Vanadium Derivative Exposure Promotes Functional Alterations of VSMCs and Consequent Atherosclerosis via ROS/p38/NF-κB-Mediated IL-6 Production

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
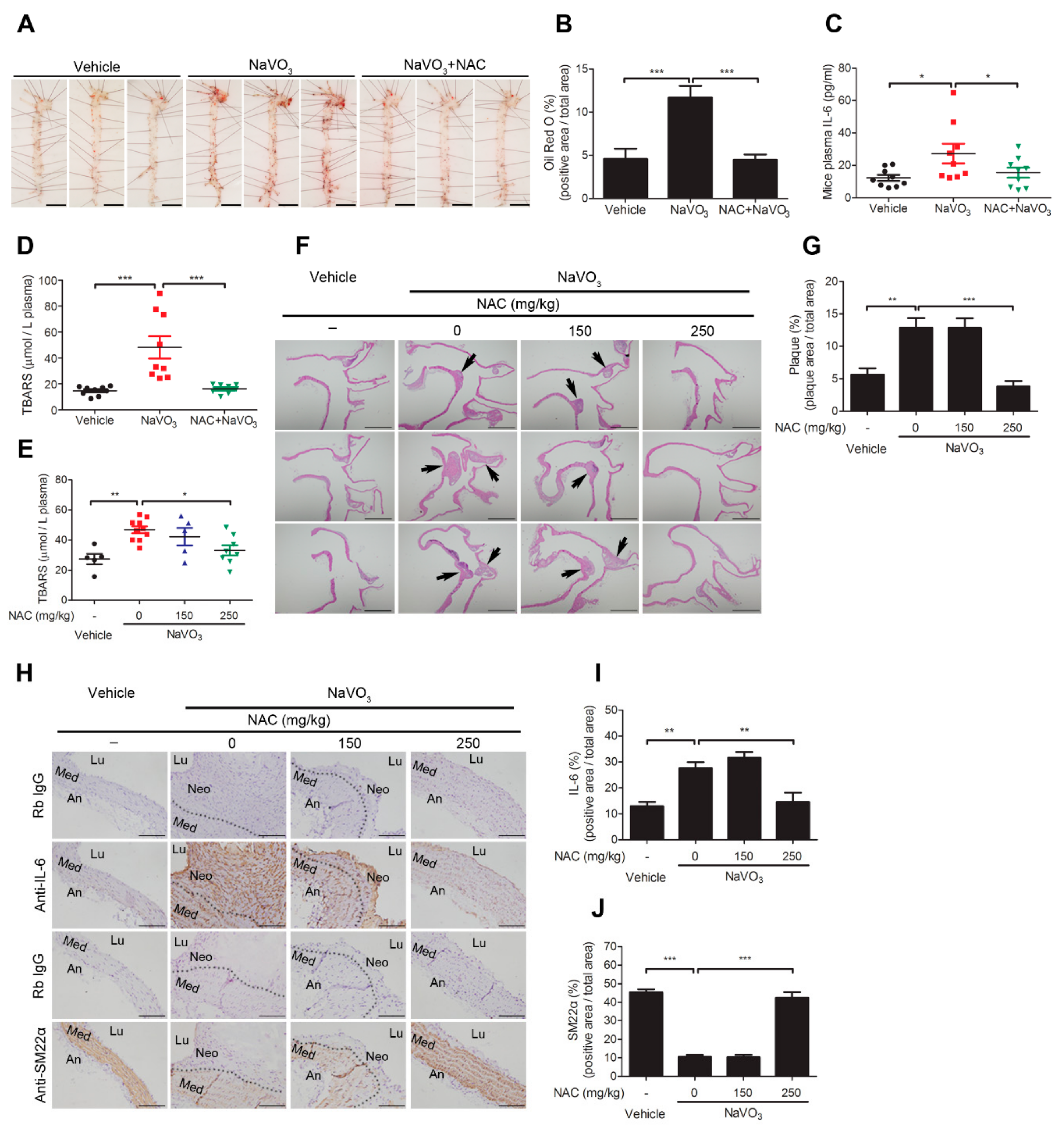
2.1. Intranasal Administration of NaVO3 Induces Atherosclerosis in ApoE−/− Mice
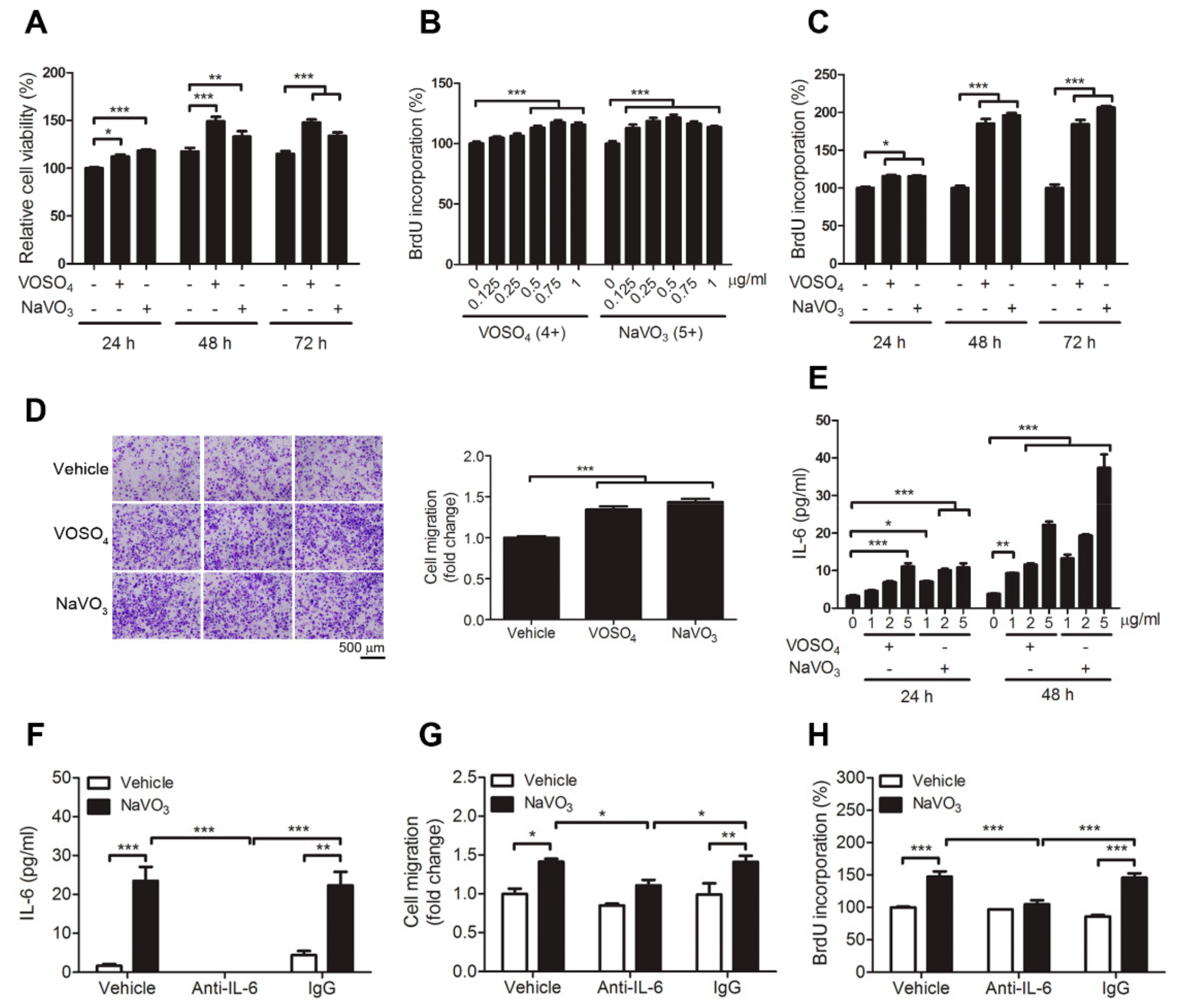
2.2. VOSO4 and NaVO3 Promote Pathophysiology of VSMC In Vitro
2.3. IL-6 Is Essential for VOSO4- and NaVO3-Induced VSMC Proliferation and Migration
2.4. ROS-Mediated IL-6 Is Essential for NaVO3-Mediated VSMC Functions
2.5. Involvement of Signaling Kinases in NaVO3-Mediated VSMC Migration
2.6. Anti-Oxidant N-Acetylcysteine Prevents NaVO3-Induced Atherosclerosis in ApoE−/− Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of NaVO3 and VOSO4 Solutions
4.3. Animals
4.4. Induction of Atherosclerosis in a Mouse Model
4.5. Cell Culture and Treatment
4.6. Measurement of Vanadium Levels in Urine and Plasma
4.7. TBARS Assay
4.8. Oil Red O Staining
4.9. Histology and Immunohistochemistry
4.10. Western Blot Analysis
4.11. Cell Viability and Cell Proliferation Assays
4.12. Migration Assays
4.13. IL-6 ELISA
4.14. Cellular ROS and Mitochondria ROS Assay
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| VSMC | Vascular smooth muscle cell |
| NAC | N-acetylcysteine |
| ROS | Reactive oxygen species |
| FBS | Fetal bovine serum (FBS) |
References
- Anderson, J.O.; Thundiyil, J.G.; Stolbach, A. Clearing the air: A review of the effects of particulate matter air pollution on human health. J. Med. Toxicol. 2012, 8, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G.; Lippmann, M. Ambient particulate matter air pollution and cardiopulmonary diseases. Semin. Respir. Crit. Care Med. 2015, 36, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Kelly, F.J.; Fussell, J.C. Linking ambient particulate matter pollution effects with oxidative biology and immune responses. Ann. N. Y. Acad. Sci. 2015, 1340, 84–94. [Google Scholar] [CrossRef]
- Kelly, F.J.; Fussell, J.C. Role of oxidative stress in cardiovascular disease outcomes following exposure to ambient air pollution. Free Radic. Biol. Med. 2017, 110, 345–367. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Mutlu, G.M. Particulate Matter Air Pollution: Effects on the Cardiovascular System. Front. Endocrinol. 2018, 9, 680. [Google Scholar] [CrossRef]
- Kelly, F.J.; Fussell, J.C. Size, source and chemical composition as determinants of toxicity attributable to ambient particulate matter. Atmos. Environ. 2012, 60, 504–526. [Google Scholar] [CrossRef]
