Deciphering Genetic Architecture of Adventitious Root and Related Shoot Traits in Populus Using QTL Mapping and RNA-Seq Data

Abstract
:1. Introduction
2. Results
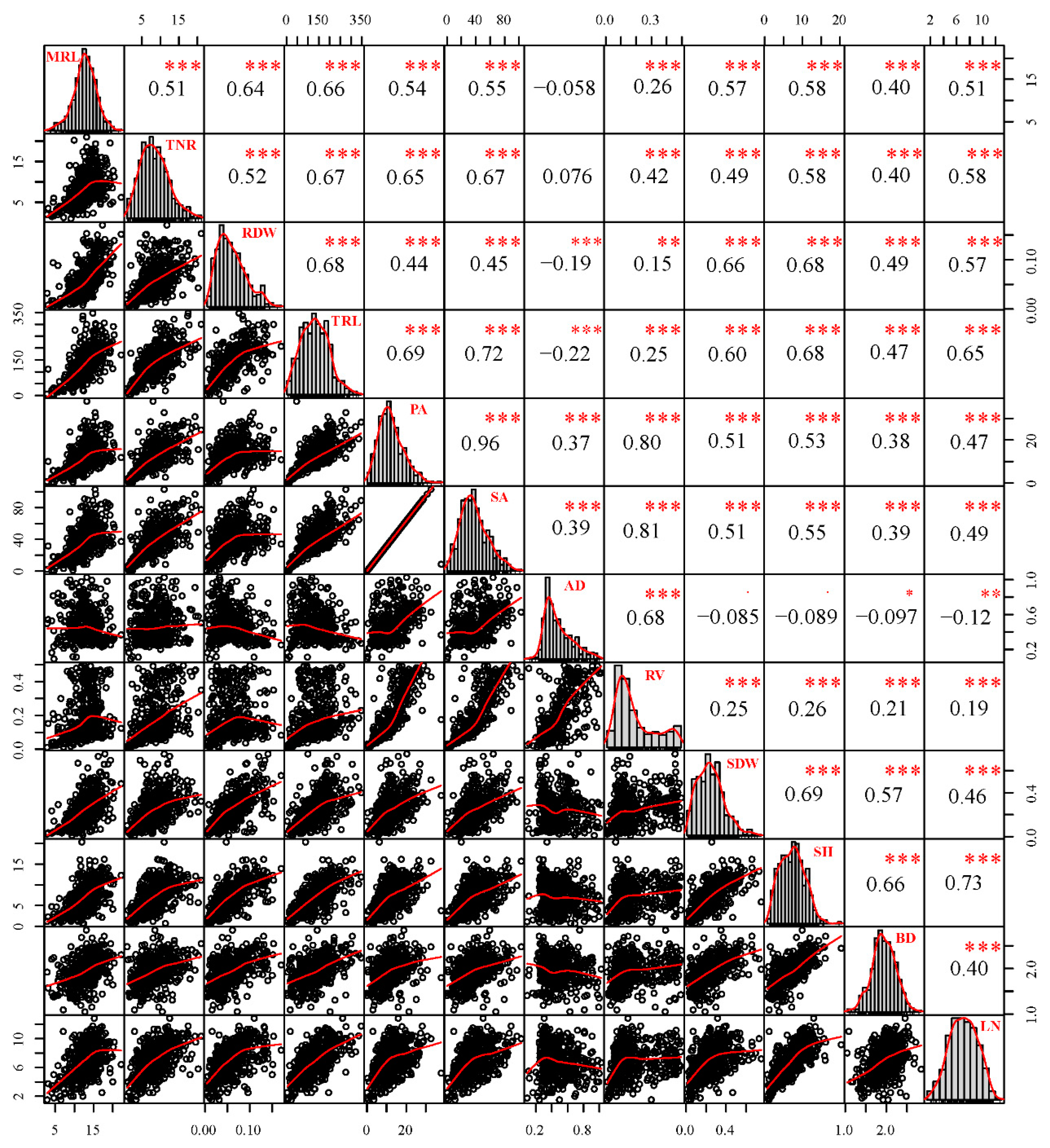
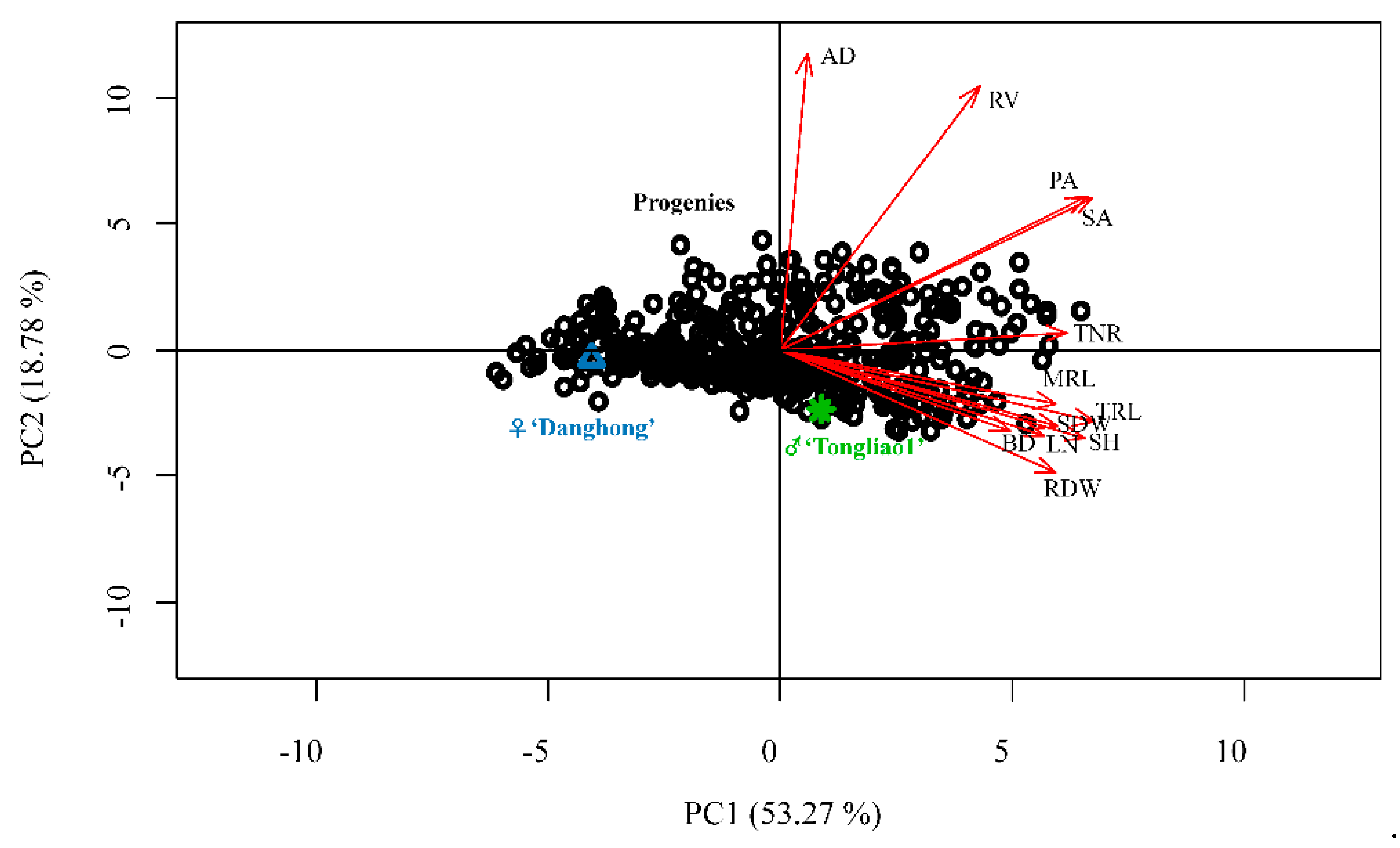
2.1. Analysis of Adventitious Root and Related Shoot Traits in the F1 Population
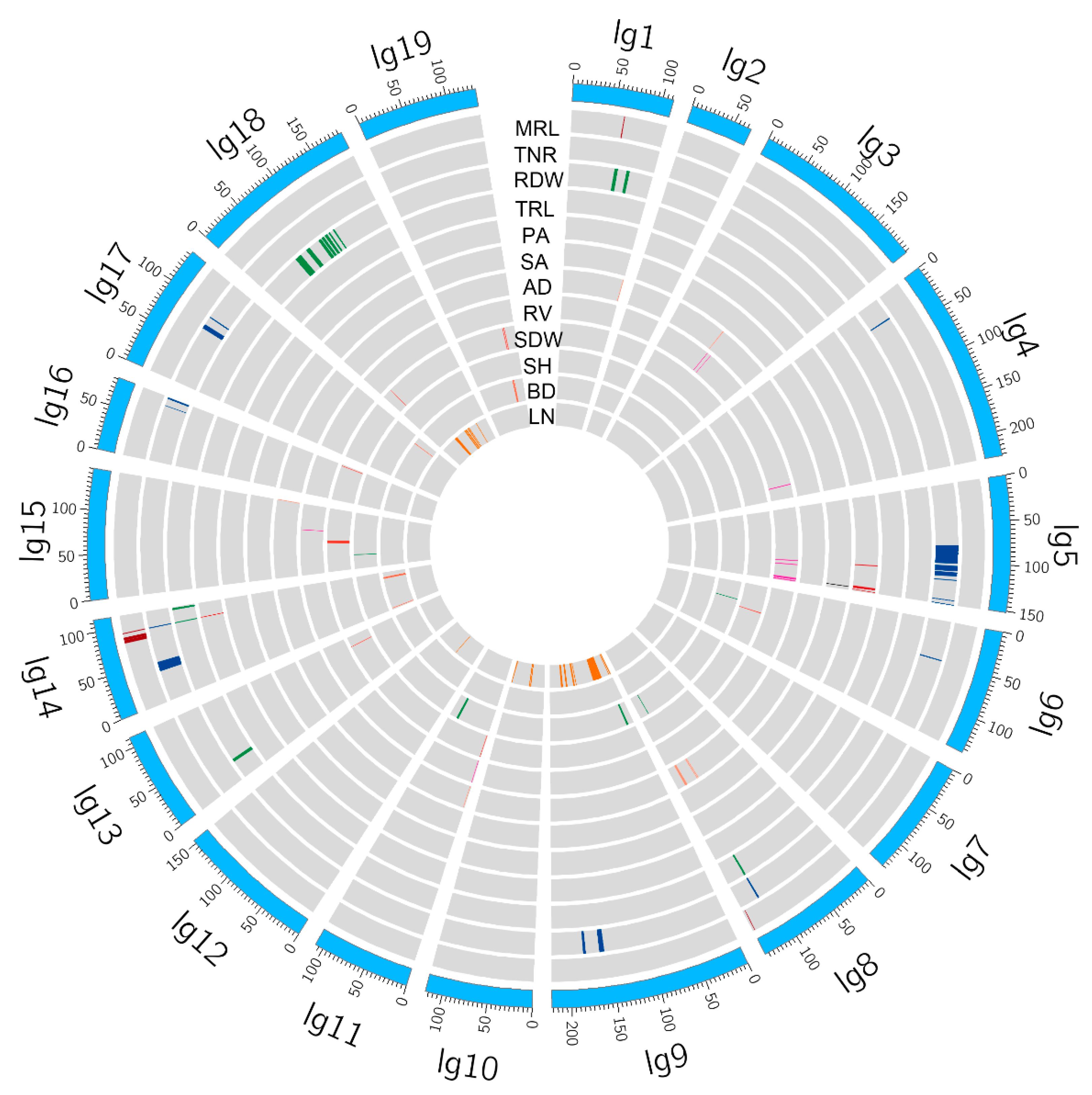
2.2. QTL Mapping and Candidate Gene Identification
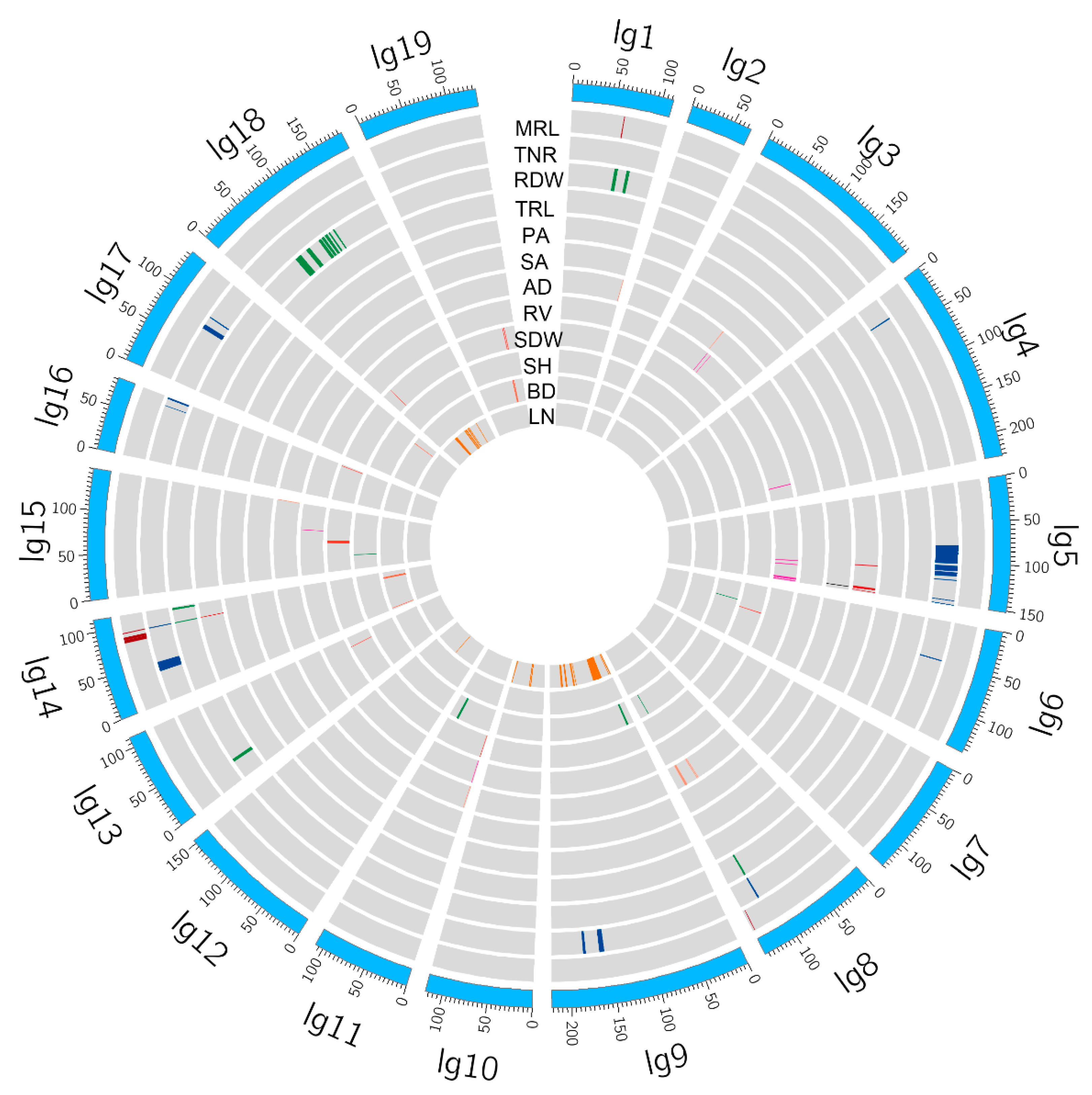
2.3. QTL Clusters and Hotspots
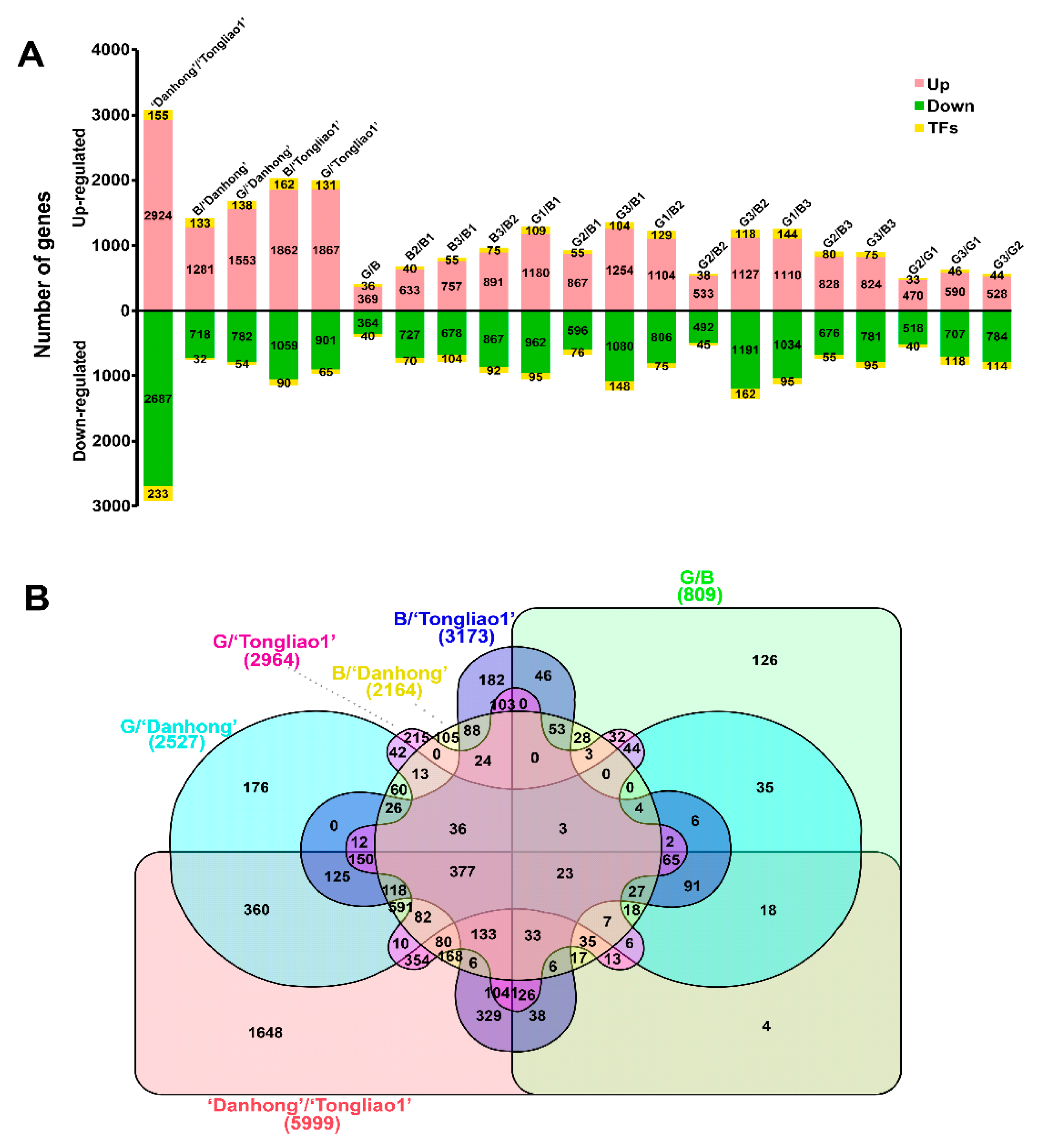
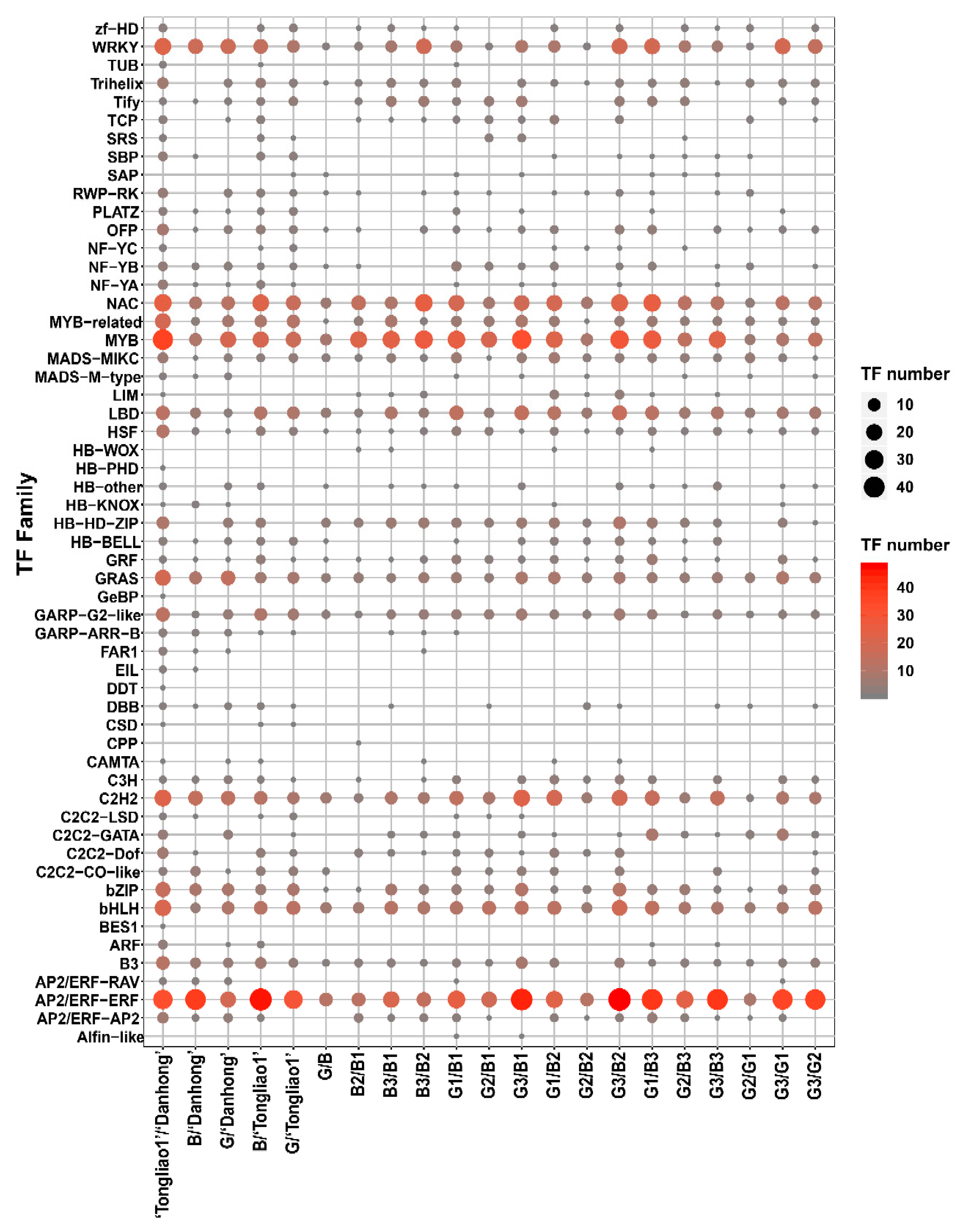
2.4. RNA-Seq Profiling
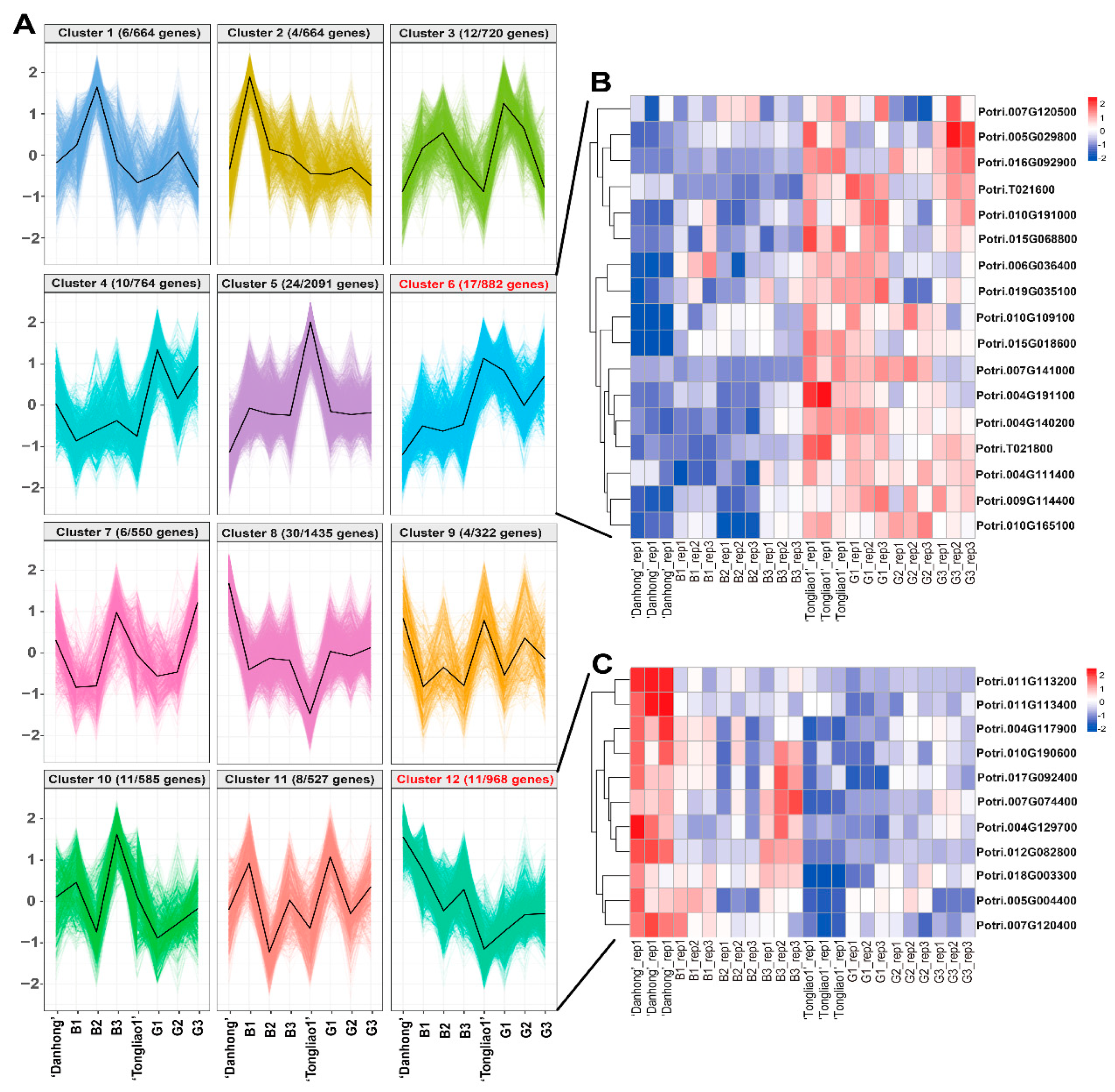
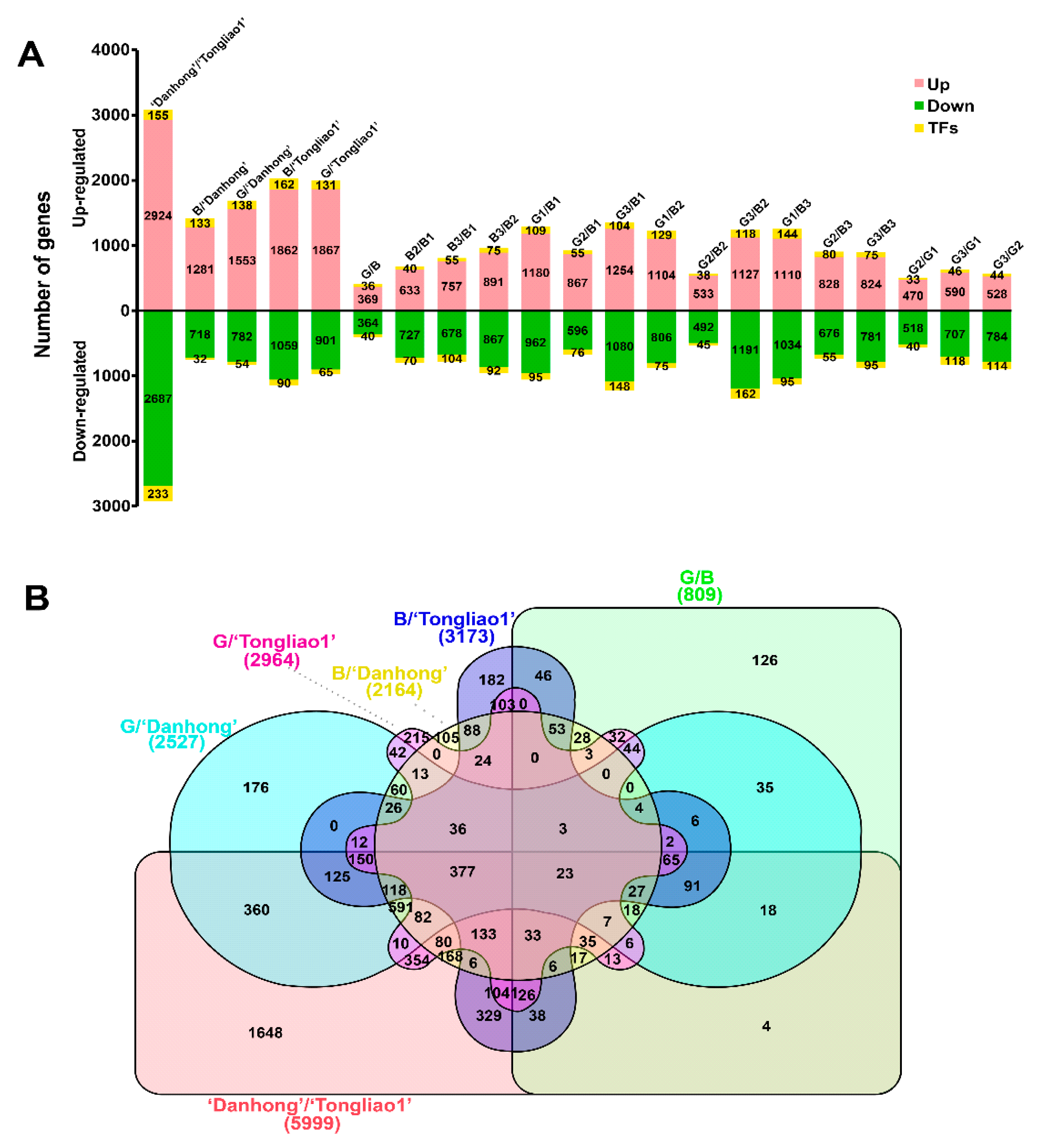
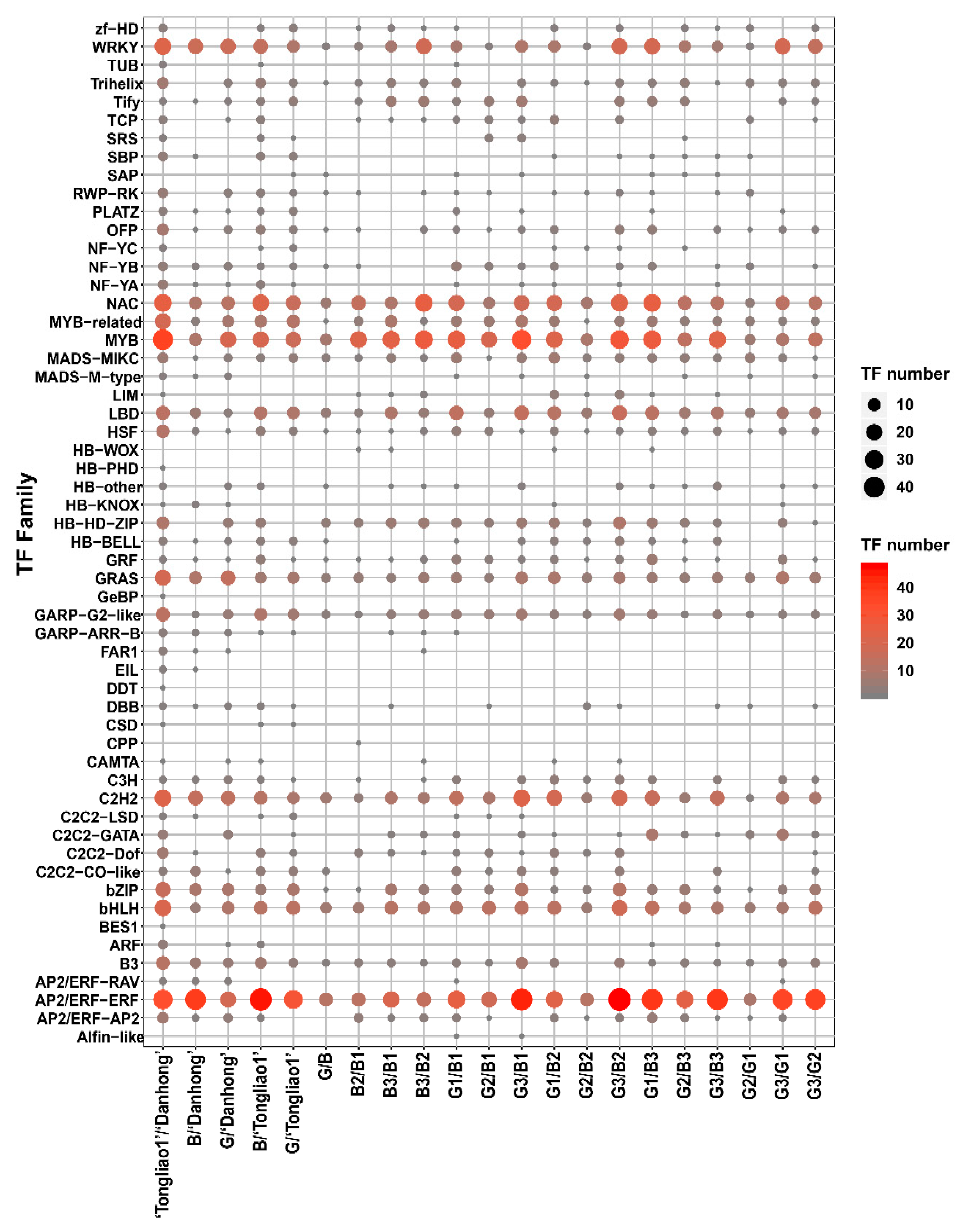
2.5. Differentially Expressed Gene (DEG) Analysis
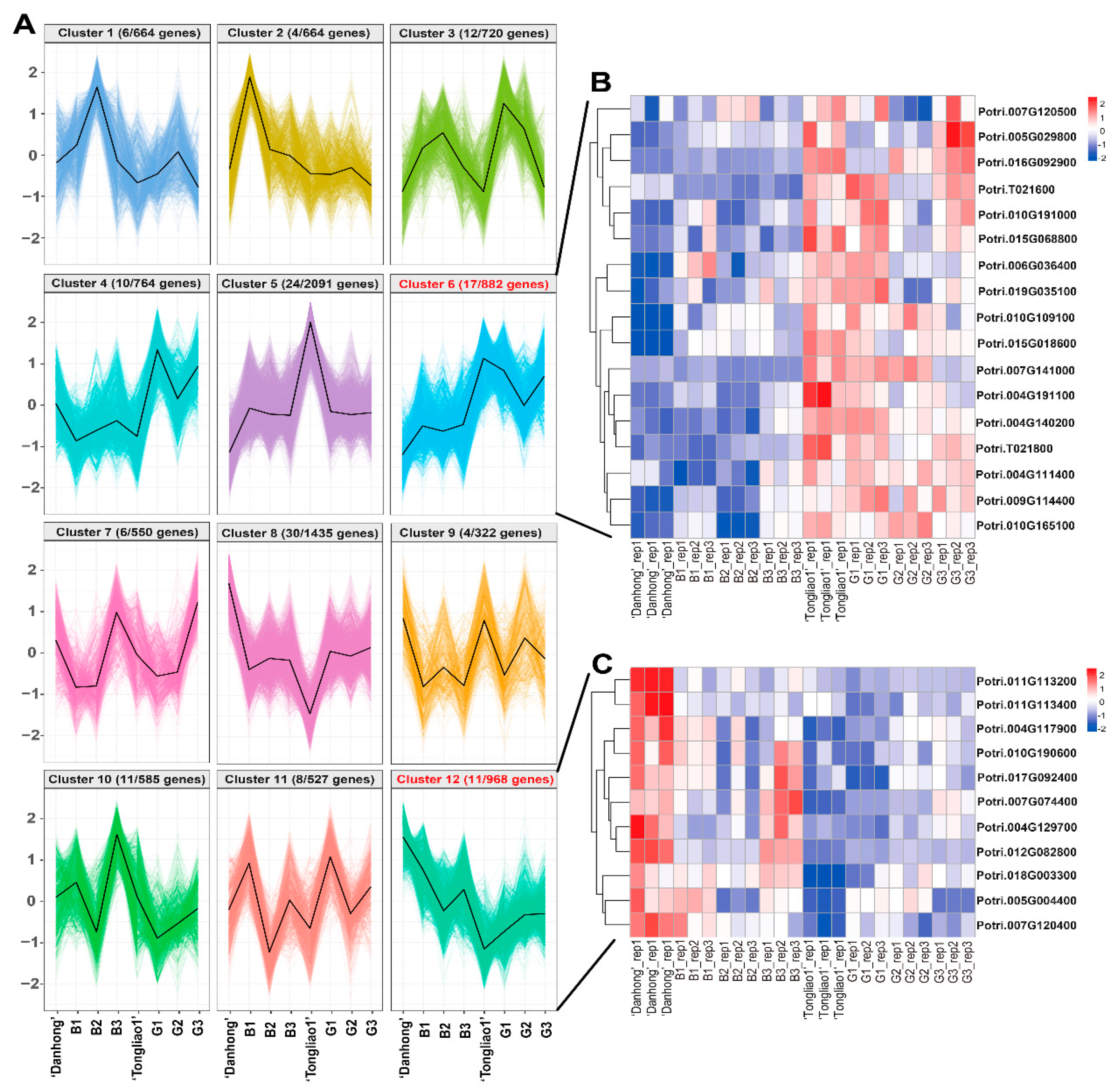
2.6. Clustering Analysis of DEGs in Different Genotypes
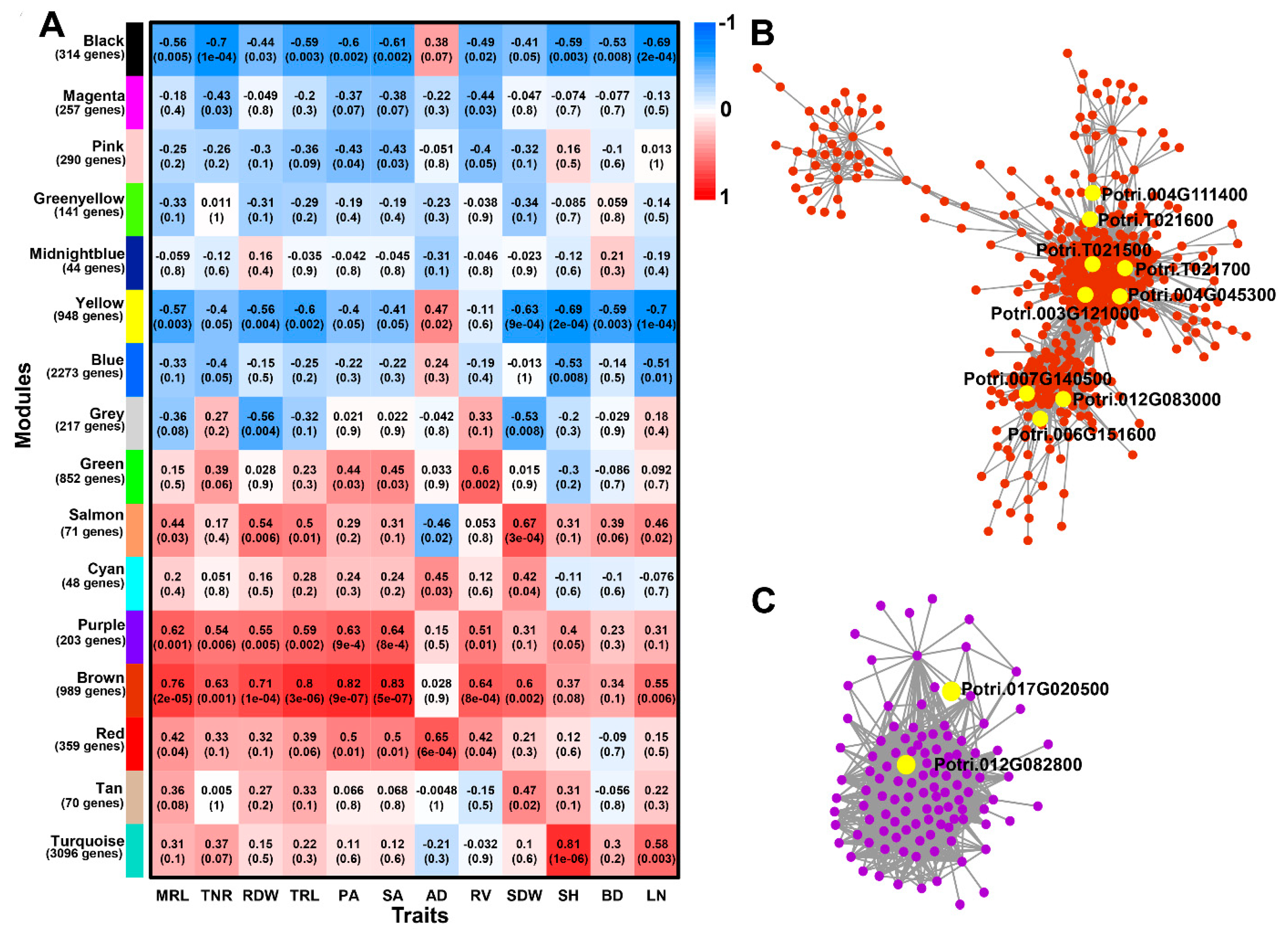
2.7. Identification of WGCNA Modules Associated with Phenotype Traits
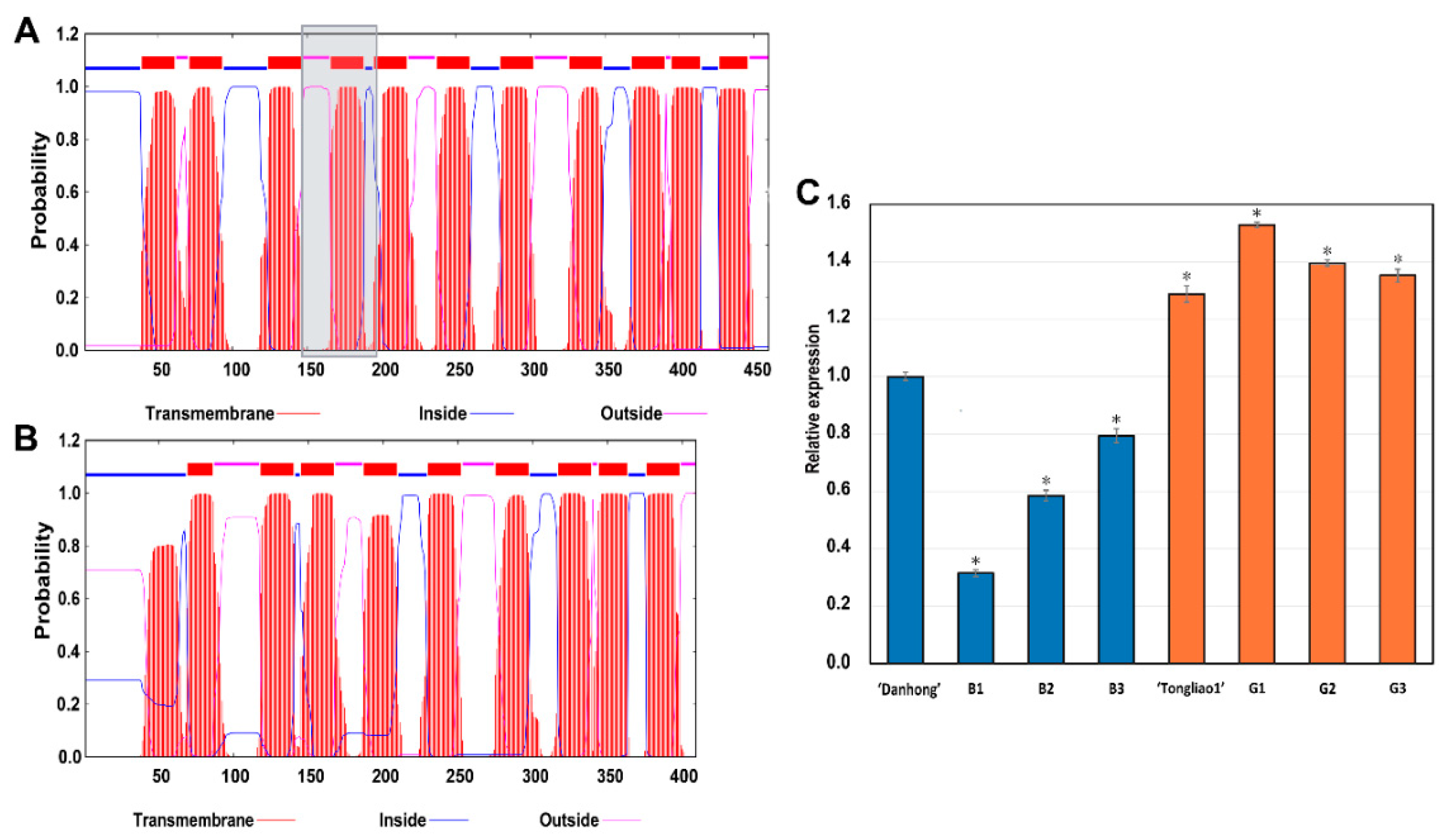
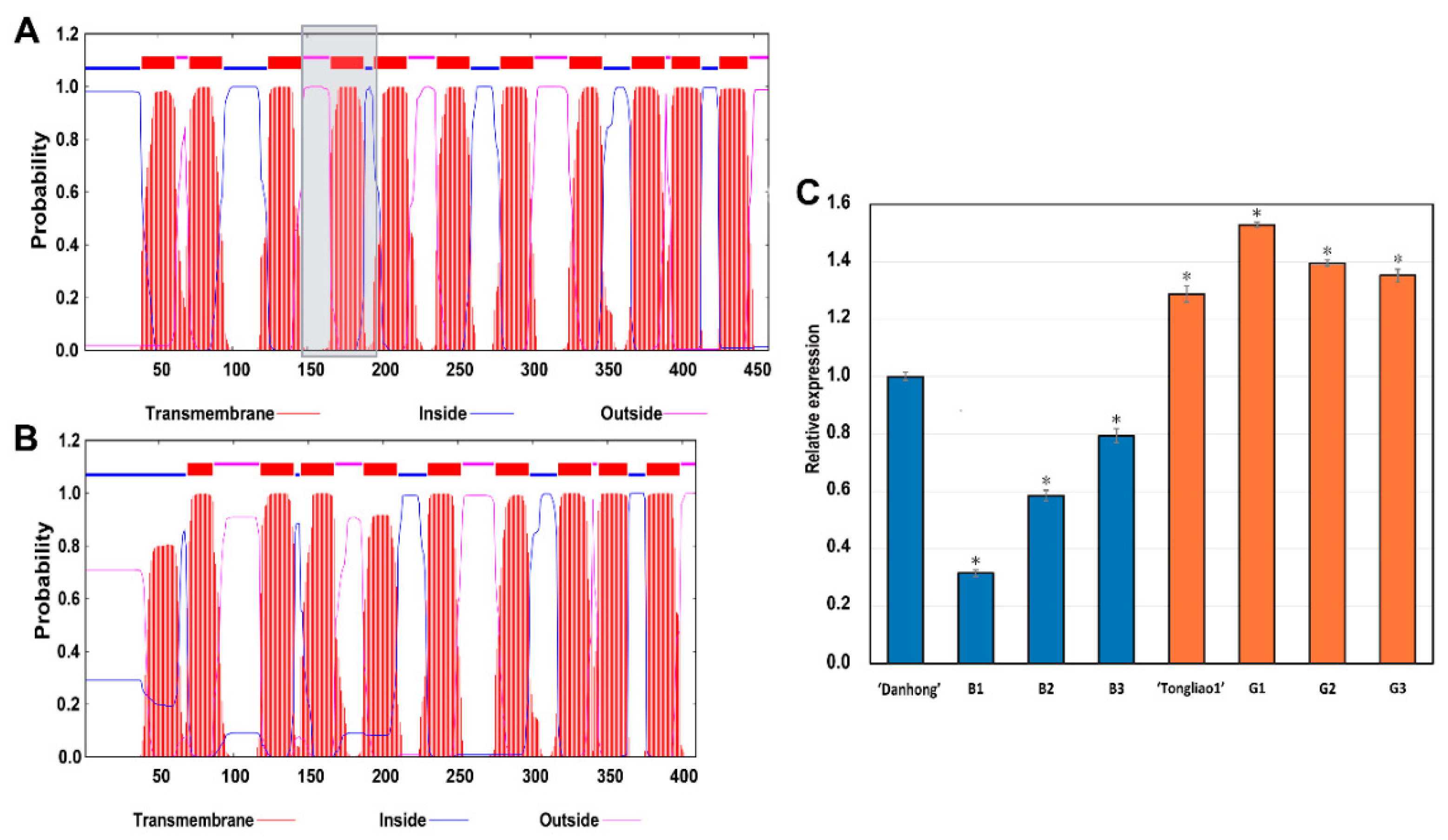
2.8. Genes Involved Potentially in Regulating Adventitious Root and Related Shoot Traits
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotypic Measurements
4.3. Statistical Analysis
4.4. QTL mapping and Candidate Gene Analysis
4.5. RNA Isolation, Library Construction, and Illumina Sequencing
4.6. Identification of Differentially Expressed Genes and K-Means Cluster Analysis
4.7. Weighted Gene Co-Expression Network Analysis
4.8. Quantitative Real-Time PCR
4.9. Gene Cloning and Protein Structure Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rae, A.M.; Tricker, P.J.; Bunn, S.M.; Taylor, G. Adaptation of tree growth to elevated CO2: Quantitative trait loci for biomass in Populus. New Phytol. 2010, 1, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouteillé, M.; Rolland, G.; Balsera, C.; Loudet, O.; Muller, B. Disentangling the intertwined genetic bases of root and shoot growth in Arabidopsis. PLoS ONE 2012, 7, e32319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reighard, G.L.; Hanover, J.W. Shoot and root development and dry matter partitioning in Populus grandidentata, P. tremuloides and P. simonii. Can. J. For. Res. 1990, 6, 849–852. [Google Scholar] [CrossRef]
