon bonds. J. Biol. Chem. 2006, 281, 7533–7545. [Google Scholar] [CrossRef] [PubMed]
- Hemmerlin, A.; Huchelmann, A.; Tritsch, D.; Schaller, H.; Bach, T.J. The specific molecular architecture of plant 3-hydroxy-3-methylglutaryl-CoA lyase. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Puisac, B.; Arnedo, M.; Casale, C.H.; Ribate, M.P.; Castiella, T.; Ramos, F.J.; Ribes, A.; Perez-Cerda, C.; Casals, N.; Hegardt, F.G.; et al. Differential HMG-CoA lyase expression in human tissues provides clues about 3-hydroxy-3-methylglutaric aciduria. J. Inherit. Metab. Dis. 2010, 33, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Arnedo, M.; Menao, S.; Puisac, B.; Teresa-Rodrigo, M.E.; Gil-Rodriguez, M.C.; Lopez-Vinas, E.; Gomez-Puertas, P.; Casals, N.; Casale, C.H.; Hegardt, F.G.; et al. Characterization of a novel HMG-CoA lyase enzyme with a dual location in endoplasmic reticulum and cytosol. J. Lipid Res. 2012, 53, 2046–2056. [Google Scholar] [CrossRef]
- Montgomery, C.; Pei, Z.; Watkins, P.A.; Miziorko, H.M. Identification and characterization of an extramitochondrial human 3-hydroxy-3-methylglutaryl-CoA lyase. J. Biol. Chem. 2012, 287, 33227–33236. [Google Scholar] [CrossRef]
- Ashmarina, L.I.; Rusnak, N.; Miziorko, H.M.; Mitchell, G.A. 3-Hydroxy-3-methylglutaryl-CoA lyase is present in mouse and human liver peroxisomes. J. Biol. Chem. 1994, 269, 31929–31932. [Google Scholar]
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Ashmarina, L.I.; Robert, M.F.; Elsliger, M.A.; Mitchell, G.A. Characterization of the hydroxymethylglutaryl-CoA lyase precursor, a protein targeted to peroxisomes and mitochondria. Biochem. J. 1996, 315 (Pt 1), 71–75. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. beta-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Puisac, B.; Ramos, M.; Arnedo, M.; Menao, S.; Gil-Rodriguez, M.C.; Teresa-Rodrigo, M.E.; Pie, A.; de Karam, J.C.; Wesselink, J.J.; Gimenez, I.; et al. Characterization of splice variants of the genes encoding human mitochondrial HMG-CoA lyase and HMG-CoA synthase, the main enzymes of the ketogenesis pathway. Mol. Biol. Rep. 2012, 39, 4777–4785. [Google Scholar] [CrossRef] [PubMed]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A. Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 265–275. [Google Scholar] [CrossRef]
- Morris, A.A. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005, 28, 109–121. [Google Scholar] [CrossRef]
- Bedi, K.C., Jr.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Kruger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. Jci Insight 2019, 4. [Google Scholar] [CrossRef]
- Ho, K.L.; Zhang, L.; Wagg, C.; Al Batran, R.; Gopal, K.; Levasseur, J.; Leone, T.; Dyck, J.R.B.; Ussher, J.R.; Muoio, D.M.; et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 2019, 115, 1606–1616. [Google Scholar] [CrossRef]
- Abdurrachim, D.; Woo, C.C.; Teo, X.Q.; Chan, W.X.; Radda, G.K.; Lee, P.T.H. A new hyperpolarized (13)C ketone body probe reveals an increase in acetoacetate utilization in the diabetic rat heart. Sci. Rep. 2019, 9, 5532. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.; Canfield, J.; Copes, N.; Rehan, M.; Lipps, D.; Bradshaw, P.C. D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging 2014, 6, 621–644. [Google Scholar] [CrossRef] [PubMed]