- Crans, D.C.; Smee, J.J.; Gaidamauskas, E.; Yang, L. The chemistry and biochemistry of vanadium and the biological activities exerted by vanadium compounds. Chem. Rev. 2004, 104, 849–902. [Google Scholar] [CrossRef]
- Fortoul, T.I.; Rodriguez-Lara, V.; Gonzalez-Villalva, A.; Rojas-Lemus, M.; Cano-Gutierrez, G.; Ustarroz-Cano, M.; Colin-Barenque, L.; Montano, L.F.; Garcia-Pelez, I.; Bizarro-Nevares, P.; et al. Vanadium inhalation in a mouse model for the understanding of air-suspended particle systemic repercussion. J. Biomed. Biotechnol. 2011, 2011, 951043. [Google Scholar] [CrossRef]
- Fortoul, T.I.; Rodriguez-Lara, V.; Gonzalez-Villalva, A.; Rojas-Lemus, M.; Cano-Gutierrez, G.; Ustarroz-Cano, M.; Colin-Barenque, L.; Bizarro-Nevares, P.; Garcia-Pealez, I.; Montano, L.F.; et al. Inhalation of vanadium pentoxide and its toxic effects in a mouse model. Inorg. Chim. Acta 2014, 420, 8–15. [Google Scholar] [CrossRef]
- Espinosa-Zurutuza, M.; Gonzalez-Villalva, A.; Albarran-Alonso, J.C.; Colin-Barenque, L.; Bizarro-Nevares, P.; Rojas-Lemus, M.; Lopez-Valdez, N.; Fortoul, T.I. Oxidative Stress as a Mechanism Involved in Kidney Damage After Subchronic Exposure to Vanadium Inhalation and Oral Sweetened Beverages in a Mouse Model. Int. J. Toxicol. 2018, 37, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.D.; Sisco, M.; Prophete, C.; Chen, L.C.; Zelikoff, J.T.; Ghio, A.J.; Stonehuerner, J.D.; Smee, J.J.; Holder, A.A.; Crans, D.C. Pulmonary immunotoxic potentials of metals are governed by select physicochemical properties: Vanadium agents. J. Immunotoxicol. 2007, 4, 49–60. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cohen, M.D.; Sisco, M.; Prophete, C.; Yoshida, K.; Chen, L.C.; Zelikoff, J.T.; Smee, J.; Holder, A.A.; Stonehuerner, J.; Crans, D.C.; et al. Effects of metal compounds with distinct physicochemical properties on iron homeostasis and antibacterial activity in the lungs: Chromium and vanadium. Inhal. Toxicol. 2010, 22, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Barceloux, D.G. Vanadium. J. Toxicol. Clin. Toxicol. 1999, 37, 265–278. [Google Scholar] [CrossRef]
- Willsky, G.R.; Chi, L.H.; Godzala, M., 3rd; Kostyniak, P.J.; Smee, J.J.; Trujillo, A.M.; Alfano, J.A.; Ding, W.; Hu, Z.; Crans, D.C. Anti-diabetic effects of a series of vanadium dipicolinate complexes in rats with streptozotocin-induced diabetes. Coord. Chem. Rev. 2011, 255, 2258–2269. [Google Scholar] [CrossRef]
- Selman, M.; Rousso, C.; Bergeron, A.; Son, H.H.; Krishnan, R.; El-Sayes, N.A.; Varette, O.; Chen, A.; Le Boeuf, F.; Tzelepis, F.; et al. Multi-modal Potentiation of Oncolytic Virotherapy by Vanadium Compounds. Mol. Ther. 2018, 26, 56–69. [Google Scholar] [CrossRef]
- Goulopoulou, S.; McCarthy, C.G.; Webb, R.C. Toll-like Receptors in the Vascular System: Sensing the Dangers Within. Pharmacol. Rev. 2016, 68, 142–167. [Google Scholar] [CrossRef]
- Wang, Y.F.; Hsu, Y.J.; Wu, H.F.; Lee, G.L.; Yang, Y.S.; Wu, J.Y.; Yet, S.F.; Wu, K.K.; Kuo, C.C. Endothelium-Derived 5-Methoxytryptophan Is a Circulating Anti-Inflammatory Molecule That Blocks Systemic Inflammation. Circ. Res. 2016, 119, 222–236. [Google Scholar] [CrossRef]
- Katakami, N. Mechanism of Development of Atherosclerosis and Cardiovascular Disease in Diabetes Mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef]
- Nowak, W.N.; Deng, J.; Ruan, X.Z.; Xu, Q. Reactive Oxygen Species Generation and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e41–e52. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ren, Z.; Zhang, J.; Chuang, C.C.; Kandaswamy, E.; Zhou, T.; Zuo, L. Role of ROS and Nutritional Antioxidants in Human Diseases. Front. Physiol. 2018, 9, 477. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Lichtman, A.H.; Hansson, G.K. Immune effector mechanisms implicated in atherosclerosis: From mice to humans. Immunity 2013, 38, 1092–1104. [Google Scholar] [CrossRef]
- Pfeiler, S.; Gerdes, N. Atherosclerosis: Cell biology and lipoproteins - focus on anti-inflammatory therapies. Curr. Opin. Lipidol. 2018, 29, 53–55. [Google Scholar] [CrossRef]
- Lee, G.L.; Chang, Y.W.; Wu, J.Y.; Wu, M.L.; Wu, K.K.; Yet, S.F.; Kuo, C.C. TLR 2 induces vascular smooth muscle cell migration through cAMP response element-binding protein-mediated interleukin-6 production. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2751–2760. [Google Scholar] [CrossRef]
- Lee, G.L.; Wu, J.Y.; Tsai, C.S.; Lin, C.Y.; Tsai, Y.T.; Lin, C.S.; Wang, Y.F.; Yet, S.F.; Hsu, Y.J.; Kuo, C.C. TLR4-Activated MAPK-IL-6 Axis Regulates Vascular Smooth Muscle Cell Function. Int. J. Mol. Sci. 2016, 17, 1394. [Google Scholar] [CrossRef]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jorgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef]
- Lee, G.L.; Yeh, C.C.; Wu, J.Y.; Lin, H.C.; Wang, Y.F.; Kuo, Y.Y.; Hsieh, Y.T.; Hsu, Y.J.; Kuo, C.C. TLR2 Promotes Vascular Smooth Muscle Cell Chondrogenic Differentiation and Consequent Calcification via the Concerted Actions of Osteoprotegerin Suppression and IL-6-Mediated RANKL Induction. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 432–445. [Google Scholar] [CrossRef]