- Poorter, H.; Nagel, O. The role of biomass allocation in the growth response of plants to different levels of light, CO2, nutrients and water: A quantitative review. Funct. Plant Biol. 2000, 27, 1191. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.R.; Song, C.H.; Zhang, Y.L.; Dang, H.S.; Cheng, X.L.; Zhang, Q.F. Global scale patterns of nutrient density and partitioning in forests in relation to climate. Glob. Chang. Boil. 2018, 1, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Geiss, G.; Gutierrez, L.; Bellini, C. Adventitious root formation: New insight and perspectives. Ann. Plant Rev. 2009, 37, 127–156. [Google Scholar]
- Tuskan, G.A. Short-rotation woody crop supply systems in the United States: What do we know and what do we need to know? Biomass Bioenergy 1998, 14, 307–315. [Google Scholar] [CrossRef]
- Gren, G.I.; Ingestad, T. Root: Shoot ratio as a balance between nitrogen productivity and photosynthesis. Plant Cell Environ. 1987, 7, 579–586. [Google Scholar]
- Heilman, P.E.; Ekuan, G.; Fogle, D. Above-and below-ground biomass and fine roots of 4-year-old hybrids of Populus trichocarpa × Populus deltoides and parental species in short-rotation culture. Can. J. For. Res. 1994, 6, 1186–1192. [Google Scholar] [CrossRef]
- Wan, X.C.; Landhausser, S.M.; Lieffers, V.J.; Zwiazek, J.J. Signals controlling root suckering and adventitious shoot formation in aspen (Populus tremuloides). Tree Physiol. 2006, 5, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Carvajal, G.A.; Morse, A.M.; Dervinis, C.; Davis, J.M. The cytokinin type-B response regulator PtRR13 is a negative regulator of adventitious root development in Populus. Plant Physiol. 2009, 2, 759–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trupiano, D.; Yordanov, Y.; Regan, S.; Meilan, R.; Tschaplinski, T.; Scippa, G.S.; Busov, V. Identification, characterization of an AP2/ERF transcription factor that promotes adventitious, lateral root formation in Populus. Planta 2013, 2, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Li, J.B.; Zhang, J.; Jia, H.X.; Liu, B.B.; Sun, P.; Hu, J.J.; Wang, L.J.; Lu, M.Z. The WUSCHEL-related homeobox 5a (PtoWOX5a) is involved in adventitious root development in poplar. Tree Physiol. 2018, 1, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Rae, A.M.; Street, N.R.; Robinson, K.M.; Harris, N.; Taylor, G. Five QTL hotspots for yield in short rotation coppice bioenergy poplar: The poplar biomass loci. BMC Plant Biol. 2009, 1, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Xu, S.Z.; Ehdaie, B.; Hoops, A.; Close, T.J.; Lukaszewski, A.J.; Waines, J.G. Dissection of QTL effects for root traits using a chromosome arm-specific mapping population in bread wheat. Theor. Appl. Genet. 2011, 122, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchadier, E.; Hanemian, M.; Tisné, S.; Bach, L.; Bazakos, C.; Gilbault, E.; Haddadi, P.; Virlouvet, L.; Loudet, O. The complex genetic architecture of shoot growth natural variation in Arabidopsis thaliana. PLoS Genet. 2019, 4, e1007954. [Google Scholar] [CrossRef] [Green Version]
