Multifocal Signal Modulation Therapy by Celecoxib: A Strategy for Managing Castration-Resistant Prostate Cancer

 ,
,  , ,
, ,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
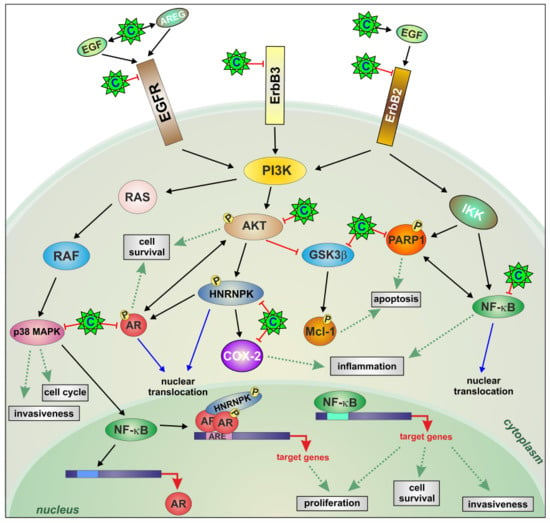
1. Introduction
2. Results
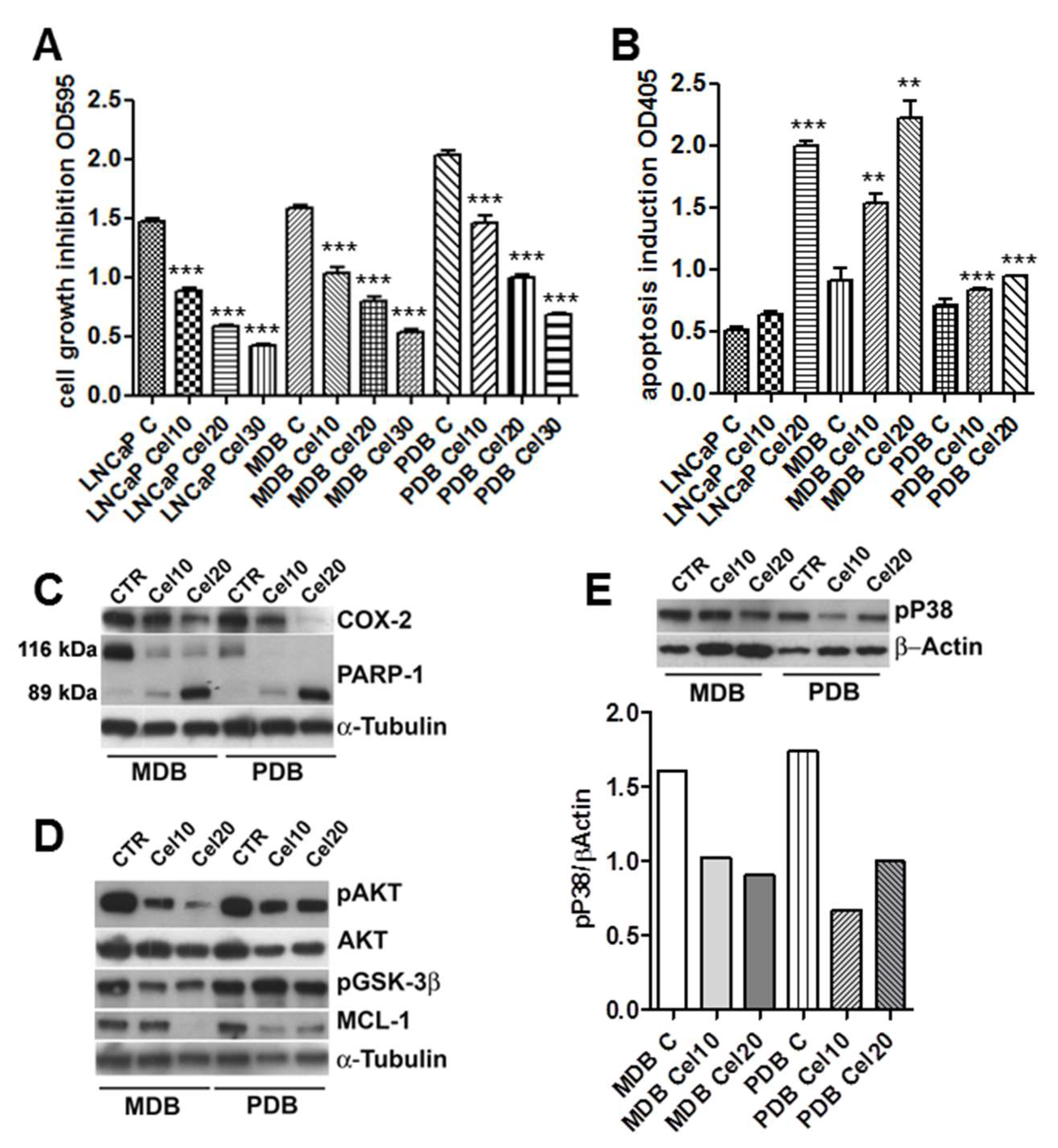
2.1. Effects of Celecoxib on the Growth and Apoptosis of the CRPC Cell Lines MDB and PDB
2.2. AKT Phosphorylation Is Inhibited by Celecoxib
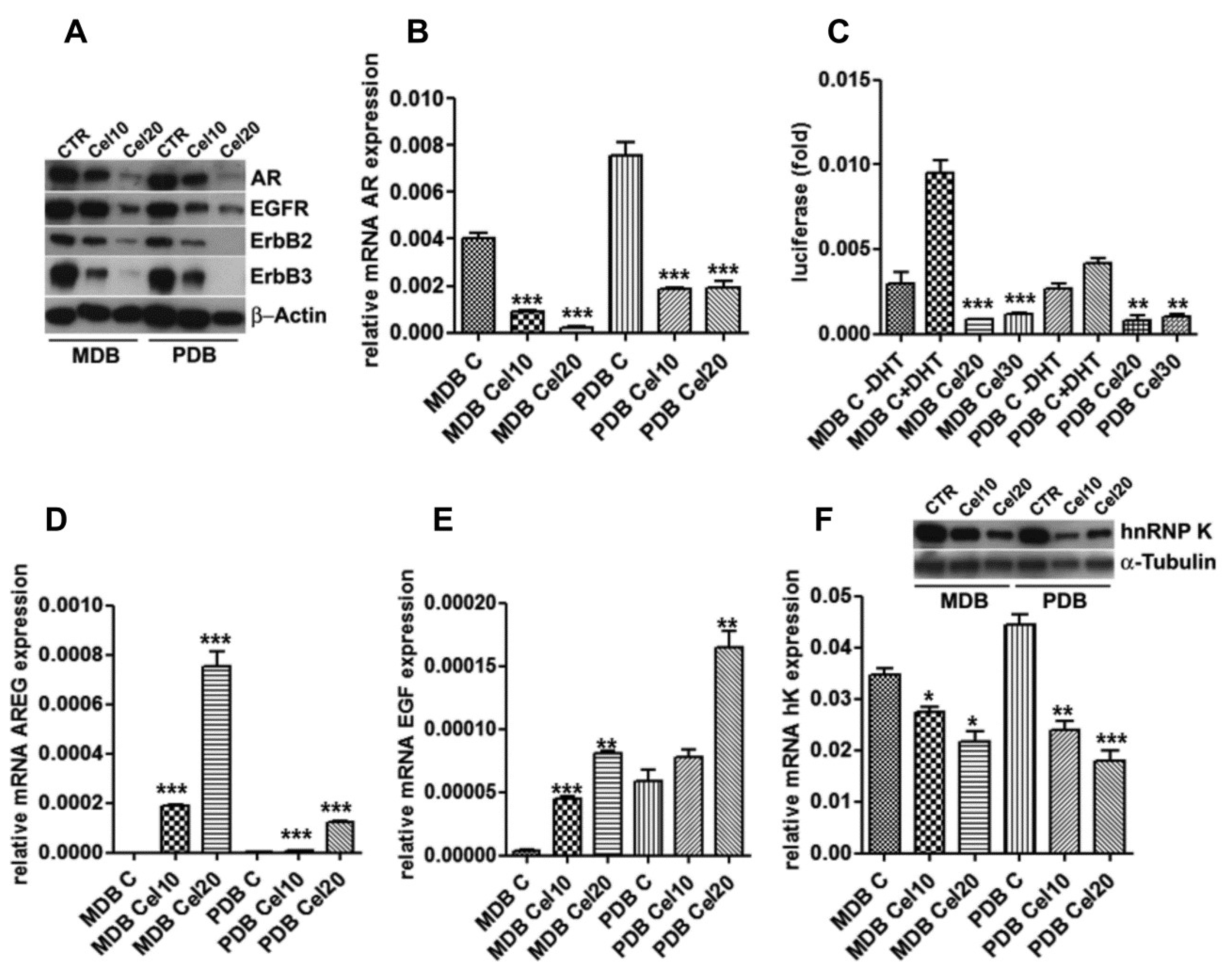
2.3. Celecoxib Attenuates AR Expression and Function in Resistant Cells through ErbB Receptor Inhibition and Epidermal Growth Factor (EGF) and Amphiregulin (AREG) Induction