- Garcia-Redondo, A.B.; Aguado, A.; Briones, A.M.; Salaices, M. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharmacol. Res. 2016, 114, 110–120. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, S.; Tapping, R.I. Toll-like receptor signaling in cell proliferation and survival. Cytokine 2010, 49, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hime, N.J.; Marks, G.B.; Cowie, C.T. A Comparison of the Health Effects of Ambient Particulate Matter Air Pollution from Five Emission Sources. Int. J. Environ. Res. Public Health 2018, 15, 1206. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.L.; Lehmann, J.R.; Winsett, D.; Richards, J.; Ledbetter, A.D.; Dreher, K.L. Comparative pulmonary toxicological assessment of oil combustion particles following inhalation or instillation exposure. Toxicol. Sci. 2006, 91, 237–246. [Google Scholar] [CrossRef]
- Beyersmann, D.; Hartwig, A. Carcinogenic metal compounds: Recent insight into molecular and cellular mechanisms. Arch. Toxicol. 2008, 82, 493–512. [Google Scholar] [CrossRef]
- Kelly, F.J.; Fuller, G.W.; Walton, H.A.; Fussell, J.C. Monitoring air pollution: Use of early warning systems for public health. Respirology 2012, 17, 7–19. [Google Scholar] [CrossRef]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006, 71, 216–225. [Google Scholar] [CrossRef]
- Chava, K.R.; Karpurapu, M.; Wang, D.; Bhanoori, M.; Kundumani-Sridharan, V.; Zhang, Q.; Ichiki, T.; Glasgow, W.C.; Rao, G.N. CREB-mediated IL-6 expression is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 809–815. [Google Scholar] [CrossRef]
- Lee, G.L.; Wu, J.Y.; Yeh, C.C.; Kuo, C.C. TLR4 induces CREB-mediated IL-6 production via upregulation of F-spondin to promote vascular smooth muscle cell migration. Biochem. Biophys. Res. Commun. 2016, 473, 1205–1210. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Ghio, A.J.; Silbajoris, R.; Carson, J.L.; Samet, J.M. Biologic effects of oil fly ash. Environ. Health Perspect. 2002, 110 (Suppl. 1), 89–94. [Google Scholar] [CrossRef] [PubMed]
- Orecchio, S.; Amorello, D.; Barreca, S.; Pettignano, A. Speciation of vanadium in urban, industrial and volcanic soils by a modified Tessier method. Environ. Sci. Process. Impacts 2016, 18, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.B.; MacGregor, J.A.; Ehman, K.D.; Nikiforov, A.I. Vanadium pentoxide: Use of relevant historical control data shows no evidence for a carcinogenic response in F344/N rats. Regul. Toxicol. Pharm. 2012, 64, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.M.; Alessi, D.S.; Tack, F.M.G.; Ok, Y.S.; Kim, K.H.; Gustafsson, J.P.; Sparks, D.L.; Rinklebe, J. Redox chemistry of vanadium in soils and sediments: Interactions with colloidal materials, mobilization, speciation, and relevant environmental implications—A review. Adv. Colloid Interface Sci. 2019, 265, 1–13. [Google Scholar] [CrossRef]
- Daugherty, A.; Tall, A.R.; Daemen, M.; Falk, E.; Fisher, E.A.; Garcia-Cardena, G.; Lusis, A.J.; Owens, A.P., 3rd; Rosenfeld, M.E.; Virmani, R.; et al. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e131–e157. [Google Scholar] [CrossRef]
- Wu, J.Y.; Kuo, C.C. ADP-Ribosylation Factor 3 Mediates Cytidine-Phosphate-Guanosine Oligodeoxynucleotide-Induced Responses by Regulating Toll-Like Receptor 9 Trafficking. J. Innate Immun. 2015, 7, 623–636. [Google Scholar] [CrossRef]
- Wu, J.Y.; Kuo, C.C. Pivotal role of ADP-ribosylation factor 6 in Toll-like receptor 9-mediated immune signaling. J. Biol. Chem. 2012, 287, 4323–4334. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, C.-C.; Wu, J.-Y.; Lee, G.-L.; Wen, H.-T.; Lin, P.; Kuo, C.-C. Vanadium Derivative Exposure Promotes Functional Alterations of VSMCs and Consequent Atherosclerosis via ROS/p38/NF-κB-Mediated IL-6 Production. Int. J. Mol. Sci. 2019, 20, 6115. https://doi.org/10.3390/ijms20246115
Yeh C-C, Wu J-Y, Lee G-L, Wen H-T, Lin P, Kuo C-C. Vanadium Derivative Exposure Promotes Functional Alterations of VSMCs and Consequent Atherosclerosis via ROS/p38/NF-κB-Mediated IL-6 Production. International Journal of Molecular Sciences. 2019; 20(24):6115. https://doi.org/10.3390/ijms20246115
Chicago/Turabian StyleYeh, Chang-Ching, Jing-Yiing Wu, Guan-Lin Lee, Hsiu-Ting Wen, Pinpin Lin, and Cheng-Chin Kuo. 2019. "Vanadium Derivative Exposure Promotes Functional Alterations of VSMCs and Consequent Atherosclerosis via ROS/p38/NF-κB-Mediated IL-6 Production" International Journal of Molecular Sciences 20, no. 24: 6115. https://doi.org/10.3390/ijms20246115
APA StyleYeh, C.-C., Wu, J.-Y., Lee, G.-L., Wen, H.-T., Lin, P., & Kuo, C.-C. (2019). Vanadium Derivative Exposure Promotes Functional Alterations of VSMCs and Consequent Atherosclerosis via ROS/p38/NF-κB-Mediated IL-6 Production. International Journal of Molecular Sciences, 20(24), 6115. https://doi.org/10.3390/ijms20246115