- Dignat, G.; Welcker, C.; Sawkins, M.; Ribaut, J.M.; Tardieu, F. The growths of leaves, shoots, roots and reproductive organs partly share their genetic control in maize plants. Plant Cell Environ. 2013, 6, 1105–1119. [Google Scholar] [CrossRef]
- Canè, M.A.; Maccaferri, M.; Nazemi, G.; Salvi, S.; Colalongo, R.C.; Tuberosa, R. Association mapping for root architectural traits in durum wheat seedlings as related to agronomic performance. Mol. Breed. 2014, 4, 1629–1645. [Google Scholar] [CrossRef] [Green Version]
- Naz, A.A.; Arifuzzaman, M.; Muzammil, S.; Pillen, K.; Léon, J. Wild barley introgression lines revealed novel QTL alleles for root and related shoot traits in the cultivated barley (Hordeum vulgare L.). BMC Genet. 2014, 15, 107. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Saravia, D.; Munive, S.; Lozano, F.; Farfan, E.; Eyzaguirre, R.; Bonierbale, M. Multiple QTLs linked to agro-morphological and physiological traits related to drought tolerance in potato. Plant Mol. Biol. Rep. 2015, 33, 1286–1298. [Google Scholar] [CrossRef] [Green Version]
- El-Lithy, M.E.; Reymond, M.; Stich, B.M.; Koornneef, M.; Vreugdenhil, D. Relation among plant growth, carbohydrates and flowering time in the Arabidopsis Landsberg erecta × Kondara recombinant inbred line population. Plant Cell Environ. 2010, 33, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.D.; Yin, T.M.; Difazio, S.P.; Tschaplinski, T.J.; Gunter, L.E.; Davis, M.F.; Tuskan, G.A. Phenotypic variation in growth and biomass distribution for two advanced-generation pedigrees of hybrid poplar. Can. J. For. Res. 2005, 8, 1779–1789. [Google Scholar] [CrossRef]
- Novaes, E.; Osorio, L.; Drost, D.R.; Miles, B.L.; Boaventura-Novaes, C.R.D.; Benedict, C.; Dervinis, C.; Yu, Q.B.; Sykes, R.; Davis, M.; et al. Quantitative genetic analysis of biomass and wood chemistry of Populus under different nitrogen levels. New Phytol. 2009, 4, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Bo, W.H.; Xu, F.; Li, H.; Ye, M.X.; Jiang, L.B.; Shi, C.Z.; Fu, Y.R.; Zhang, G.M.; Huang, Y.J.; et al. The genetic architecture of shoot-root covariation during seedling emergence of a desert tree, Populus euphratica. Plant J. 2017, 5, 918–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickmann, D.; Hendrick, R. Modeling adventitious root system development in trees: Clonal poplars. In Biology of Adventitious Root Formation; Davis, T., Haissig, B., Eds.; Plenum Press: New York, NY, USA, 1994; pp. 203–218. [Google Scholar]