2.4. Celecoxib Regulates AR through hnRNP K
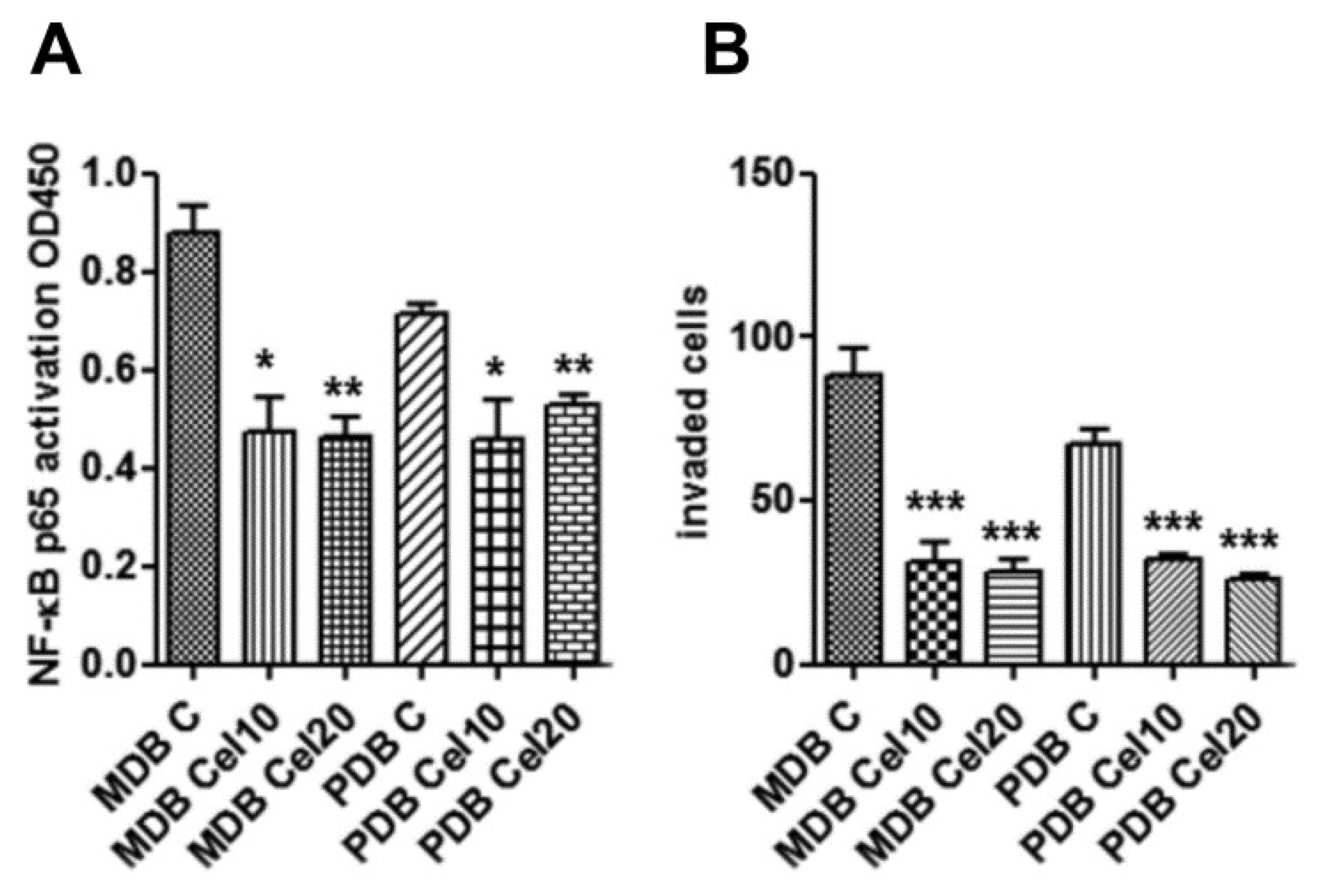
2.5. Downregulation of Constitutive NF-κB Activity by Celecoxib Reduces Resistant Cell Invasion
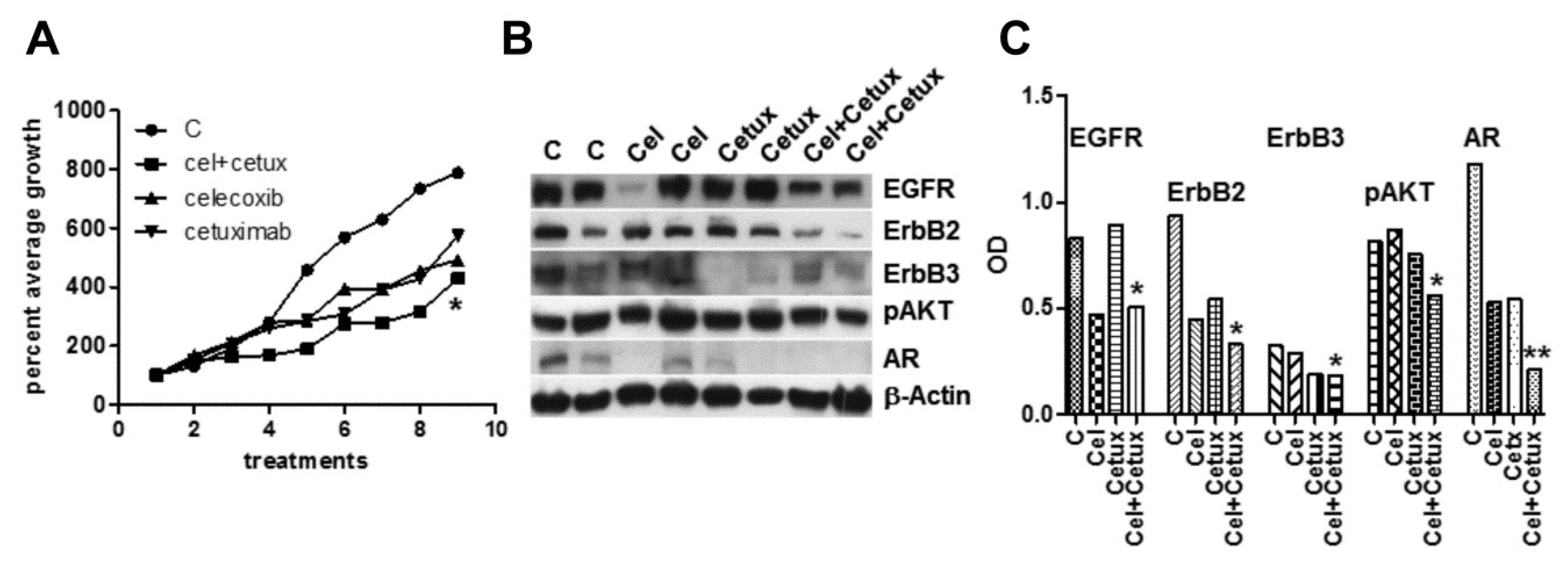
2.6. Effects of Celecoxib and Cetuximab Alone or in Combination on the Growth of MDB Xenografts in Immunodeficient Mice
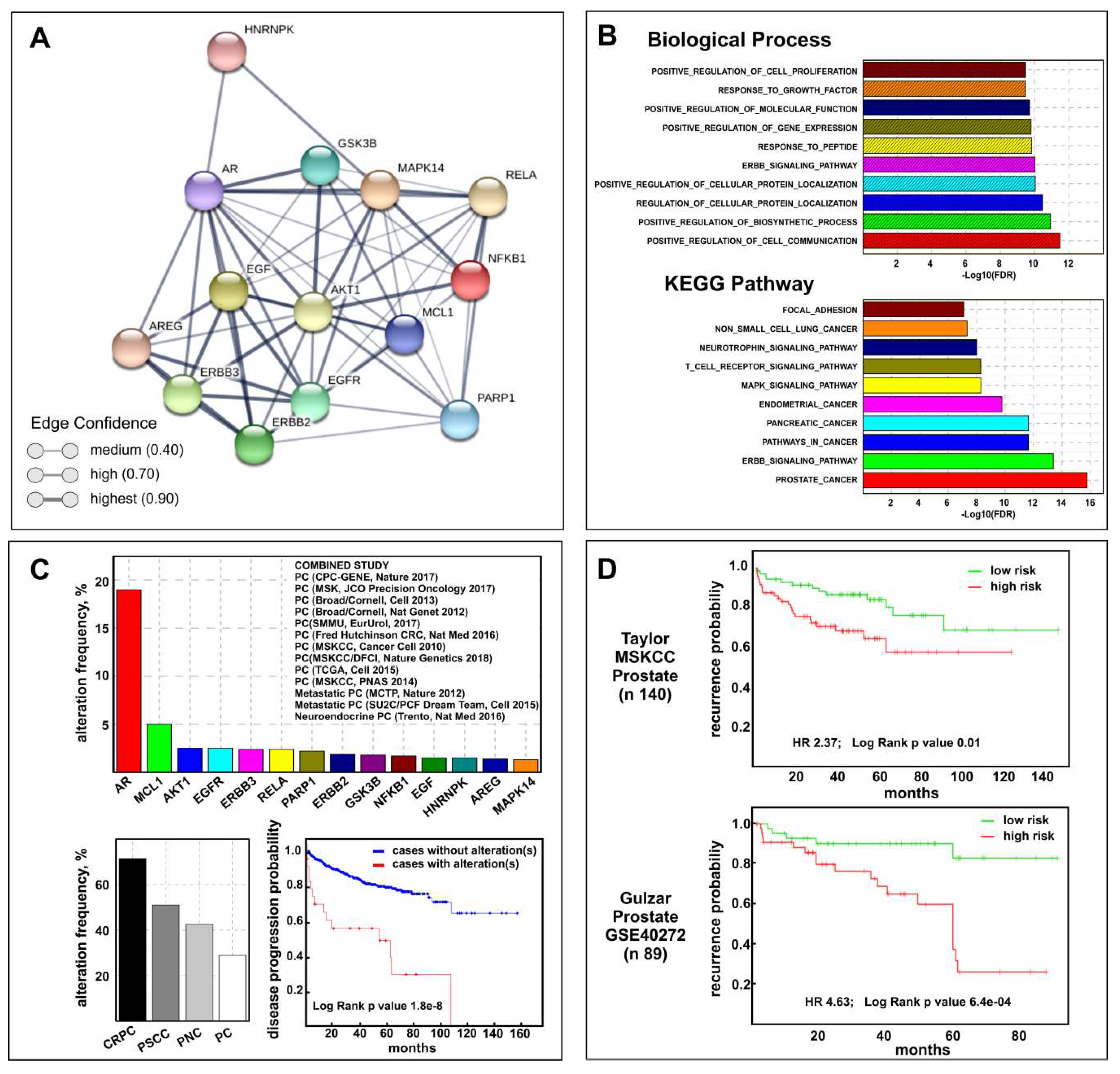
2.7. Biological and Clinical Relevance of the Celecoxib-Controlled Gene Set (CGS) in Prostate Cancer: A Bioinformatics Analysis