- Julio-Amilpas, A.; Montiel, T.; Soto-Tinoco, E.; Geronimo-Olvera, C.; Massieu, L. Protection of hypoglycemia-induced neuronal death by beta-hydroxybutyrate involves the preservation of energy levels and decreased production of reactive oxygen species. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2015, 35, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.R.; Ribeiro, L.C.; Hagenn, M.; Siqueira, I.R.; Araujo, E.; Torres, I.L.; Gottfried, C.; Netto, C.A.; Goncalves, C.A. Ketogenic diet increases glutathione peroxidase activity in rat hippocampus. Neurochem. Res. 2003, 28, 1793–1797. [Google Scholar] [CrossRef]
- Achanta, L.B.; Rae, C.D. beta-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef]
- Steriade, C.; Andrade, D.M.; Faghfoury, H.; Tarnopolsky, M.A.; Tai, P. Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) may respond to adjunctive ketogenic diet. Pediatr. Neurol. 2014, 50, 498–502. [Google Scholar] [CrossRef]
- Frey, S.; Geffroy, G.; Desquiret-Dumas, V.; Gueguen, N.; Bris, C.; Belal, S.; Amati-Bonneau, P.; Chevrollier, A.; Barth, M.; Henrion, D.; et al. The addition of ketone bodies alleviates mitochondrial dysfunction by restoring complex I assembly in a MELAS cellular model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 284–291. [Google Scholar] [CrossRef]
- Ahmed, K.; Tunaru, S.; Offermanns, S. GPR109A, GPR109B and GPR81, a family of hydroxy-carboxylic acid receptors. Trends Pharmacol. Sci. 2009, 30, 557–562. [Google Scholar] [CrossRef]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Muller-Fielitz, H.; Pokorna, B.; Vollbrandt, T.; Stolting, I.; Nadrowitz, R.; et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef]
- Yang, H.; Shan, W.; Zhu, F.; Wu, J.; Wang, Q. Ketone Bodies in Neurological Diseases: Focus on Neuroprotection and Underlying Mechanisms. Front. Neurol. 2019, 10, 585. [Google Scholar] [CrossRef]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Board, M.; Lopez, C.; van den Bos, C.; Callaghan, R.; Clarke, K.; Carr, C. Acetoacetate is a more efficient energy-yielding substrate for human mesenchymal stem cells than glucose and generates fewer reactive oxygen species. Int. J. Biochem. Cell Biol. 2017, 88, 75–83. [Google Scholar] [CrossRef]
- Han, Y.M.; Bedarida, T.; Ding, Y.; Somba, B.K.; Lu, Q.; Wang, Q.; Song, P.; Zou, M.H. beta-Hydroxybutyrate Prevents Vascular Senescence through hnRNP A1-Mediated Upregulation of Oct4. Mol. Cell 2018, 71, 1064–1078 e1065. [Google Scholar] [CrossRef]
- Puchalska, P.; Martin, S.E.; Huang, X.; Lengfeld, J.E.; Daniel, B.; Graham, M.J.; Han, X.; Nagy, L.; Patti, G.J.; Crawford, P.A. Hepatocyte-Macrophage Acetoacetate Shuttle Protects against Tissue Fibrosis. Cell Metab. 2019, 29, 383–398. [Google Scholar] [CrossRef]
- Cheng, C.W.; Biton, M.; Haber, A.L.; Gunduz, N.; Eng, G.; Gaynor, L.T.; Tripathi, S.; Calibasi-Kocal, G.; Rickelt, S.; Butty, V.L.; et al. Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell 2019, 178, 1115–1131. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine beta-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef]
- Liu, K.; Li, F.; Sun, Q.; Lin, N.; Han, H.; You, K.; Tian, F.; Mao, Z.; Li, T.; Tong, T.; et al. p53 beta-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019, 10, 243. [Google Scholar] [CrossRef]
- Ruan, H.B.; Crawford, P.A. Ketone bodies as epigenetic modifiers. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 260–266. [Google Scholar] [CrossRef]