- Lin, H.X.; Qian, H.R.; Zhuang, J.Y.; Lu, J.; Min, S.K.; Xiong, Z.M.; Huang, N.; Zheng, K.L. RFLP mapping of QTLs for yield and related characters in rice (Oryza sativa L.). Theor. Appl. Genet. 1996, 92, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Dong, Y.T.; Zhao, T.L.; Li, L.; Li, C.; Yu, E.; Mei, L.; Daud, M.K.; He, Q.L.; Chen, J.H.; et al. Genome-wide SNP linkage mapping and QTL analysis for fiber quality and yield traits in the upland cotton recombinant inbred lines population. Front. Plant Sci. 2016, 7, 1356. [Google Scholar] [CrossRef] [PubMed]
- Said, J.I.; Lin, Z.X.; Zhang, X.L.; Song, M.Z.; Zhang, J.F. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genomics 2013, 14, 776. [Google Scholar] [CrossRef] [Green Version]
- Peltier, C.; Schmidlin, L.; Klein, E.; Taconnat, L.; Prinsen, E.; Erhardt, M.; Heintz, D.; Weyens, G.; Lefebvre, M.; Renou, J.P.; et al. Expression of the Beet necrotic yellow vein virus p25 protein induces hormonal changes and a root branching phenotype in Arabidopsis thaliana. Transgenic Res. 2011, 3, 443–466. [Google Scholar] [CrossRef]
- Sun, J.Q.; Xu, Y.X.; Ye, S.Q.; Jiang, H.L.; Chen, Q.; Liu, F.; Zhou, W.K.; Chen, R.; Li, X.G.; Tietz, O.; et al. Arabidopsis ASA1 is important for jasmonate-mediated regulation of auxin biosynthesis and transport during lateral root formation. Plant Cell. 2009, 21, 1495–1511. [Google Scholar] [CrossRef] [Green Version]
- Brenner, W.G.; Schmülling, T. Transcript profiling of cytokinin action in Arabidopsis roots and shoots discovers largely similar but also organ-specific responses. BMC Plant Biol. 2012, 1, 112. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Wu, S.; Chen, Z.; Dong, Q.; Yan, H.; Xiang, Y. Genome-wide survey and expression analysis of the amino acid transporter gene family in poplar. Tree Genet. Genomes 2015, 11, 83. [Google Scholar] [CrossRef]
- Lee, Y.H.; Foster, J.; Chen, J.; Voll, L.M.; Weber, A.P.M.; Tegeder, M. AAP1 transports uncharged amino acids into roots of Arabidopsis. Plant J. 2007, 50, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Enquist, B.J.; Niklas, K.J. Global allocation rules for patterns of biomass partitioning in seed plants. Science 2002, 5559, 1517–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauh, B.L.; Basten, C.; Buckler, E.S. Quantitative trait loci analysis of growth response to varying nitrogen sources in Arabidopsis thaliana. Theor. Appl. Genet. 2002, 104, 743–750. [Google Scholar] [CrossRef]
