microRNAs Tune Oxidative Stress in Cancer Therapeutic Tolerance and Resistance


Abstract
1. Reactive Oxygen Species (ROSs)
2. microRNAs (miRNAs)
3. Therapeutic Tolerance and Resistance
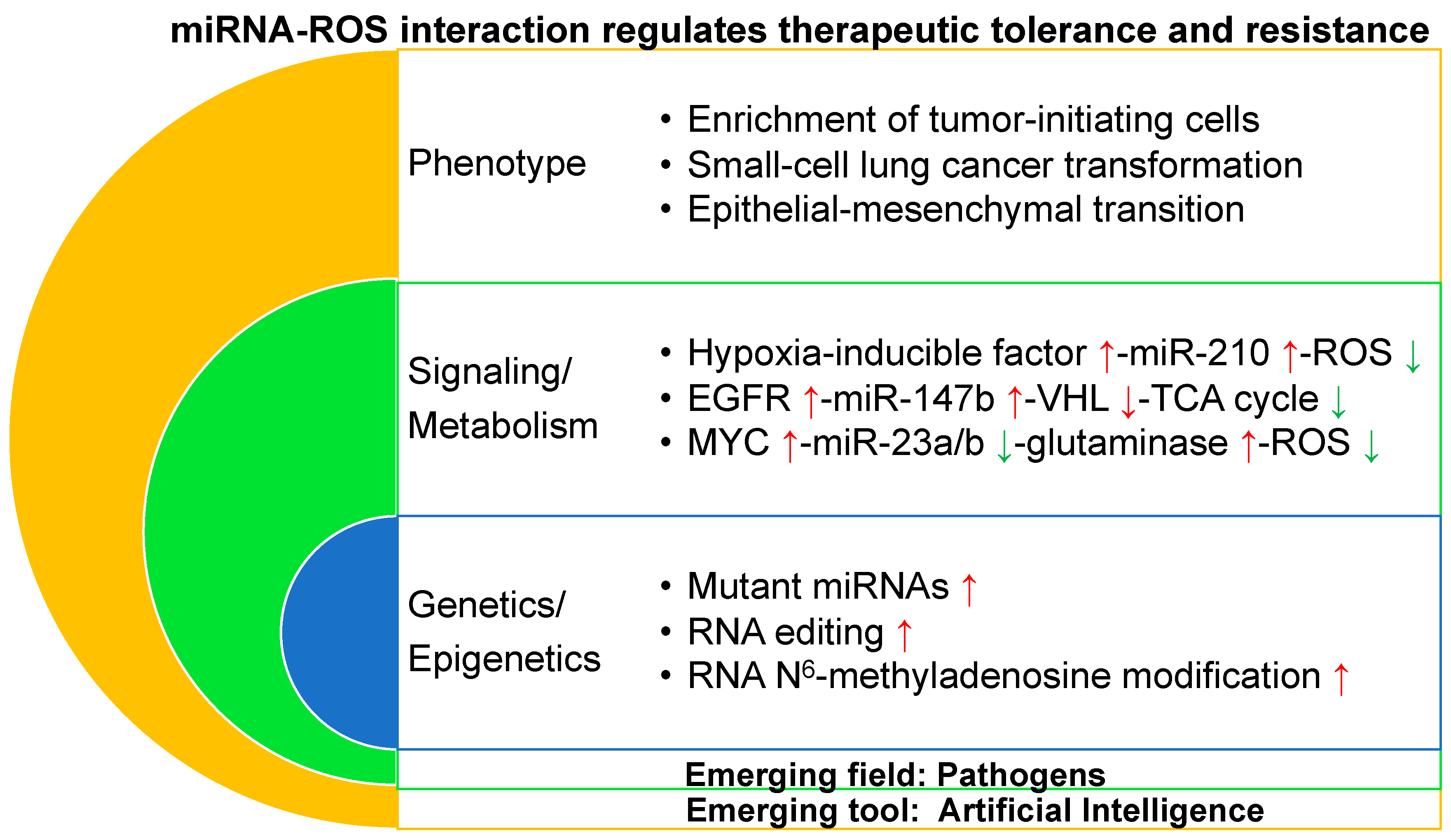
4. miRNA–ROS Interaction Regulates Therapeutic Tolerance/Resistance at the Phenotypic Level
4.1. Enrichment of Tumor-Initiating Cells
4.2. Small Cell Lung Cancer Transformation
4.3. Epithelial–Mesenchymal Transition
5. miRNA–ROS Interaction Regulates Therapeutic Tolerance/Resistance at a Signaling/Metabolic Level
5.1. HIF-miR-210-ROS
5.2. EGFR-miR-147b-VHL-TCA Cycle
5.3. Myc-miR-23a/b-Glutaminase-ROS
6. miRNA–ROS Interaction Regulates Therapeutic Tolerance/Resistance at a Genetic/Epigenetic Level
6.1. Mutant miRNAs
6.2. RNA Editing
6.3. RNA m6A Modification
7. Emerging Fields and Tools in Preventing and Overcoming Therapeutic Tolerance/Resistance
7.1. Artificial Intelligence (AI)
7.2. Pathogens
8. Concluding Remarks and Future Directions
Acknowledgments
Conflicts of Interest
References
- Harman, S.M.; Liang, L.; Tsitouras, P.D.; Gucciardo, F.; Heward, C.B.; Reaven, P.D.; Ping, W.; Ahmed, A.; Cutler, R.G. Urinary excretion of three nucleic acid oxidation adducts and isoprostane F2α measured by liquid chromatography-mass spectrometry in smokers, ex-smokers, and nonsmokers. Free Radic. Biol Med. 2003, 1301, 35–1309. [Google Scholar] [CrossRef]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant. Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Dupré-Crochet, S.; Erard, M.; Nüße, O. ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in biology and medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Yeung, A.W.K.; Tzvetkov, N.T.; El-Tawil, O.S.; Bungau, S.G.; Abdel-Daim, M.M.; Atanasov, A.G. Antioxidants: Scientific literature landscape analysis. Oxid. Med. Cell Longev. 2019, 8278, 2019454. [Google Scholar] [CrossRef]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. Pgc-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Raimundo, N.; Baysal, B.E.; Shadel, G.S. Revisiting the TCA cycle: Signaling to tumor formation. Trends Mol. Med. 2011, 17, 641–649. [Google Scholar] [CrossRef]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Dahia, P.L. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef]
- Krieg, M.; Haas, R.; Brauch, H.; Acker, T.; Flamme, I.; Plate, K.H. Up-regulation of hypoxia-inducible factors HIF-1α and HIF-2α under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 2000, 19, 5435–5443. [Google Scholar] [CrossRef]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef]
- Gottlieb, E.; Tomlinson, I.P.M. Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 2005, 5, 857–866. [Google Scholar] [CrossRef]
- Wagner, A.J.; Remillard, S.P.; Zhang, Y.X.; Doyle, L.A.; George, S.; Hornick, J.L. Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod. Pathol. 2013, 26, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.M.; Douglas, F.; George, E.; Skoldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Niemann, S.; Muller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.M.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef]
- Zhang, W.C.; Lim, B. Targeting metabolic enzyme with locked nucleic acids in non-small cell lung cancer. Cancer Res. 2014, 74, 1438. [Google Scholar]
- Lim, B.; Zhang, W. Targeting Metabolic Enzymes in Human Cancer. U.S. Patent 9,297,813,B2, 11 November 2011. [Google Scholar]
- Go, M.K.; Zhang, C W.; Lim, B.; Yew, W.S. Glycine decarboxylase is an unusual amino acid decarboxylase involved in tumorigenesis. Biochemistry 2014, 53, 947–956. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-Myc suppression of miR-23a/B enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Testa, U.; Labbaye, C.; Castelli, G.; Pelosi, E. Oxidative stress and hypoxia in normal and leukemic stem cells. Exp. Hematol. 2016, 44, 540–560. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kasinski, A.L.; Slack, F.J. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 2011, 11, 849–864. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Fan, M.; Krutilina, R.; Sun, J.; Sethuraman, A.; Yang, H.C.; Wu, H.Z.; Yue, J.; Pfeffer, L.M. Comprehensive analysis of microRNA (miRNA) targets in breast cancer cells. J. Biol. Chem. 2013, 288, 27480–27493. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, A.E.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Nouraee, N.; Calin, G.A. MicroRNAs as cancer biomarkers. MicroRNA 2013, 2, 102–117. [Google Scholar] [CrossRef]
- Tentori, A.M.; Nagarajan, M.B.; Kim, J.J.; Zhang, W.C.; Slack, F.J.; Doyle, P.S. Quantitative and multiplex microRNA assays from unprocessed cells in isolated nanoliter well arrays. Lab Chip 2018, 18, 2410–2424. [Google Scholar] [CrossRef]
- Nagarajan, M.B.; Tentori, A.M.; Zhang, W.C.; Slack, F.J.; Doyle, P.S. Nonfouling, encoded hydrogel microparticles for multiplex microRNA profiling directly from formalin-fixed, paraffin-embedded tissue. Anal. Chem. 2018, 90, 10279–10285. [Google Scholar] [CrossRef]
- Anfossi, S.; Babayan, A.; Pantel, K.; Calin, G.A. Clinical utility of circulating non-coding RNAs—An update. Nat. Rev. Clin. Oncol. 2018, 15, 541–563. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Nishida, N.; Calin, G.A.; Pantel, K. clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef]
- Cortez, M.A.; Bueso-Ramos, C.; Ferdin, J.; Lopez-Berestein, G.; Sood, A.K.; Calin, G.A. MicroRNAs in body fluids—The mix of hormones and biomarkers. Nat. Rev. Clin. Oncol. 2011, 8, 467–477. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Medina, P.P.; Nolde, M.; Slack, F.J. Oncomir addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 2010, 467, 86–90. [Google Scholar] [CrossRef]
- Edmonds, M.D.; Boyd, K.L.; Moyo, T.; Mitra, R.; Duszynski, R.; Arrate, M.P.; Chen, X.; Zhao, Z.; Blackwell, T.S.; Andl, T.; et al. MicroRNA-31 initiates lung tumorigenesis and promotes mutant KRAS-driven lung cancer. J. Clin. Investig. 2016, 126, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015, 518, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar] [CrossRef]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef]
- Kasinski, A.L.; Slack, F.J. MiRNA-34 prevents cancer initiation and progression in a therapeutically resistant k-RAS and P53-induced mouse model of lung adenocarcinoma. Cancer Res. 2012, 72, 5576–5587. [Google Scholar] [CrossRef]
- Krzeszinski, J.Y.; Wei, W.; Huynh, H.; Jin, Z.; Wang, X.; Chang, T.C.; Xie, X.J.; He, L.; Mangala, L.S.; Lopez-Berestein, G.; et al. MiR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature 2014, 512, 431–435. [Google Scholar] [CrossRef]
- Banerjee, J.; Khanna, S.; Bhattacharya, A. MicroRNA regulation of oxidative stress. Oxid. Med. Cell Longev. 2017, 2872, 2017156. [Google Scholar] [CrossRef]
- Hatley, M.E.; Patrick, D.M.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; van Rooij, E.; Olson, E.N. Modulation of K-Ras-dependent lung tumorigenesis by microRNA-21. Cancer Cell 2010, 18, 282–293. [Google Scholar] [CrossRef]
- Seike, M.; Goto, A.; Okano, T.; Bowman, D.E.; Schetter, J.A.; Horikawa, I.; Mathe, A.E.; Jen, J.; Yang, P.; Sugimura, H.; et al. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc. Natl. Acad. Sci. USA 2009, 106, 12085–12090. [Google Scholar] [CrossRef]
- Zhang, X.; Ng, W.L.; Wang, P.; Tian, L.; Werner, E.; Wang, H.; Doetsch, P.; Wang, Y. MicroRNA-21 modulates the levels of reactive oxygen species by targeting Sod3 Tnfα. Cancer Res. 2012, 72, 4707–4713. [Google Scholar] [CrossRef]
- Engedal, N.; Zerovnik, E.; Rudov, A.; Galli, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Monsurro, V.; Betti, M.; Albertini, M.C. From oxidative stress damage to pathways, networks, and autophagy via microRNAs. Oxid. Med. Cell Longev. 2018, 2018, 4968321. [Google Scholar] [CrossRef]
- Poyil, P.; Son, Y.-O.; Divya, S.P.; Wang, L.; Zhang, Z.; Shi, H. Oncogenic transformation of human lung bronchial epithelial cells induced by arsenic involves ROS-dependent activation of STAT3-miR-21-PDCD4 mechanism. Sci. Rep. 2016, 6, 37227. [Google Scholar]