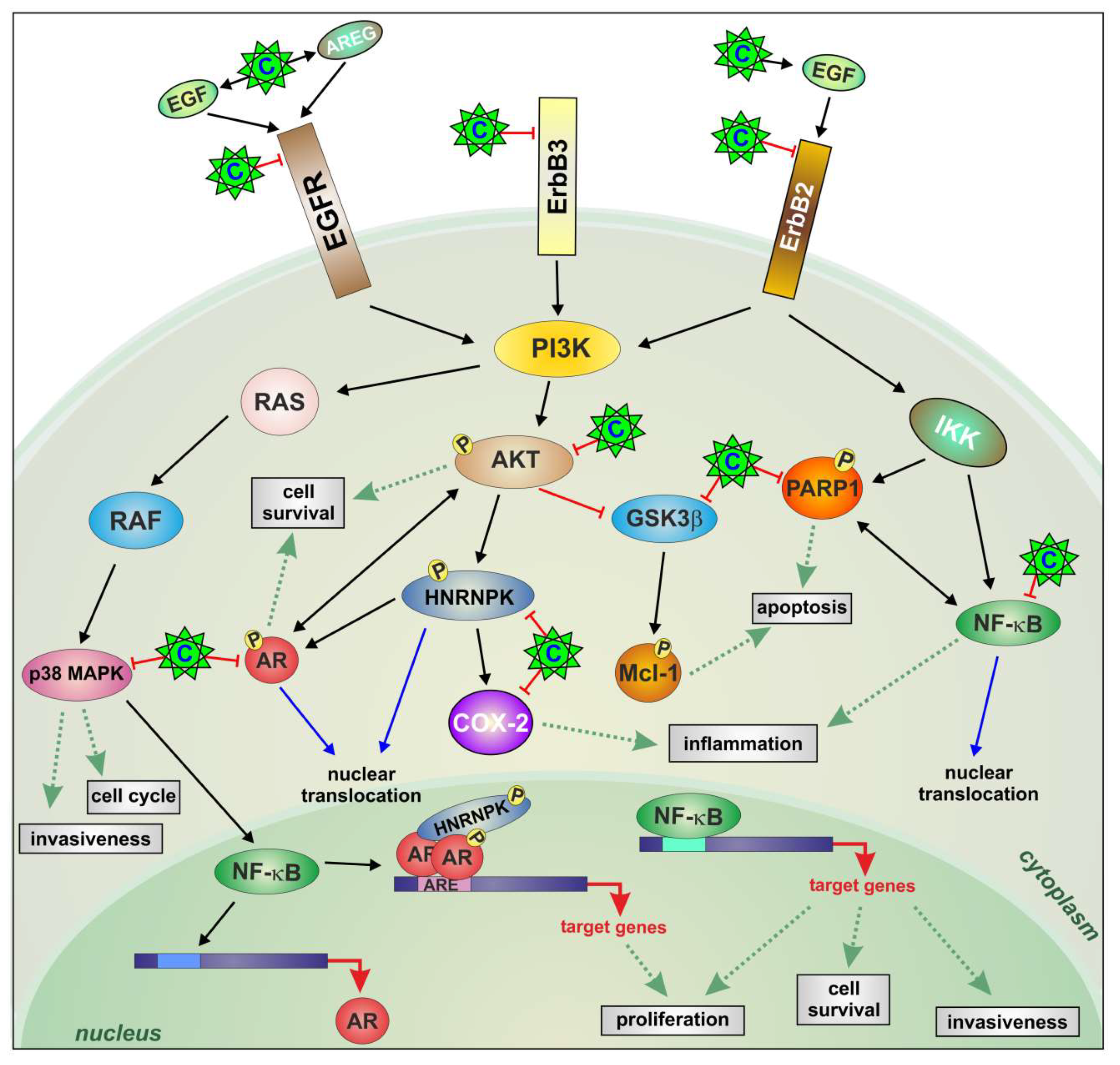
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Cell Proliferation, Apoptosis, Invasion, and ELISA Assays
4.3. Reporter Assay
4.4. RNA Isolation and Real-Time RT-PCR
4.5. Protein Extraction and Western Blot
4.6. In Vivo Studies
4.7. Immunohistochemistry (IHC)
4.8. Bioinformatics Analyses
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethics Approval and Consent to Participate
Abbreviations
| ADT | androgen deprivation therapy |
| AR | androgen receptor |
| AREG | amphiregulin |
| BPH | benign prostatic hyperplasia |
| BIC | bicalutamide |