- Hund, A.; Fracheboud, Y.; Soldati, A.; Frascaroli, E.; Salvi, S.; Stamp, P. QTL controlling root and shoot traits of maize seedlings under cold stress. Theor. Appl. Genet. 2004, 3, 618–629. [Google Scholar] [CrossRef]
- Laperche, A.; Devienne-Barret, F.; Maury, O.; le Gouis, J.; Ney, B. A simplified conceptual model of carbon/nitrogen functioning for QTL analysis of winter wheat adaptation to nitrogen deficiency. Theor. Appl. Genet. 2006, 113, 1131–1146. [Google Scholar] [CrossRef]
- Han, K.H.; Bradshaw, H.D.; Gordon, M.P. Adventitious root and shoot regeneration in vitro is under major gene control in an F2 family of hybrid poplar (Populus trichocarpa × P. deltoides). For. Genet. 1994, 1, 139–146. [Google Scholar]
- Zhang, B.; Tong, C.F.; Yin, T.M.; Zhang, X.Y.; Zhuge, Q.; Huang, M.R.; Wang, M.X.; Wu, R.L. Detection of quantitative trait loci influencing growth trajectories of adventitious roots in Populus using functional mapping. Tree Genet. Genomes 2009, 3, 539–552. [Google Scholar] [CrossRef]
- Ribeiro, C.L.; Silva, C.M.; Drost, D.R.; Novaes, E.; Novaes, C.R.D.B.; Dervinis, C.; Kirst, M. Integration of genetic, genomic and transcriptomic information identifies putative regulators of adventitious root formation in Populus. BMC Plant Biol. 2016, 1, 66. [Google Scholar] [CrossRef] [Green Version]
- Malamy, J. Intrinsic and environmental response pathways that regulate root system architecture. Plant Cell Environ. 2005, 28, 67–77. [Google Scholar] [CrossRef]
- Okumoto, S.; Koch, W.; Tegeder, M.; Fischer, W.N.; Biehl, A.; Leister, D.; Stierhof, Y.D.; Frommer, W.B. Root phloem-specific expression of the plasma membrane amino acid proton co-transporter AAP3. J. Exp. Bot. 2004, 55, 2155–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [PubMed] [Green Version]
- Zhao, X.B.; Huang, J.Y.; Yu, H.; Lei, W.; Xie, W. Genomic survey, characterization and expression profile analysis of the peptide transporter family in rice (Oryza sativa L.). BMC Plant Biol. 2010, 10, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Street, N.R.; Skogström, O.; Sjodin, A.; Tucker, J.; Rodriduez-Acosta, M.; Nilsson, P.; Jansson, S.; Taylor, G. The genetics and genomics of the drought response in Populus. Plant J. 2006, 48, 321–341. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, W.J.; Lu, N.; Wang, Z.; Wang, N.; Zhai, W.J.; Kong, L.S.; Qu, G.Z.; Wang, Q.X.; Wang, J.H. Genetic variation of growth traits and genotype-by-environment interactions in clones of Catalpa bungei and Catalpa fargesii f. duclouxii. Forests 2019, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Tisne, S.; Reymond, M.; Vile, D.; Fabre, J.; Dauzat, M.; Koornneef, M.; Granier, C. Combined genetic and modeling approaches reveal that epidermal cell area and number in leaves are controlled by leaf and plant developmental processes in Arabidopsis. Plant Physiol. 2008, 148, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- van Ooijen, J.W. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet. Res. 2011, 93, 343–349. [Google Scholar] [CrossRef]
- Parkhomchuk, D.; Borodina, D.T.; Amstislavskiy, V.; Banaru, M.; Hallen, L.; Krobitsch, S.; Lehrach, H.; Soldatov, A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009, 18, e123. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Baggerly, K.A.; Deng, L.; Morris, J.S.; Aldaz, C.M. Differential expression in SAGE: Accounting for normal between-library variation. Bioinformatics 2003, 19, 1477–1483. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Parents | F1 population | |||||||
|---|---|---|---|---|---|---|---|---|---|
| P. deltoides ‘Danhong’ | P. simonii ‘Tongliao1’ | Min | Max | Mean ± SD | CV | Kurtosis | Skewness | Repeatability | |
| Maximum root length (MRL)/cm | 4.10 | 11.93 ** | 2.910 | 22.200 | 12.74 ± 3.12 | 0.245 | 0.361 | −0.231 | 0.84 |
| Total root length (TNR)/n | 2.00 | 11.00 ** | 1.000 | 21.000 | 8 ± 3.74 | 0.442 | 0.146 | 0.557 | 0.89 |
| Root dry weight (RDW)/g | 0.02 | 0.05 ** | 0.005 | 0.170 | 0.06 ± 0.03 | 0.563 | −0.017 | 0.724 | 0.81 |
| Total number of root (TRL)/cm | 22.30 | 88.31 ** | 3.111 | 347.042 | 135.94 ± 65.96 | 0.485 | −0.145 | 0.325 | 0.84 |
| Projected area (PA)/cm2 | 2.46 | 6.30 ** | 0.061 | 37.999 | 12.32 ± 6.20 | 0.503 | 0.533 | 0.700 | 0.89 |
| Surface area (SA)/cm2 | 7.74 | 19.81 ** | 0.193 | 102.960 | 38.50 ± 19.15 | 0.497 | 0.094 | 0.595 | 0.89 |
| Average diameter (AD)/mm | 0.53 | 0.34 | 0.089 | 1.025 | 0.47 ± 0.18 | 0.388 | 0.255 | 0.921 | 0.76 |
| Root volume (RV)/cm3 | 0.04 | 0.07 ** | 0.010 | 0.496 | 0.19 ± 0.13 | 0.697 | −0.325 | 0.962 | 0.88 |
| Shoot dry weight (SDW)/g | 0.23 | 0.26 | 0.010 | 0.753 | 0.25 ± 0.14 | 0.559 | 0.387 | 0.667 | 0.86 |
| Shoot height (SH)/cm | 4.77 | 20.33 ** | 0.65 | 16.17 | 7.13 ± 3.35 | 0.470 | −0.634 | 0.191 | 0.84 |
| Basal diameter (BD)/mm | 2.20 | 2.45 | 1.05 | 2.84 | 1.94 ± 0.30 | 0.153 | 0.011 | −0.068 | 0.72 |
| Leaf number of shoot (LN)/n | 4.00 | 9.00 ** | 2.00 | 13.00 | 7 ± 2.21 | 0.32 | −0.562 | −0.015 | 0.87 |
| QTL Clusters | Location/cM | Marker | QTLs |
|---|---|---|---|
| LG1 | 61.517 | np1680 | qMRL-LG1-2; qRDW-LG1-4 |
| LG3 | 94.464 | lm5516 | qAD-LG3-1; qRV-LG3-1 |
| LG5 | 108.269 | np5635/np3887 | qRV-LG5-1; qTNR-LG5-5 to 6; qPA-LG5-1 |
| LG5 | 109.465 | lm4991 | qTNR-LG5-7; qPA-LG5-2 |
| LG5 | 144.026 | lm1039 | qRV-LG5-2; qPA-LG5-3 |
| LG5 | 145.220 | np3795 | qRV-LG5-3; qPA-LG5-4 |
| LG5 | 145.598 | lm4903 | qRV-LG5-4; qPA-LG5-5 |
| LG5 | 146.390 | lm1038 | qRV-LG5-5; qPA-LG5-6; qSA-LG5-1 |
| LG5 | 146.547 | lm4886/lm4889/lm4897/lm4895 | qRV-LG5-7 to 10; qPA-LG5-7 to 10; qTNR-LG5-13 to 14; qSA-LG5-2 |
| LG5 | 150.040 | hk421/np2628/np3744/np5403 | qRV-LG5-9 to 12; qPA-LG5-11 |
| LG8 | 113.645 | np5178 | qTNR-LG8-1; qRDW-LG8-1 |
| LG8 | 114.080 | lm4584/lm6091 | qTNR-LG8-2; qRDW-LG8-2 to 3 |
| LG9 | 181.569 | lm6429/hk339 | qTNR-LG9-3; qLN-LG9-32 |
| LG9 | 16.719 | hk321 | qSH-LG9-1; qBD-LG9-1 |
| LG11 | 1.838 | lm1575 | qAD-LG11-1; qRV-LG11-1 |
| LG14 | 93.110 | lm2588 | qMRL-LG14-7; qTNR-LG14-1 to 2; qRDW-LG14-1 |
| LG18 | 61.454 | hk178 | qRDW-LG18-2; qLN-LG18-1 |
| LG18 | 63.535 | lm1912 | qRDW-LG18-3; qLN-LG18-2 |
| LG18 | 63.546 | hk181 | qRDW-LG18-4; qLN-LG18-3 |
| LG18 | 63.554 | lm1961 | qRDW-LG18-5; qLN-LG18-4 |
| LG18 | 103.390 | np1865 | qRDW-LG18-11; qLN-LG18-5 |
| LG18 | 103.474 | np2428 | qRDW-LG18-12; qLN-LG18-6 |
| LG18 | 104.882 | np2706 | qRDW-LG18-17; qLN-LG18-7 |
| LG18 | 108.381 | hk190 | qRDW-LG18-22; qLN-LG18-8 |
| LG18 | 120.773 | np1530 | qRDW-LG18-27; qLN-LG18-13 |
| QTL Hotspots | Location/cM | QTLs |
|---|---|---|
| LG1-hotspot | 60.187–61.536 | qMRL-LG1-1 to qMRL-LG1-3 |
| LG1-hotspot | 111.46–111.724 | qTNR-LG1-1 to qTNR-LG1-3 |
| LG1-hotspot | 60.826–78.527 | qRDW-LG1-1 to qRDW-LG1-6 |
| LG1-hotspot | 110.921 | qAD-LG1-1; qAD-LG1-2 |
| LG3-hotspot | 94.464–97.935 | qRV-LG3-1 to qRV-LG3-4 |
| LG5-hotspot | 83.835–94.992 | qTNR-LG5-1 to qTNR-LG5-4 |
| LG5-hotspot | 108.269–120.75 | qTNR-LG5-5 to qTNR-LG5-12 |
| LG5-hotspot | 146.547–151.667 | qTNR-LG5-13 to qTNR-LG5-16 |
| LG5-hotspot | 108.269–109.465 | qPA-LG5-1; qPA-LG5-2 |
| LG5-hotspot | 144.026–146.547 | qPA-LG5-3 to qPA-LG5-11 |
| LG5-hotspot | 146.39–146.547 | qSA-LG5-1; qSA-LG5-2 |
| LG5-hotspot | 108.269–116.987 | qRV-LG5-1; qRV-LG5-2 |
| LG5-hotspot | 144.026–150.04 | qRV-LG5-3 to qRV-LG5-14 |
| LG6-hotspot | 48.932–48.967 | qTNR-LG6-1; qTNR-LG6-2 |
| LG8-hotspot | 136.892 | qMRL-LG8-1; qMRL-LG8-2 |
| LG8-hotspot | 113.645–114.08 | qTNR-LG8-1; qTNR-LG8-2 |
| LG8-hotspot | 113.645–115.205 | qRDW-LG8-1 to qRDW-LG8-6 |
| LG8-hotspot | 96.906–99.318 | qAD-LG8-1 to qAD-LG8-5 |
| LG9-hotspot | 16.194–161.708 | qTNR-LG9-1; qTNR-LG9-2 |
| LG9-hotspot | 181.569–181.707 | qTNR-LG9-3; qTNR-LG9-4 |
| LG9-hotspot | 1.005–5.783 | qLN-LG9-1 to qLN-LG9-6 |
| LG9-hotspot | 39.995–59.604 | qLN-LG9-7 to qLN-LG9-19 |
| LG9-hotspot | 60.121–64.262 | qLN-LG9-20 to qLN-LG9-26 |
| LG9-hotspot | 128.667–137.152 | qLN-LG9-27 to qLN-LG9-29 |
| LG9-hotspot | 162.57–181.569 | qLN-LG9-30 to qLN-LG9-32 |
| LG10-hotspot | 48.46–52.626 | qLN-LG10-1 to qLN-LG10-5 |
| LG11-hotspot | 88.512 | qSH-LG11-1; qSH-LG11-2 |
| LG14-hotspot | 82.879–93.11 | qMRL-LG14-1 to qMRL-LG14-7 |
| LG14-hotspot | 93.11–111.965 | qRDW-LG14-1 to qRDW-LG14-7 |
| LG14-hotspot | 96.276–98.586 | qBD-LG14-1 to qBD-LG14-6 |
| LG15-hotspot | 66.466 | qSDW-LG15-1; qSDW-LG15-2 |
| LG16-hotspot | 70.358–80.56 | qTNR-LG16-1 to qTNR-LG16-6 |
| LG17-hotspot | 75.914–89.987 | qTNR-LG17-1 to qTNR-LG17-7 |
| LG18-hotspot | 58.039–65.532 | qRDW-LG18-1 to qRDW-LG18-6 |
| LG18-hotspot | 79.845–97.932 | qRDW-LG18-7 to qRDW-LG18-10 |
| LG18-hotspot | 103.39–121.726 | qRDW-LG18-11 to qRDW-LG18-29 |
| LG18-hotspot | 61.454–63.554 | qLN-LG18-1 to qLN-LG18-4 |
| LG18-hotspot | 103.39–120.773 | qLN-LG18-5 to qLN-LG18-13 |
| LG19-hotspot | 107.005–110.402 | qSDW-LG19-1; qSDW-LG19-2 |
| LG19-hotspot | 103.889–107.006 | qBD-LG19-1 to qBD-LG19-3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, P.; Jia, H.; Zhang, Y.; Li, J.; Lu, M.; Hu, J. Deciphering Genetic Architecture of Adventitious Root and Related Shoot Traits in Populus Using QTL Mapping and RNA-Seq Data. Int. J. Mol. Sci. 2019, 20, 6114. https://doi.org/10.3390/ijms20246114
Sun P, Jia H, Zhang Y, Li J, Lu M, Hu J. Deciphering Genetic Architecture of Adventitious Root and Related Shoot Traits in Populus Using QTL Mapping and RNA-Seq Data. International Journal of Molecular Sciences. 2019; 20(24):6114. https://doi.org/10.3390/ijms20246114
Chicago/Turabian StyleSun, Pei, Huixia Jia, Yahong Zhang, Jianbo Li, Mengzhu Lu, and Jianjun Hu. 2019. "Deciphering Genetic Architecture of Adventitious Root and Related Shoot Traits in Populus Using QTL Mapping and RNA-Seq Data" International Journal of Molecular Sciences 20, no. 24: 6114. https://doi.org/10.3390/ijms20246114
APA StyleSun, P., Jia, H., Zhang, Y., Li, J., Lu, M., & Hu, J. (2019). Deciphering Genetic Architecture of Adventitious Root and Related Shoot Traits in Populus Using QTL Mapping and RNA-Seq Data. International Journal of Molecular Sciences, 20(24), 6114. https://doi.org/10.3390/ijms20246114