- Thulasingam, S.; Massilamany, C.; Gangaplara, A.; Dai, H.; Yarbaeva, S.; Subramaniam, S.; Riethoven, J.J.; Eudy, J.; Lou, M.; Reddy, J. MiR-27b*, an oxidative stress-responsive microRNA modulates nuclear factor-kB pathway in Raw 264.7 cells. Mol. Cell Biochem. 2011, 352, 181–188. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Papaioannou, A.I.; Fenwick, P.; Donnelly, L.; Ito, K.; Barnes, P.J. Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci. Rep. 2016, 6, 35871. [Google Scholar] [CrossRef]
- Lin, Z.; Fang, D. The roles of SIRT1 in cancer. Genes Cancer 2013, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Bussing, I.; Slack, F.J.; Grosshans, H. Let-7 microRNAs in development, stem cells and cancer. Trends Mol. Med. 2008, 14, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.D.; Savage, J.E.; Cao, L.; Soule, B.P.; Ly, D.; DeGraff, W.; Harris, C.C.; Mitchell, J.B.; Simone, N.L. Cellular stress induced alterations in microRNA let-7a and let-7b expression are dependent on P53. PLoS ONE 2011, 6, e24429. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef]
- Adams, B.D.; Parsons, C.; Walker, L.; Zhang, W.C.; Slack, F.J. Targeting noncoding RNAs in disease. J. Clin. Investig. 2017, 127, 761–771. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Van Zandwijk, N.; McDiarmid, J.; Brahmbhatt, H.; Reid, G. Response to an innovative mesothelioma treatment based on miR-16 mimic loaded EGFR targeted minicells (TargomiRs). Transl. Lung Cancer Res. 2018, 7, S60–S61. [Google Scholar] [CrossRef]
- Fennell, D. MiR-16: Expanding the range of molecular targets in mesothelioma. Lancet Oncol. 2017, 18, 1296–1297. [Google Scholar] [CrossRef]
- Slack, F.J.; Chinnaiyan, A.M. The role of non-coding RNAs in oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, L.; Rodriguez-Aguayo, C.; Yuan, Y.; Debeb, B.G.; Chen, D.; Sun, Y.; You, M.J.; Liu, Y.; Dean, C.D.; et al. MiR-205 acts as a tumour radiosensitizer by targeting ZEB1 and Ubc13. Nat. Commun. 2014, 5, 5671. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, R.; Claret, F.X.; Yang, H. Involvement of microRNA-24 and DNA methylation in resistance of nasopharyngeal carcinoma to ionizing radiation. Mol. Cancer Ther. 2014, 13, 3163–3174. [Google Scholar] [CrossRef] [PubMed]
- Babar, I.A.; Czochor, J.; Steinmetz, A.; Weidhaas, J.B.; Glazer, P.M.; Slack, F.J. Inhibition of hypoxia-induced miR-155 radiosensitizes hypoxic lung cancer cells. Cancer Biol. Ther. 2011, 12, 908–914. [Google Scholar] [CrossRef]
- Zhang, W.C.; Chin, T.M.; Yang, H.; Nga, M.E.; Lunny, D.P.; Lim, E.K.; Sun, L.L.; Pang, Y.H.; Leow, Y.N.; Malusay, S.R.; et al. Tumour-initiating cell-specific miR-1246 and miR-1290 expression converge to promote non-small cell lung cancer progression. Nat. Commun. 2016, 7, 11702. [Google Scholar] [CrossRef]
- Zhang, W.C.; Wells, J.M.; Chow, K.H.; Huang, H.; Yuan, M.; Saxena, T.; Melnick, M.A.; Politi, K.; Asara, J.M.; Costa, D.B.; et al. MiR-147b-mediated TCA cycle dysfunction and pseudohypoxia initiate drug tolerance to EGFR inhibitors in lung adenocarcinoma. Nat. Metab. 2019, 1, 460–474. [Google Scholar] [CrossRef]
- Costinean, S.; Zanesi, N.; Pekarsky, Y.; Tili, E.; Volinia, S.; Heerema, N.; Croce, M.C. Pre-B Cell Proliferation and Lymphoblastic Leukemia/High-Grade Lymphoma in E(Mu)-Mir155 Transgenic Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7024–7029. [Google Scholar] [CrossRef]
- Mikamori, M.; Yamada, D.; Eguchi, H.; Hasegawa, S.; Kishimoto, T.; Tomimaru, Y.; Asaoka, T.; Noda, T.; Wada, H.; Kawamoto, K.; et al. MicroRNA-155 controls exosome synthesis and promotes gemcitabine resistance in pancreatic ductal adenocarcinoma. Sci. Rep. 2017, 7, 42339. [Google Scholar] [CrossRef]
- Moriyama, T.; Ohuchida, K.; Mizumoto, K.; Yu, J.; Sato, N.; Nabae, T.; Takahata, S.; Toma, H.; Nagai, E.; Tanaka, M. MicroRNA-21 modulates biological functions of pancreatic cancer cells including their proliferation, invasion, and chemoresistance. Mol. Cancer Ther. 2009, 8, 1067–1074. [Google Scholar] [CrossRef]
- Giovannetti, E.; Funel, N.; Peters, J G.; Del Chiaro, M.; Erozenci, L.A.; Vasile, E.; Leon, L.G.; Pollina, L.E.; Groen, A.; Falcone, A.; et al. MicroRNA-21 in pancreatic cancer: correlation with clinical outcome and pharmacologic aspects underlying its role in the modulation of gemcitabine activity. Cancer Res. 2010, 70, 4528–4538. [Google Scholar] [CrossRef]
- Jiang, S.; Wang, R.; Yan, H.; Jin, L.; Dou, X.; Chen, D. MicroRNA-21 modulates radiation resistance through upregulation of hypoxia-inducible factor-1α-promoted glycolysis in non-small cell lung cancer cells. Mol. Med. Rep. 2016, 13, 4101–4107. [Google Scholar] [CrossRef]
- Zhang, W.C.; Slack, F.J. MicroRNA-21 mediates resistance to EGFR tyrosine kinase inhibitors in lung cancer. J. Thorac. Oncol. 2017, 12, S1536. [Google Scholar] [CrossRef]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Hu, W.; Pan, L.; Fu, W.; Dai, L.; Jiang, Z.; Zhang, F.; Zhao, J. Let-7 and miR-17 promote self-renewal and drive gefitinib resistance in non-small cell lung cancer. Oncol. Rep. 2019, 42, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Deng, H.; Yao, H.; Liu, Q.; Su, F.; Song, E. MiR-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene 2010, 29, 4194–4204. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.S.; Yu, F.; Zhong, X.M.; Lu, G.X.; Cong, X.L.; Xue, S.B.; Xie, W.T.; Hou, L.K.; Pang, L.J.; Wu, W.; et al. MiR-30 family reduction maintains self-renewal and promotes tumorigenesis in NSCLC-initiating cells by targeting oncogene TM4SF1. Mol. Ther. 2018, 26, 2751–2765. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Romano, G.; Di Leva, G.; Nuovo, G.; Jeon, Y.J.; Ngankeu, A.; Sun, J.; Lovat, F.; Alder, H.; Condorelli, G.; et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat. Med. 2012, 18, 74–82. [Google Scholar] [CrossRef]
- Du, X.; Liu, B.; Luan, X.; Cui, Q.; Li, L. MiR-30 decreases multidrug resistance in human gastric cancer cells by modulating cell autophagy. Exp. Ther. Med. 2018, 15, 599–605. [Google Scholar] [CrossRef]
- Jiang, L.; Hermeking, H. MiR-34a and miR-34b/c suppress intestinal tumorigenesis. Cancer Res. 2017, 77, 2746–2758. [Google Scholar] [CrossRef]
- Zenz, T.; Mohr, J.; Eldering, E.; Kater, A.P.; Bühler, A.; Kienle, D.; Winkler, D.; Dürig, J.; van Oers, M.H.; Mertens, D.; et al. MiR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood 2009, 113, 3801–3808. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Politi, K.; Herbst, R.S. Lung cancer in the era of precision medicine. Clin. Cancer Res. 2015, 21, 2213–2220. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Politi, K.; Ayeni, D.; Lynch, T. The next wave of EGFR tyrosine kinase inhibitors enter the clinic. Cancer Cell 2015, 27, 751–753. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting drivers of non-mutational drug tolerance is a salvage strategy for targeted melanoma therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, J.M.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, L.S.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef]
- Sebolt-Leopold, J.S.; English, J.M. Mechanisms of drug inhibition of signalling molecules. Nature 2006, 441, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-j.; Zheng, B.; Wang, H.-y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef]
- Mendez-Blanco, C.; Fondevila, F.; Garcia-Palomo, A.; Gonzalez-Gallego, J.; Mauriz, L.J. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 134. [Google Scholar] [CrossRef]
- Vanecek, J. Cellular mechanisms of melatonin action. Physiol. Rev. 1998, 78, 687–721. [Google Scholar] [CrossRef]
- Prieto-Domínguez, N.; Ordóñez, R.; Fernández, A.; Méndez-Blanco, C.; Baulies, A.; Garcia-Ruiz, C.; Fernández-Checa, J.C.; Mauriz, J.L.; González-Gallego, J. Melatonin-induced increase in sensitivity of human hepatocellular carcinoma cells to sorafenib is associated with reactive oxygen species production and mitophagy. J. Pineal Res. 2016, 61, 396–407. [Google Scholar] [CrossRef]
- Prieto-Dominguez, N.; Mendez-Blanco, C.; Carbajo-Pescador, S.; Fondevila, F.; Garcia-Palomo, A.; Gonzalez-Gallego, J.; Mauriz, J.L. Melatonin enhances sorafenib actions in human hepatocarcinoma cells by inhibiting mTORC1/p70S6K/HIF-1α and hypoxia-mediated mitophagy. Oncotarget 2017, 8, 91402–91414. [Google Scholar] [CrossRef]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Zhang, W.C.; Yang, H.; Soh, B.S.; Sun, L.L.; Chin, T.M.; Lim, E.H.; Lim, B. Abstract 487: Evidence for tumor initiating stem cells in lung cancer. Cancer Res. 2011, 71, 487. [Google Scholar]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 2013, 24, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Hoe, S.L.L.; Tan, L.P.; Jamal, J.; Peh, S.C.; Ng, C.C.; Zhang, W.C.; Ahmad, M.; Khoo, A.S.B. Evaluation of stem-like side population cells in a recurrent nasopharyngeal carcinoma cell line. Cancer Cell Int. 2014, 14, 101. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Dando, I.; Cordani, M.; Dalla Pozza, E.; Biondani, G.; Donadelli, M.; Palmieri, M. Antioxidant mechanisms and ROS-related microRNAs in cancer stem cells. Oxid. Med. Cell Longev. 2015, 4257, 201508. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Curtis, S.J.; Sinkevicius, K.W.; Li, D.; Lau, A.N.; Roach, R.R.; Zamponi, R.; Woolfenden, A.E.; Kirsch, D.G.; Wong, K.K.; Kim, C.F. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell 2010, 7, 127–133. [Google Scholar] [CrossRef]
- Houessinon, A.; Francois, C.; Sauzay, C.; Louandre, C.; Mongelard, G.; Godin, C.; Bodeau, S.; Takahashi, S.; Saidak, Z.; Gutierrez, L.; et al. Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol. Cancer 2016, 15, 38. [Google Scholar] [CrossRef]
- Li, L.; Abdel Fattah, E.; Cao, G.; Ren, C.; Yang, G.; Goltsov, A.A.; Chinault, A.C.; Cai, W.W.; Timme, T.L.; Thompson, T.C. Glioma pathogenesis-related protein 1 exerts tumor suppressor activities through proapoptotic reactive oxygen species-c-Jun-NH2 kinase signaling. Cancer Res. 2008, 68, 434–443. [Google Scholar] [CrossRef]
- Yuan, D.; Xu, J.; Wang, J.; Pan, Y.; Fu, J.; Bai, Y.; Zhang, J.; Shao, C. Extracellular miR-1246 promotes lung cancer cell proliferation and enhances radioresistance by directly targeting DR5. Oncotarget 2016, 7, 32707–32722. [Google Scholar] [CrossRef]
- Golestaneh, A.F.; Atashi, A.; Langroudi, L.; Shafiee, A.; Ghaemi, N.; Soleimani, M. MiRNAs expressed differently in cancer stem cells and cancer cells of human gastric cancer cell line MKN-45. Cell Biochem. Funct. 2012, 30, 411–418. [Google Scholar] [CrossRef]
- Jackman, D.M.; Johnson, B.E. Small-cell lung cancer. Lancet 2005, 366, 1385–1396. [Google Scholar] [CrossRef]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: clinical outcomes. J. Clin. Oncol. 2018, 37, 278–285. [Google Scholar] [CrossRef]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef]
- Ebi, H.; Sato, T.; Sugito, N.; Hosono, Y.; Yatabe, Y.; Matsuyama, Y.; Yamaguchi, T.; Osada, H.; Suzuki, M.; Takahashi, T. Counterbalance between RB inactivation and miR-17-92 overexpression in reactive oxygen species and DNA damage induction in lung cancers. Oncogene 2009, 28, 3371–3379. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Lindahl-Allen, M.; Polytarchou, C.; Hirsch, H.A.; Tsichlis, P.N.; Struhl, K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol. Cell 2010, 39, 761–772. [Google Scholar] [CrossRef]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Mateescu, B.; Batista, L.; Cardon, M.; Gruosso, T.; de Feraudy, Y.; Mariani, O.; Nicolas, A.; Meyniel, J.-P.; Cottu, P.; Sastre-Garau, X.; et al. MiR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat. Med. 2011, 17, 1627–1635. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. MiR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via Zeb1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ding, L.; Bennewith, K.L.; Tong, R.T.; Welford, S.M.; Ang, K.K.; Story, M.; Le, Q.T.; Giaccia, A.J. Hypoxia-inducible miR-210 regulates normoxic gene expression involved in tumor initiation. Mol. Cell 2009, 35, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Favaro, E.; Ramachandran, A.; McCormick, R.; Gee, H.; Blancher, C.; Crosby, M.; Devlin, C.; Blick, C.; Buffa, F.; Li, J.L.; et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS ONE 2010, 5, e10345. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef]
- Yang, W.; Sun, T.; Cao, J.; Liu, F.; Tian, Y.; Zhu, W. Downregulation of miR-210 expression inhibits proliferation, induces apoptosis and enhances radiosensitivity in hypoxic human hepatoma cells in vitro. Exp. Cell Res. 2012, 318, 944–954. [Google Scholar] [CrossRef]
- Yang, W.; Wei, J.; Guo, T.; Shen, Y.; Liu, F. Knockdown of miR-210 decreases hypoxic glioma stem cells stemness and radioresistance. Exp. Cell Res. 2014, 326, 22–35. [Google Scholar] [CrossRef]
- Ivan, M.; Huang, X. MiR-210: Fine-tuning the hypoxic response. Adv. Exp. Med. Biol. 2014, 772, 205–227. [Google Scholar]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer 2008, 8, 865–873. [Google Scholar] [CrossRef]
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 2016, 353, 929–932. [Google Scholar] [CrossRef]
- Lee, S.B.; Frattini, V.; Bansal, M.; Castano, A.M.; Sherman, D.; Hutchinson, K.; Bruce, J.N.; Califano, A.; Liu, G.; Cardozo, T.; et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature 2016, 529, 172–177. [Google Scholar] [CrossRef]
- MacKenzie, E.D.; Selak, M.A.; Tennant, D.A.; Payne, L.J.; Crosby, S.; Frederiksen, C.M.; Watson, D.G.; Gottlieb, E. Cell-permeating α-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell Biol. 2007, 27, 3282–3289. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef]
- Niederst, M.J.; Engelman, J.A. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci. Signal. 2013, 6, re6. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic heterogeneity in human lung tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef]
- Imielinski, M.; Guo, G.; Meyerson, M. Insertions and deletions target lineage-defining genes in human cancers. Cell 2017, 168, 460.e14–472.e14. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ren, S.; Li, X.; Wang, Y.; Garfield, D.; Zhou, S.; Chen, X.; Su, C.; Chen, M.; Kuang, P.; et al. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer 2014, 83, 146–153. [Google Scholar] [CrossRef]
- Zheng, G.; Li, N.; Jia, X.; Peng, C.; Luo, L.; Deng, Y.; Yin, J.; Song, Y.; Liu, H.; Lu, M.; et al. MYCN-mediated miR-21 overexpression enhances chemo-resistance via targeting CADM1 in tongue cancer. J. Mol. Med. (Berl.) 2016, 94, 1129–1141. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef]
- Lazzari, E.; Crews, L.A.; Wu, C.; Leu, H.; Ali, S.; Chiaramonte, R.; Minden, M.; Costello, C.; Jamieson, C.H.M. Abstract 2414: ADAR1-dependent RNA editing is a mechanism of therapeutic resistance in human plasma cell malignancies. Cancer Res. 2016, 76, 2414. [Google Scholar]
- Zhang, W.C.; Slack, F.J. ADARs edit microRNAs to promote leukemic stem cell activity. Cell Stem Cell 2016, 19, 141–142. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Delos Santos, N.P.; Balaian, L.; Chun, H.J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I.; et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell 2016, 19, 177–191. [Google Scholar] [CrossRef]
- Jiang, Q.; Isquith, J.; Zipeto, M.A.; Diep, R.H.; Pham, J.; Delos Santos, N.; Reynoso, E.; Chau, J.; Leu, H.; Lazzari, E.; et al. Hyper-editing of cell-cycle regulatory and tumor suppressor RNA promotes malignant progenitor propagation. Cancer Cell 2019, 35, 81.e7–94.e7. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Ishizuka, J.J.; Manguso, R.T.; Cheruiyot, C.K.; Bi, K.; Panda, A.; Iracheta-Vellve, A.; Miller, B.C.; Du, P.P.; Yates, K.B.; Dubrot, J.; et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2019, 565, 43–48. [Google Scholar] [CrossRef]
- Chen, L.; Rio, D.C.; Haddad, G.G.; Ma, E. Regulatory role of dADAR in ROS metabolism in drosophila CNS. Brain Res. Mol. Brain Res. 2004, 131, 93–100. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. M6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef]
- Choe, J.; Lin, S.; Zhang, W.; Liu, Q.; Wang, L.; Ramirez-Moya, J.; Du, P.; Kim, W.; Tang, S.; Sliz, P.; et al. MRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 2018, 561, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Liu, P.Y.; Haase, J.; Bell, J.L.; Huttelmaier, S.; Liu, T. The critical role of RNA m6A methylation in cancer. Cancer Res. 2019, 79, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wei, J.; He, C. Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, A.; Patil, V.; Arora, A.; Hegde, S.A.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Essential Role of Mettl3-Mediated M6a Modification in Glioma Stem-Like Cells Maintenance and Radioresistance. Oncogene 2018, 37, 522–533. [Google Scholar] [CrossRef]
- Taketo, K.; Konno, M.; Asai, A.; Koseki, J.; Toratani, M.; Satoh, T.; Doki, Y.; Mori, M.; Ishii, H.; Ogawa, K. The Epitranscriptome M6a Writer Mettl3 Promotes Chemo- and Radioresistance in Pancreatic Cancer Cells. Int. J. Oncol. 2018, 52, 621–629. [Google Scholar] [CrossRef]
- Yan, F.; Al-Kali, A.; Zhang, Z.; Liu, J.; Pang, J.; Zhao, N.; He, C.; Litzow, M.R.; Liu, S. A dynamic N 6-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 2018, 28, 1062–1076. [Google Scholar] [CrossRef]
- Wang, J.; Ishfaq, M.; Xu, L.; Xia, C.; Chen, C.; Li, J. METTL3/m6A/miRNA-873–5p attenuated oxidative stress and apoptosis in colistin-induced kidney injury by modulating Keap1/Nrf2 pathway. Front. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Topol, E.J. High-performance medicine: The convergence of human and artificial intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef]
- Romm, E.L.; Tsigelny, I.F. Artificial intelligence in drug treatment. Annu. Rev. Pharmacol. Toxicol. 2020, 60. [Google Scholar] [CrossRef]
- Mason, D.J.; Eastman, R.T.; Lewis, R.P.I.; Stott, I.P.; Guha, R.; Bender, A. Using machine learning to predict synergistic antimalarial compound combinations with novel structures. Front. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Li, X.; Xu, Y.; Cui, H.; Huang, T.; Wang, D.; Lian, B.; Li, W.; Qin, G.; Chen, L.; Xie, L. Prediction of synergistic anti-cancer drug combinations based on drug target network and drug induced gene expression profiles. Artif. Intell. Med. 2017, 83, 35–43. [Google Scholar] [CrossRef]
- Xia, F.; Shukla, M.; Brettin, T.; Garcia-Cardona, C.; Cohn, J.; Allen, J.E.; Maslov, S.; Holbeck, S.L.; Doroshow, J.H.; Evrard, Y.A.; et al. Predicting tumor cell line response to drug pairs with deep learning. BMC Bioinform. 2018, 19, 486. [Google Scholar] [CrossRef] [PubMed]
- McQuade, J.L.; Daniel, C.R.; Helmink, B.A.; Wargo, J.A. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. 2019, 20, e77–e91. [Google Scholar] [CrossRef]
- Peek, R.M., Jr.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef]
- Sam, Q.H.; Chang, M.W.; Chai, L.Y. The fungal mycobiome and its interaction with gut bacteria in the host. Int. J. Mol. Sci. 2017, 18, 330. [Google Scholar] [CrossRef]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017, 170, 548.e16–563.e16. [Google Scholar] [CrossRef] [PubMed]
- Tsang, W.P.; Kwok, T.T. The miR-18a* microRNA functions as a potential tumor suppressor by targeting on K-Ras. Carcinogenesis 2009, 30, 953–959. [Google Scholar] [CrossRef]
- Lievre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Cote, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Haqshenas, G.; Doerig, C. Targeting of host cell receptor tyrosine kinases by intracellular pathogens. Sci. Signal. 2019, 12, eaau9894. [Google Scholar] [CrossRef]
- Schor, S.; Einav, S. Combating intracellular pathogens with repurposed host-targeted drugs. ACS Infect. Dis. 2018, 4, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.C.; Gay, C.M.; Xi, Y.; Fujimoto, J.; Kalhor, N.; Hartsfield, P.M.; Tran, H.; Fernandez, L.; Lu, D.; Wang, Y.; et al. Abstract 2899: Single-cell analyses reveal increasing intratumoral heterogeneity as an essential component of treatment resistance in small cell lung cancer. Cancer Res. 2019, 79, 2899. [Google Scholar]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward minimal residual disease-directed therapy in melanoma. Cell 2018, 174, 843.e19–855.e19. [Google Scholar] [CrossRef]
- Bivona, T.G.; Doebele, R.C. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat. Med. 2016, 22, 472–478. [Google Scholar] [CrossRef]
- Ponz-Sarvise, M.; Corbo, V.; Tiriac, H.; Engle, D.D.; Frese, K.K.; Oni, T.E.; Hwang, C.I.; Ohlund, D.; Chio, I.I.C.; Baker, L.A.; et al. Identification of resistance pathways specific to malignancy using organoid models of pancreatic cancer. Clin. Cancer Res. 2019, 25, 6742–6755. [Google Scholar] [CrossRef]
- Usui, T.; Sakurai, M.; Umata, K.; Elbadawy, M.; Ohama, T.; Yamawaki, H.; Hazama, S.; Takenouchi, H.; Nakajima, M.; Tsunedomi, R.; et al. Hedgehog signals mediate anti-cancer drug resistance in three-dimensional primary colorectal cancer organoid culture. Int. J. Mol. Sci. 2018, 19, 1098. [Google Scholar] [CrossRef]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A E.; Rich, N.J. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, J.; Holokai, L.; Syu, L.; Steele, N.; Chang, J.; Dlugosz, A.; Zavros, Y. Mouse-derived gastric organoid and immune cell co-culture for the study of the tumor microenvironment. Methods Mol. Biol. 2018, 1817, 157–168. [Google Scholar] [PubMed]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid modeling of the tumor immune microenvironment. Cell 2018, 175, 1972.e16–1988.e16. [Google Scholar] [CrossRef] [PubMed]
- Sozzi, G.; Boeri, M.; Rossi, M.; Verri, C.; Suatoni, P.; Bravi, F.; Roz, L.; Conte, D.; Grassi, M.; Sverzellati, N.; et al. Clinical utility of a plasma-based miRNA signature classifier within computed tomography lung cancer screening: A correlative mild trial study. J. Clin. Oncol. 2014, 32, 768–773. [Google Scholar] [CrossRef]
- Siravegna, G.; Bardelli, A. Genotyping cell-free tumor DNA in the blood to detect residual disease and drug resistance. Genome Biol. 2014, 15, 449. [Google Scholar] [CrossRef]


{kind=link}
| miRNA | Signaling Involved in Tumorigenesis | Signaling Involved in Therapeutic Tolerance and Resistance |
|---|---|---|
| miR-1246 and miR-1290 ↑ | (+) tumorigenesis via repressing metallothioneins in human non-small cell lung cancer [75] | (+) resistance to EGFR tyrosine kinase inhibitor gefitinib via repressing metallothioneins in human non-small cell lung cancer [75] |
| miR-147b ↑ | N.A. | (+) tolerance to EGFR tyrosine kinase inhibitor osimertinib through activating pseudohypoxia signaling pathways via repressing VHL and succinate dehydrogenase in human non-small cell lung cancer [76] |
| miR-155 ↑ | (+) tumorigenesis in mouse miR155 transgenic B cell lymphomas [77] | (+) chemoresistance to gemcitabine through decreasing apoptosis in human pancreatic cancer [78] |
| miR-21 ↑ | (+) Ras/MEK/ERK signaling via repressing negative regulators of the Ras/MEK/ERK pathway and inhibition of apoptosis in mouse KRAS transgenic non-small cell lung cancer [54] | (+) chemoresistance to gemcitabine through decreasing apoptosis and activating Akt phosphorylation in human pancreatic cancer [79,80] (+) radioresistance through upregulation of hypoxia-inducible factor 1α in human non-small cell lung cancer [81] (+) resistance to EGFR tyrosine kinase inhibitors through activating PI3K-AKT signaling pathway in human non-small cell lung cancer [82] |
| miR-31 ↑ | (+) tumorigenesis through activating RAS/MAPK signaling via repressing negative regulators of RAS/MAPK signaling in mouse KRAS transgenic non-small cell lung cancer [46] | N.A. |
| let-7 family ↓ | (+) tumorigenesis in human breast cancer through repressing H-RAS and high mobility group AT-hook 2 [83] | (+) resistance to EGFR tyrosine kinase inhibitor gefitinib through upregulation of MYC in human non-small cell lung cancer [84] |
| miR-30 ↓ | ||
| miR-34a/b/c ↓ | (+) chemoresistance to fludarabine through p53 inactivation and apoptosis resistance in human chronic lymphocytic leukemia [90] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.C. microRNAs Tune Oxidative Stress in Cancer Therapeutic Tolerance and Resistance. Int. J. Mol. Sci. 2019, 20, 6094. https://doi.org/10.3390/ijms20236094
Zhang WC. microRNAs Tune Oxidative Stress in Cancer Therapeutic Tolerance and Resistance. International Journal of Molecular Sciences. 2019; 20(23):6094. https://doi.org/10.3390/ijms20236094
Chicago/Turabian StyleZhang, Wen Cai. 2019. "microRNAs Tune Oxidative Stress in Cancer Therapeutic Tolerance and Resistance" International Journal of Molecular Sciences 20, no. 23: 6094. https://doi.org/10.3390/ijms20236094
APA StyleZhang, W. C. (2019). microRNAs Tune Oxidative Stress in Cancer Therapeutic Tolerance and Resistance. International Journal of Molecular Sciences, 20(23), 6094. https://doi.org/10.3390/ijms20236094