| CGS | celecoxib-controlled gene set |
| COX-2 | cyclooxygenase-2 |
| CRPC | castration-resistant prostate cancer |
| DHT | 5-α-dihydrotestosterone |
| DSigDB | Drug Signature Database |
| EGF | epidermal growth factor |
| EGFR-Erb | epidermal growth factor receptor |
| GO-BP | gene ontology-biological process |
| GSK3β | glycogen synthase 3β |
| H&E | hematoxylin and eosin |
| hnRNP K | heterogeneous nuclear ribonucleoprotein K |
| mHSPC | metastatic hormone-sensitive PCa |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| PARP-1 | poly (ADP-ribose) polymerase-1 |
| PCa | prostate cancer |
| P38 | p38MAPK |
| PPI | protein–protein interaction |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Mousses, S.; Wagner, U.; Chen, Y.; Kim, J.W.; Bubendorf, L.; Bittner, M.; Pretlow, T.; Elkahloun, A.G.; Trepel, J.B.; Kallioniemi, O.P. Failure of hormone therapy in prostate cancer involves systematic restoration of androgen responsive genes and activation of rapamycin sensitive signaling. Oncogene 2001, 20, 6718–6723. [Google Scholar] [CrossRef] [PubMed]
- Leversha, M.A.; Han, J.; Asgari, Z.; Danila, D.C.; Lin, O.; Gonzalez-Espinoza, R.; Anand, A.; Lilja, H.; Heller, G.; Fleisher, M.; et al. Fluorescence in situ hybridization analysis of circulating tumor cells in metastatic prostate cancer. Clin. Cancer Res. 2009, 15, 2091–2097. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocr. Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef]
- Sciarra, A.; Di Silverio, F.; Salciccia, S.; Autran Gomez, A.M.; Gentilucci, A.; Gentile, V. Inflammation and chronic prostatic diseases: Evidence for a link? Eur. Urol. 2007, 52, 964–972. [Google Scholar] [CrossRef]
- Palapattu, G.S.; Sutcliffe, S.; Bastian, P.J.; Platz, E.A.; De Marzo, A.M.; Isaacs, W.B.; Nelson, W.G. Prostate carcinogenesis and inflammation: Emerging insights. Carcinogenesis 2005, 26, 1170–1181. [Google Scholar] [CrossRef]
- Di Silverio, F.; Bosman, C.; Salvatori, M.; Albanesi, L.; Proietti Pannunzi, L.; Ciccariello, M.; Cardi, A.; Salvatori, G.; Sciarra, A. Combination therapy with rofecoxib and finasteride in the treatment of men with lower urinary tract symptoms (LUTS) and benign prostatic hyperplasia (BPH). Eur. Urol. 2005, 47, 72–78, discussion 78–79. [Google Scholar] [CrossRef]
- Brizzolara, A.; Benelli, R.; Vene, R.; Barboro, P.; