DNA Oxidation and Excision Repair Pathways



{kind=link}
Abstract
1. Introduction
2. DNA Excision Repairs and Implication on Human Health
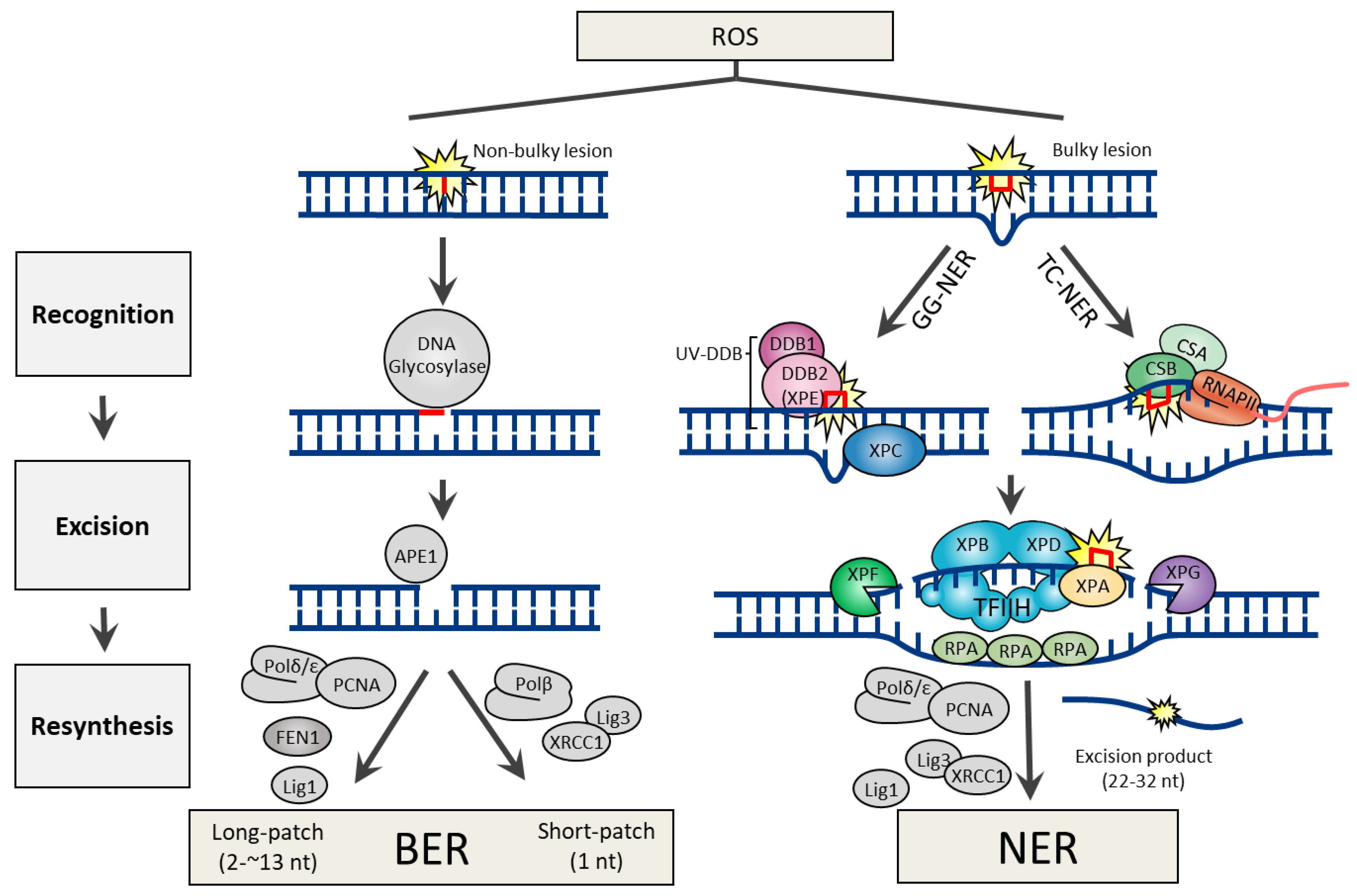
2.1. Base Excision Repair
2.2. General Features of BER Defect
2.3. Nucleotide Excision Repair
2.4. General Features of NER Defect
3. Facilitated BER Kinetics by the NER Lesion Recognition Factors
3.1. XPC
3.2. UV-DDB
3.3. CSB
4. Oxidative DNA Damages that Can Be Readily Repaired by a Canonical NER
4.1. Bulky Lesions
4.2. Non-Bulky Lesions
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5′-dRP | 5′-2-deoxyribose-5′-phosphate |
| 6-4PP | Pyrimidine-(6,4)-pyrimidone product |
| 8-oxoG | 8-oxo-7,8-dihydroguanine |
| Abasic site | Apurinic/apyrimidinic site or AP site |
| AD | Alzheimer’s disease |
| APE1 | AP endonuclease 1 |
| BER | Base excision repair |
| cdA | 5′,8-cyclo-2′-deoxyadenosine |
| cdG | 5′,8-cyclo-2′-deoxyguanosine |
| CPD | Cyclobutane pyrimidine dimer |
| CSB | Cockayne syndrome B |
| dL | 2-deoxyribonolactone |
| DPCs | DNA–protein crosslinks |
| FEN1 | Flap endonuclease 1 |
| GG-NER | Global genome NER |
| Gh | Guanidinohydantoin |
| HR23B | Human RAD23 homolog B |
| ICL | Interstrand crosslink |
| MBD4 | Methyl-binding domain glycosylase 4 |
| MPG | Methylpurine glycosylase |
| MYH | MutY homolog DNA glycosylase |
| NEIL1 | Endonuclease VIII-like glycosylase |
| NER | Nucleotide excision repair |
| NTH1 | Endonuclease III-like glycosylase |
| OGG1 | 8-oxoguanine DNA glycosylase |
| PCNA | Proliferating cell nuclear antigen |
| Polβ | DNA polymerase β |
| RNAPII | RNA polymerase II |
| ROS | Reactive oxygen species |
| SMUG1 | Single-strand-specific monofunctional uracil DNA glycosylase |
| Sp | Spiroiminodihydantoin |
| TC-NER | Transcription-coupled NER |
| TDG | Thymine DNA glycosylase |
| TFIIH | Transcription Factor II H; complexed with the XPB and XPD helicases |
| TTD | Trichothiodystrophy |
| UNG | Uracil-N glycosylase |
| UV-DDB | UV-damaged DNA binding protein; a heterodimeric complex with DDB1 and DDB2 |
| XPC | Xeroderma pigmentosum C |
| XRCC1 | X-ray repair cross-complementing protein 1 |
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, I.; Bender, C.; Romieu, A.; Cadet, J.; Wood, R.D.; Lindahl, T. Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3832–3837. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Joh, H.M.; Park, J.M.; Kim, M.J.; Chung, T.H.; Kang, T.H. Non-thermal plasma-induced apoptosis is modulated by ATR- and PARP1-mediated DNA damage responses and circadian clock. Oncotarget 2016, 7, 32980–32989. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Choi, J.Y.; Yi, J.M.; Chung, J.W.; Leem, S.H.; Koh, S.S.; Kang, T.H. NDR1 modulates the UV-induced DNA-damage checkpoint and nucleotide excision repair. Biochem. Biophys. Res. Commun. 2015, 461, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Leem, S.H. Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res. 2014, 42, 4427–4434. [Google Scholar] [CrossRef]
- Choi, J.Y.; Park, J.M.; Yi, J.M.; Leem, S.H.; Kang, T.H. Enhanced nucleotide excision repair capacity in lung cancer cells by preconditioning with DNA-damaging agents. Oncotarget 2015, 6, 22575–22586. [Google Scholar] [CrossRef]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell. Mol. Life Sci. CMLS 2009, 66, 981–993. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Smeyne, R.J.; Wallace, J.D.; Curran, T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 8919–8923. [Google Scholar] [CrossRef]
- Gu, H.; Marth, J.D.; Orban, P.C.; Mossmann, H.; Rajewsky, K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science 1994, 265, 103–106. [Google Scholar] [CrossRef]
- Kucherlapati, M.; Yang, K.; Kuraguchi, M.; Zhao, J.; Lia, M.; Heyer, J.; Kane, M.F.; Fan, K.; Russell, R.; Brown, A.M.; et al. Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proc. Natl. Acad. Sci. USA 2002, 99, 9924–9929. [Google Scholar] [CrossRef] [PubMed]
- Puebla-Osorio, N.; Lacey, D.B.; Alt, F.W.; Zhu, C. Early embryonic lethality due to targeted inactivation of DNA ligase III. Mol. Cell. Biol. 2006, 26, 3935–3941. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, D.; Kunz, C.; Selfridge, J.; Lettieri, T.; Saito, Y.; MacDougall, E.; Wirz, A.; Schuermann, D.; Jacobs, A.L.; Siegrist, F.; et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 2011, 470, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Croteau, D.L.; Souza-Pinto, N.; Pitta, M.; Tian, J.; Wu, C.; Jiang, H.; Mustafa, K.; Keijzers, G.; Bohr, V.A.; et al. Evidence that OGG1 glycosylase protects neurons against oxidative DNA damage and cell death under ischemic conditions. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2011, 31, 680–692. [Google Scholar] [CrossRef]
- Lillenes, M.S.; Rabano, A.; Stoen, M.; Riaz, T.; Misaghian, D.; Mollersen, L.; Esbensen, Y.; Gunther, C.C.; Selnes, P.; Stenset, V.T.; et al. Altered DNA base excision repair profile in brain tissue and blood in Alzheimer’s disease. Mol. Brain 2016, 9, 61. [Google Scholar] [CrossRef]
- Weissman, L.; Jo, D.G.; Sorensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007, 35, 5545–5555. [Google Scholar] [CrossRef]
- Sykora, P.; Misiak, M.; Wang, Y.; Ghosh, S.; Leandro, G.S.; Liu, D.; Tian, J.; Baptiste, B.A.; Cong, W.N.; Brenerman, B.M.; et al. DNA polymerase beta deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes. Nucleic Acids Res. 2015, 43, 943–959. [Google Scholar] [CrossRef]
- Klungland, A.; Rosewell, I.; Hollenbach, S.; Larsen, E.; Daly, G.; Epe, B.; Seeberg, E.; Lindahl, T.; Barnes, D.E. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13300–13305. [Google Scholar] [CrossRef]
- Minowa, O.; Arai, T.; Hirano, M.; Monden, Y.; Nakai, S.; Fukuda, M.; Itoh, M.; Takano, H.; Hippou, Y.; Aburatani, H.; et al. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4156–4161. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, H.; Cunanan, C.; Okamoto, K.; Shibata, D.; Pan, J.; Barnes, D.E.; Lindahl, T.; McIlhatton, M.; Fishel, R.; et al. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res. 2004, 64, 3096–3102. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Kusakabe, M.; Onishi, Y.; Tada, H.; Kurihara, F.; Kusao, K.; Furukawa, M.; Iwai, S.; Yokoi, M.; Sakai, W.; Sugasawa, K. Mechanism and regulation of DNA damage recognition in nucleotide excision repair. Genes Environ. Off. J. Jpn. Environ. Mutagen Soc. 2019, 41, 2. [Google Scholar] [CrossRef] [PubMed]
- Scrima, A.; Konickova, R.; Czyzewski, B.K.; Kawasaki, Y.; Jeffrey, P.D.; Groisman, R.; Nakatani, Y.; Iwai, S.; Pavletich, N.P.; Thoma, N.H. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell 2008, 135, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Hoeijmakers, J.H.J.; Vermeulen, W.; Marteijn, J.A. The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 2019, 20, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Kang, T.H. Transcriptional and Posttranslational Regulation of Nucleotide Excision Repair: The Guardian of the Genome against Ultraviolet Radiation. Int. J. Mol. Sci. 2016, 17, 1840. [Google Scholar] [CrossRef]
- Lehmann, A.R.; McGibbon, D.; Stefanini, M. Xeroderma pigmentosum. Orphanet J. Dis. 2011, 6, 70. [Google Scholar] [CrossRef]
- Wang, Y.; Chakravarty, P.; Ranes, M.; Kelly, G.; Brooks, P.J.; Neilan, E.; Stewart, A.; Schiavo, G.; Svejstrup, J.Q. Dysregulation of gene expression as a cause of Cockayne syndrome neurological disease. Proc. Natl. Acad. Sci. USA 2014, 111, 14454–14459. [Google Scholar] [CrossRef]
- Pascucci, B.; Lemma, T.; Iorio, E.; Giovannini, S.; Vaz, B.; Iavarone, I.; Calcagnile, A.; Narciso, L.; Degan, P.; Podo, F.; et al. An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell 2012, 11, 520–529. [Google Scholar] [CrossRef]
- Andrade, L.N.; Nathanson, J.L.; Yeo, G.W.; Menck, C.F.; Muotri, A.R. Evidence for premature aging due to oxidative stress in iPSCs from Cockayne syndrome. Hum. Mol. Genet. 2012, 21, 3825–3834. [Google Scholar] [CrossRef]
- Cleaver, J.E.; Bezrookove, V.; Revet, I.; Huang, E.J. Conceptual developments in the causes of Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Stevnsner, T.; Muftuoglu, M.; Aamann, M.D.; Bohr, V.A. The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mech. Ageing Dev. 2008, 129, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Troelstra, C.; van Gool, A.; de Wit, J.; Vermeulen, W.; Bootsma, D.; Hoeijmakers, J.H. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell 1992, 71, 939–953. [Google Scholar] [CrossRef]
- van Hoffen, A.; Kalle, W.H.; de Jong-Versteeg, A.; Lehmann, A.R.; van Zeeland, A.A.; Mullenders, L.H. Cells from XP-D and XP-D-CS patients exhibit equally inefficient repair of UV-induced damage in transcribed genes but different capacity to recover UV-inhibited transcription. Nucleic Acids Res. 1999, 27, 2898–2904. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soltys, D.T.; Rocha, C.R.; Lerner, L.K.; de Souza, T.A.; Munford, V.; Cabral, F.; Nardo, T.; Stefanini, M.; Sarasin, A.; Cabral-Neto, J.B.; et al. Novel XPG (ERCC5) mutations affect DNA repair and cell survival after ultraviolet but not oxidative stress. Hum. Mutat. 2013, 34, 481–489. [Google Scholar] [CrossRef]
- Pascucci, B.; D’Errico, M.; Parlanti, E.; Giovannini, S.; Dogliotti, E. Role of nucleotide excision repair proteins in oxidative DNA damage repair: An updating. Biochem. Biokhimiia 2011, 76, 4–15. [Google Scholar] [CrossRef]
- Menoni, H.; Hoeijmakers, J.H.; Vermeulen, W. Nucleotide excision repair-initiating proteins bind to oxidative DNA lesions in vivo. J. Cell Biol. 2012, 199, 1037–1046. [Google Scholar] [CrossRef]
- Ng, J.M.; Vermeulen, W.; van der Horst, G.T.; Bergink, S.; Sugasawa, K.; Vrieling, H.; Hoeijmakers, J.H. A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev. 2003, 17, 1630–1645. [Google Scholar] [CrossRef]
- Araujo, S.J.; Tirode, F.; Coin, F.; Pospiech, H.; Syvaoja, J.E.; Stucki, M.; Hubscher, U.; Egly, J.M.; Wood, R.D. Nucleotide excision repair of DNA with recombinant human proteins: Definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev. 2000, 14, 349–359. [Google Scholar]
- Nishi, R.; Okuda, Y.; Watanabe, E.; Mori, T.; Iwai, S.; Masutani, C.; Sugasawa, K.; Hanaoka, F. Centrin 2 stimulates nucleotide excision repair by interacting with xeroderma pigmentosum group C protein. Mol. Cell. Biol. 2005, 25, 5664–5674. [Google Scholar] [CrossRef]
- D’Errico, M.; Parlanti, E.; Teson, M.; de Jesus, B.M.; Degan, P.; Calcagnile, A.; Jaruga, P.; Bjoras, M.; Crescenzi, M.; Pedrini, A.M.; et al. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 2006, 25, 4305–4315. [Google Scholar] [CrossRef] [PubMed]
- Bernardes de Jesus, B.M.; Bjoras, M.; Coin, F.; Egly, J.M. Dissection of the molecular defects caused by pathogenic mutations in the DNA repair factor XPC. Mol. Cell. Biol. 2008, 28, 7225–7235. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Bouziane, M.; Dammann, R.; Masutani, C.; Hanaoka, F.; Pfeifer, G.; O’Connor, T.R. 3-Methyladenine-DNA glycosylase (MPG protein) interacts with human RAD23 proteins. J. Biol. Chem. 2000, 275, 28433–28438. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Iwai, S.; Hanaoka, F.; Sugasawa, K. Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO J. 2003, 22, 164–173. [Google Scholar] [CrossRef]
- Shimizu, Y.; Uchimura, Y.; Dohmae, N.; Saitoh, H.; Hanaoka, F.; Sugasawa, K. Stimulation of DNA Glycosylase Activities by XPC Protein Complex: Roles of Protein-Protein Interactions. J. Nucleic Acid 2010, 2010. [Google Scholar] [CrossRef]
- Odell, I.D.; Wallace, S.S.; Pederson, D.S. Rules of engagement for base excision repair in chromatin. J. Cell. Physiol. 2013, 228, 258–266. [Google Scholar] [CrossRef]
- Sugasawa, K.; Okamoto, T.; Shimizu, Y.; Masutani, C.; Iwai, S.; Hanaoka, F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001, 15, 507–521. [Google Scholar] [CrossRef]
- Nishi, R.; Alekseev, S.; Dinant, C.; Hoogstraten, D.; Houtsmuller, A.B.; Hoeijmakers, J.H.; Vermeulen, W.; Hanaoka, F.; Sugasawa, K. UV-DDB-dependent regulation of nucleotide excision repair kinetics in living cells. DNA Repair 2009, 8, 767–776. [Google Scholar] [CrossRef]
- Fischer, E.S.; Scrima, A.; Bohm, K.; Matsumoto, S.; Lingaraju, G.M.; Faty, M.; Yasuda, T.; Cavadini, S.; Wakasugi, M.; Hanaoka, F.; et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 2011, 147, 1024–1039. [Google Scholar] [CrossRef]
- Esadze, A.; Rodriguez, G.; Cravens, S.L.; Stivers, J.T. AP-Endonuclease 1 Accelerates Turnover of Human 8-Oxoguanine DNA Glycosylase by Preventing Retrograde Binding to the Abasic-Site Product. Biochemistry 2017, 56, 1974–1986. [Google Scholar] [CrossRef]
- Jang, S.; Kumar, N.; Beckwitt, E.C.; Kong, M.; Fouquerel, E.; Rapic-Otrin, V.; Prasad, R.; Watkins, S.C.; Khuu, C.; Majumdar, C.; et al. Damage sensor role of UV-DDB during base excision repair. Nat. Struct. Mol. Biol. 2019, 26, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Beerens, N.; Hoeijmakers, J.H.; Kanaar, R.; Vermeulen, W.; Wyman, C. The CSB protein actively wraps DNA. J. Biol. Chem. 2005, 280, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Menoni, H.; Wienholz, F.; Theil, A.F.; Janssens, R.C.; Lans, H.; Campalans, A.; Radicella, J.P.; Marteijn, J.A.; Vermeulen, W. The transcription-coupled DNA repair-initiating protein CSB promotes XRCC1 recruitment to oxidative DNA damage. Nucleic Acids Res. 2018, 46, 7747–7756. [Google Scholar] [CrossRef] [PubMed]
- Khobta, A.; Epe, B. Repair of oxidatively generated DNA damage in Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Tuo, J.; Muftuoglu, M.; Chen, C.; Jaruga, P.; Selzer, R.R.; Brosh, R.M., Jr.; Rodriguez, H.; Dizdaroglu, M.; Bohr, V.A. The Cockayne Syndrome group B gene product is involved in general genome base excision repair of 8-hydroxyguanine in DNA. J. Biol. Chem. 2001, 276, 45772–45779. [Google Scholar] [CrossRef]
- Dianov, G.; Bischoff, C.; Sunesen, M.; Bohr, V.A. Repair of 8-oxoguanine in DNA is deficient in Cockayne syndrome group B cells. Nucleic Acids Res. 1999, 27, 1365–1368. [Google Scholar] [CrossRef]
- Tuo, J.; Jaruga, P.; Rodriguez, H.; Bohr, V.A.; Dizdaroglu, M. Primary fibroblasts of Cockayne syndrome patients are defective in cellular repair of 8-hydroxyguanine and 8-hydroxyadenine resulting from oxidative stress. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 668–674. [Google Scholar] [CrossRef]
- Ranes, M.; Boeing, S.; Wang, Y.; Wienholz, F.; Menoni, H.; Walker, J.; Encheva, V.; Chakravarty, P.; Mari, P.O.; Stewart, A.; et al. A ubiquitylation site in Cockayne syndrome B required for repair of oxidative DNA damage, but not for transcription-coupled nucleotide excision repair. Nucleic Acids Res. 2016, 44, 5246–5255. [Google Scholar] [CrossRef]
- Thorslund, T.; von Kobbe, C.; Harrigan, J.A.; Indig, F.E.; Christiansen, M.; Stevnsner, T.; Bohr, V.A. Cooperation of the Cockayne syndrome group B protein and poly(ADP-ribose) polymerase 1 in the response to oxidative stress. Mol. Cell. Biol. 2005, 25, 7625–7636. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD (+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef]
- Boetefuer, E.L.; Lake, R.J.; Dreval, K.; Fan, H.Y. Poly(ADP-ribose) polymerase 1 (PARP1) promotes oxidative stress-induced association of Cockayne syndrome group B protein with chromatin. J. Biol. Chem. 2018, 293, 17863–17874. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Bulky DNA lesions induced by reactive oxygen species. Chem. Res. Toxicol. 2008, 21, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Ravanat, J.L.; TavernaPorro, M.; Menoni, H.; Angelov, D. Oxidatively generated complex DNA damage: Tandem and clustered lesions. Cancer Lett. 2012, 327, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Melis, J.P.; van Steeg, H.; Luijten, M. Oxidative DNA damage and nucleotide excision repair. Antioxid. Redox Signal. 2013, 18, 2409–2419. [Google Scholar] [CrossRef]
- Gu, C.; Zhang, Q.; Yang, Z.; Wang, Y.; Zou, Y.; Wang, Y. Recognition and incision of oxidative intrastrand cross-link lesions by UvrABC nuclease. Biochemistry 2006, 45, 10739–10746. [Google Scholar] [CrossRef]
- Wang, J.; Cao, H.; You, C.; Yuan, B.; Bahde, R.; Gupta, S.; Nishigori, C.; Niedernhofer, L.J.; Brooks, P.J.; Wang, Y. Endogenous formation and repair of oxidatively induced G[8–5 m]T intrastrand cross-link lesion. Nucleic Acids Res. 2012, 40, 7368–7374. [Google Scholar] [CrossRef]
- Krokidis, M.G.; Terzidis, M.A.; Efthimiadou, E.; Zervou, S.K.; Kordas, G.; Papadopoulos, K.; Hiskia, A.; Kletsas, D.; Chatgilialoglu, C. Purine 5′,8-cyclo-2′-deoxynucleoside lesions: Formation by radical stress and repair in human breast epithelial cancer cells. Free Radic. Res. 2017, 51, 470–482. [Google Scholar] [CrossRef]
- Kropachev, K.; Ding, S.; Terzidis, M.A.; Masi, A.; Liu, Z.; Cai, Y.; Kolbanovskiy, M.; Chatgilialoglu, C.; Broyde, S.; Geacintov, N.E.; et al. Structural basis for the recognition of diastereomeric 5′,8-cyclo-2′-deoxypurine lesions by the human nucleotide excision repair system. Nucleic Acids Res. 2014, 42, 5020–5032. [Google Scholar] [CrossRef]
- Stingele, J.; Bellelli, R.; Boulton, S.J. Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 563–573. [Google Scholar] [CrossRef]
- DeMott, M.S.; Beyret, E.; Wong, D.; Bales, B.C.; Hwang, J.T.; Greenberg, M.M.; Demple, B. Covalent trapping of human DNA polymerase beta by the oxidative DNA lesion 2-deoxyribonolactone. J. Biol. Chem. 2002, 277, 7637–7640. [Google Scholar] [CrossRef]
- Reardon, J.T.; Cheng, Y.; Sancar, A. Repair of DNA-protein cross-links in mammalian cells. Cell Cycle 2006, 5, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.J.; Liu, S.; Andersen, N.; Yang, Z.; Johnson, K.M.; Price, N.E.; Wang, Y.; Gates, K.S. Chemical structure and properties of interstrand cross-links formed by reaction of guanine residues with abasic sites in duplex DNA. J. Am. Chem. Soc. 2015, 137, 3933–3945. [Google Scholar] [CrossRef] [PubMed]
- Price, N.E.; Johnson, K.M.; Wang, J.; Fekry, M.I.; Wang, Y.; Gates, K.S. Interstrand DNA-DNA cross-link formation between adenine residues and abasic sites in duplex DNA. J. Am. Chem. Soc. 2014, 136, 3483–3490. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Muller, J.G.; Rachlin, E.M.; Burrows, C.J. Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-Oxo-7,8-dihydroguanosine. Org. Lett. 2000, 2, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Niles, J.C.; Wishnok, J.S.; Tannenbaum, S.R. Spiroiminodihydantoin is the major product of the 8-oxo-7,8-dihydroguanosine reaction with peroxynitrite in the presence of thiols and guanosine photooxidation by methylene blue. Org. Lett. 2001, 3, 963–966. [Google Scholar] [CrossRef]
- Shafirovich, V.; Kropachev, K.; Anderson, T.; Liu, Z.; Kolbanovskiy, M.; Martin, B.D.; Sugden, K.; Shim, Y.; Chen, X.; Min, J.H.; et al. Base and Nucleotide Excision Repair of Oxidatively Generated Guanine Lesions in DNA. J. Biol. Chem. 2016, 291, 5309–5319. [Google Scholar] [CrossRef]
- Shafirovich, V.; Kropachev, K.; Kolbanovskiy, M.; Geacintov, N.E. Excision of Oxidatively Generated Guanine Lesions by Competing Base and Nucleotide Excision Repair Mechanisms in Human Cells. Chem. Res. Toxicol. 2019, 32, 753–761. [Google Scholar] [CrossRef]
- Reardon, J.T.; Bessho, T.; Kung, H.C.; Bolton, P.H.; Sancar, A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: Possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc. Natl. Acad. Sci. USA 1997, 94, 9463–9468. [Google Scholar] [CrossRef]
- Rosa, S.; Fortini, P.; Karran, P.; Bignami, M.; Dogliotti, E. Processing in vitro of an abasic site reacted with methoxyamine: A new assay for the detection of abasic sites formed in vivo. Nucleic Acids Res. 1991, 19, 5569–5574. [Google Scholar] [CrossRef]
- Kitsera, N.; Rodriguez-Alvarez, M.; Emmert, S.; Carell, T.; Khobta, A. Nucleotide excision repair of abasic DNA lesions. Nucleic Acids Res. 2019, 47, 8537–8547. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Hinshaw, H.D.; Jalal, S.I.; Sears, C.R.; Pawelczak, K.S.; Turchi, J.J. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol. Ther. 2016, 160, 65–83. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, T.-H.; Kang, T.-H. DNA Oxidation and Excision Repair Pathways. Int. J. Mol. Sci. 2019, 20, 6092. https://doi.org/10.3390/ijms20236092
Lee T-H, Kang T-H. DNA Oxidation and Excision Repair Pathways. International Journal of Molecular Sciences. 2019; 20(23):6092. https://doi.org/10.3390/ijms20236092
Chicago/Turabian StyleLee, Tae-Hee, and Tae-Hong Kang. 2019. "DNA Oxidation and Excision Repair Pathways" International Journal of Molecular Sciences 20, no. 23: 6092. https://doi.org/10.3390/ijms20236092
APA StyleLee, T.-H., & Kang, T.-H. (2019). DNA Oxidation and Excision Repair Pathways. International Journal of Molecular Sciences, 20(23), 6092. https://doi.org/10.3390/ijms20236092