- Jeffares, D.C.; Penkett, C.J.; Bahler, J. Rapidly regulated genes are intron poor. Trends Genet. 2008, 24, 375–378. [Google Scholar] [CrossRef]
- Seoighe, C.; Korir, P.K. Evidence for intron length conservation in a set of mammalian genes associated with embryonic development. BMC Bioinform. 2011, 12, S16. [Google Scholar] [CrossRef]
- Casals, N.; Gomez-Puertas, P.; Pie, J.; Mir, C.; Roca, R.; Puisac, B.; Aledo, R.; Clotet, J.; Menao, S.; Serra, D.; et al. Structural (betaalpha)8 TIM barrel model of 3-hydroxy-3-methylglutaryl-coenzyme A lyase. J. Biol. Chem. 2003, 278, 29016–29023. [Google Scholar] [CrossRef]
- Tuinstra, R.L.; Burgner, J.W., II; Miziorko, H.M. Investigation of the oligomeric status of the peroxisomal isoform of human 3-hydroxy-3-methylglutaryl-CoA lyase. Arch. Biochem. Biophys. 2002, 408, 286–294. [Google Scholar] [CrossRef]
- Tuinstra, R.L.; Miziorko, H.M. Investigation of conserved acidic residues in 3-hydroxy-3-methylglutaryl-CoA lyase: Implications for human disease and for functional roles in a family of related proteins. J. Biol. Chem. 2003, 278, 37092–37098. [Google Scholar] [CrossRef]
- Roberts, J.R.; Narasimhan, C.; Hruz, P.W.; Mitchell, G.A.; Miziorko, H.M. 3-Hydroxy-3-methylglutaryl-CoA lyase: Expression and isolation of the recombinant human enzyme and investigation of a mechanism for regulation of enzyme activity. J. Biol. Chem. 1994, 269, 17841–17846. [Google Scholar]
- Menao, S.; Lopez-Vinas, E.; Mir, C.; Puisac, B.; Gratacos, E.; Arnedo, M.; Carrasco, P.; Moreno, S.; Ramos, M.; Gil, M.C.; et al. Ten novel HMGCL mutations in 24 patients of different origin with 3-hydroxy-3-methyl-glutaric aciduria. Hum. Mutat. 2009, 30, E520–E529. [Google Scholar] [CrossRef]
- Montgomery, C.; Miziorko, H.M. Influence of multiple cysteines on human 3-hydroxy-3-methylglutaryl-CoA lyase activity and formation of inter-subunit adducts. Arch. Biochem. Biophys. 2011, 511, 48–55. [Google Scholar] [CrossRef]
- Wanders, R.J.; Schutgens, R.B.; Zoeters, P.H. 3-Hydroxy-3-methylglutaryl-CoA lyase in human skin fibroblasts: Study of its properties and deficient activity in 3-hydroxy-3-methylglutaric aciduria patients using a simple spectrophotometric method. Clin. Chim. Acta 1988, 171, 95–101. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Lang, D.; Thoma, R.; Henn-Sax, M.; Sterner, R.; Wilmanns, M. Structural evidence for evolution of the beta/alpha barrel scaffold by gene duplication and fusion. Science 2000, 289, 1546–1550. [Google Scholar] [CrossRef]
- Smith, J.J.; Keinath, M.C. The sea lamprey meiotic map improves resolution of ancient vertebrate genome duplications. Genome Res. 2015, 25, 1081–1090. [Google Scholar] [CrossRef]
- Smith, J.J.; Timoshevskaya, N.; Ye, C.; Holt, C.; Keinath, M.C.; Parker, H.J.; Cook, M.E.; Hess, J.E.; Narum, S.R.; Lamanna, F.; et al. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat. Genet. 2018, 50, 270–277. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Cardoso-Moreira, M.; Halbert, J.; Valloton, D.; Velten, B.; Chen, C.; Shao, Y.; Liechti, A.; Ascencao, K.; Rummel, C.; Ovchinnikova, S.; et al. Gene expression across mammalian organ development. Nature 2019, 571, 505–509. [Google Scholar] [CrossRef]
- Available online: https://gtexportal.org (accessed on 31 October 2019).
- Gibson, K.M.; Breuer, J.; Nyhan, W.L. 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: Review of 18 reported patients. Eur. J. Pediatr. 1988, 148, 180–186. [Google Scholar] [CrossRef]
- Grunert, S.C.; Schlatter, S.M.; Schmitt, R.N.; Gemperle-Britschgi, C.; Mrazova, L.; Balci, M.C.; Bischof, F.; Coker, M.; Das, A.M.; Demirkol, M.; et al. 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: Clinical presentation and outcome in a series of 37 patients. Mol. Genet. Metab. 2017, 121, 206–215. [Google Scholar] [CrossRef]
- Ozand, P.T.; Devol, E.B.; Gascon, G.G. Neurometabolic diseases at a national referral center: Five years experience at the King Faisal Specialist Hospital and Research Centre. J. Child. Neurol. 1992, 7, S4–S11. [Google Scholar] [CrossRef]
- Bolhuis, P.A.; Page-Christiaens, G.C. The advisory report ‘Neonatal screening’ from the Health Council of The Netherlands. Ned. Tijdschr Geneeskd 2005, 149, 2857–2860. [Google Scholar]
- Vargas, C.R.; Sitta, A.; Schmitt, G.; Ferreira, G.C.; Cardoso, M.L.; Coelho, D.; Gibson, K.M.; Wajner, M. Incidence of 3-hydroxy-3-methylglutaryl-coenzyme A lyase (HL) deficiency in Brazil, South America. J. Inherit. Metab. Dis. 2008, 31, 511–515. [Google Scholar] [CrossRef]
- Bischof, F.; Nagele, T.; Wanders, R.J.; Trefz, F.K.; Melms, A. 3-hydroxy-3-methylglutaryl-CoA lyase deficiency in an adult with leukoencephalopathy. Ann. Neurol. 2004, 56, 727–730. [Google Scholar] [CrossRef]
- Reimao, S.; Morgado, C.; Almeida, I.T.; Silva, M.; Corte Real, H.; Campos, J. 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: Initial presentation in a young adult. J. Inherit. Metab. Dis. 2009, 32, S49–S52. [Google Scholar] [CrossRef]
- Fernandes, C.G.; Rodrigues, M.D.N.; Seminotti, B.; Colin-Gonzalez, A.L.; Santamaria, A.; Quincozes-Santos, A.; Wajner, M. Induction of a Proinflammatory Response in Cortical Astrocytes by the Major Metabolites Accumulating in HMG-CoA Lyase Deficiency: The Role of ERK Signaling Pathway in Cytokine Release. Mol. Neurobiol. 2015, 53, 3586–3595. [Google Scholar] [CrossRef]
- Santosa, D.; Donner, M.G.; Vom Dahl, S.; Fleisch, M.; Hoehn, T.; Mayatepek, E.; Heldt, K.; Niehues, T.; Haussinger, D. Favourable Outcome in Two Pregnancies in a Patient with 3-Hydroxy-3-Methylglutaryl-CoA Lyase Deficiency. JIMD Rep. 2017, 37, 1–5. [Google Scholar]
- Pie, J.; Lopez-Vinas, E.; Puisac, B.; Menao, S.; Pie, A.; Casale, C.; Ramos, F.J.; Hegardt, F.G.; Gomez-Puertas, P.; Casals, N. Molecular genetics of HMG-CoA lyase deficiency. Mol. Genet. Metab. 2007, 92, 198–209. [Google Scholar] [CrossRef]
- Buesa, C.; Pie, J.; Barcelo, A.; Casals, N.; Mascaro, C.; Casale, C.H.; Haro, D.; Duran, M.; Smeitink, J.A.; Hegardt, F.G. Aberrantly spliced mRNAs of the 3-hydroxy-3-methylglutaryl coenzyme A lyase (HL) gene with a donor splice-site point mutation produce hereditary HL deficiency. J. Lipid Res. 1996, 37, 2420–2432. [Google Scholar]
- Casale, C.H.; Casals, N.; Pie, J.; Zapater, N.; Perez-Cerda, C.; Merinero, B.; Martinez-Pardo, M.; Garcia-Penas, J.J.; Garcia-Gonzalez, J.M.; Lama, R.; et al. A nonsense mutation in the exon 2 of the 3-hydroxy-3-methylglutaryl coenzyme A lyase (HL) gene producing three mature mRNAs is the main cause of 3-hydroxy-3-methylglutaric aciduria in European Mediterranean patients. Arch. Biochem. Biophys. 1998, 349, 129–137. [Google Scholar] [CrossRef]
- Casals, N.; Pie, J.; Casale, C.H.; Zapater, N.; Ribes, A.; Castro-Gago, M.; Rodriguez-Segade, S.; Wanders, R.J.; Hegardt, F.G. A two-base deletion in exon 6 of the 3-hydroxy-3-methylglutaryl coenzyme A lyase (HL) gene producing the skipping of exons 5 and 6 determines 3-hydroxy-3-methylglutaric aciduria. J. Lipid Res. 1997, 38, 2303–2313. [Google Scholar]
- Puisac, B.; Lopez-Vinas, E.; Moreno, S.; Mir, C.; Perez-Cerda, C.; Menao, S.; Lluch, D.; Pie, A.; Gomez-Puertas, P.; Casals, N.; et al. Skipping of exon 2 and exons 2 plus 3 of HMG-CoA lyase (HL) gene produces the loss of beta sheets 1 and 2 in the recently proposed (beta-alpha)8 TIM barrel model of HL. Biophys. Chem. 2005, 115, 241–245. [Google Scholar] [CrossRef]
- Li, M.; Sun, Q.; Wang, X. Transcriptional landscape of human cancers. Oncotarget 2017, 8, 34534–34551. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Kang, H.B.; Fan, J.; Lin, R.; Elf, S.; Ji, Q.; Zhao, L.; Jin, L.; Seo, J.H.; Shan, C.; Arbiser, J.L.; et al. Metabolic Rewiring by Oncogenic BRAF V600E Links Ketogenesis Pathway to BRAF-MEK1 Signaling. Mol. Cell 2015, 59, 345–358. [Google Scholar] [CrossRef]
- Luo, W.; Qin, L.; Li, B.; Liao, Z.; Liang, J.; Xiao, X.; Xiao, X.; Mo, Y.; Huang, G.; Zhang, Z.; et al. Inactivation of HMGCL promotes proliferation and metastasis of nasopharyngeal carcinoma by suppressing oxidative stress. Sci. Rep. 2017, 7, 11954. [Google Scholar] [CrossRef]
- Park, J.H.; Woo, Y.M.; Youm, E.M.; Hamad, N.; Won, H.H.; Naka, K.; Park, E.J.; Park, J.H.; Kim, H.J.; Kim, S.H.; et al. HMGCLL1 is a predictive biomarker for deep molecular response to imatinib therapy in chronic myeloid leukemia. Leukemia 2019, 33, 1439–1450. [Google Scholar] [CrossRef]
- Fang, Q.; Benedetti, A.F.; Ma, Q.; Gregory, L.; Li, J.Z.; Dattani, M.; Sadeghi-Nejad, A.; Arnhold, I.J.; Mendonca, B.B.; Camper, S.A.; et al. HESX1 mutations in patients with congenital hypopituitarism: Variable phenotypes with the same genotype. Clin. Endocrinol. 2016, 85, 408–414. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnedo, M.; Latorre-Pellicer, A.; Lucia-Campos, C.; Gil-Salvador, M.; Antoñanzas-Peréz, R.; Gómez-Puertas, P.; Bueno-Lozano, G.; Puisac, B.; Pié, J. More Than One HMG-CoA Lyase: The Classical Mitochondrial Enzyme Plus the Peroxisomal and the Cytosolic Ones. Int. J. Mol. Sci. 2019, 20, 6124. https://doi.org/10.3390/ijms20246124
Arnedo M, Latorre-Pellicer A, Lucia-Campos C, Gil-Salvador M, Antoñanzas-Peréz R, Gómez-Puertas P, Bueno-Lozano G, Puisac B, Pié J. More Than One HMG-CoA Lyase: The Classical Mitochondrial Enzyme Plus the Peroxisomal and the Cytosolic Ones. International Journal of Molecular Sciences. 2019; 20(24):6124. https://doi.org/10.3390/ijms20246124
Chicago/Turabian StyleArnedo, María, Ana Latorre-Pellicer, Cristina Lucia-Campos, Marta Gil-Salvador, Rebeca Antoñanzas-Peréz, Paulino Gómez-Puertas, Gloria Bueno-Lozano, Beatriz Puisac, and Juan Pié. 2019. "More Than One HMG-CoA Lyase: The Classical Mitochondrial Enzyme Plus the Peroxisomal and the Cytosolic Ones" International Journal of Molecular Sciences 20, no. 24: 6124. https://doi.org/10.3390/ijms20246124
APA StyleArnedo, M., Latorre-Pellicer, A., Lucia-Campos, C., Gil-Salvador, M., Antoñanzas-Peréz, R., Gómez-Puertas, P., Bueno-Lozano, G., Puisac, B., & Pié, J. (2019). More Than One HMG-CoA Lyase: The Classical Mitochondrial Enzyme Plus the Peroxisomal and the Cytosolic Ones. International Journal of Molecular Sciences, 20(24), 6124. https://doi.org/10.3390/ijms20246124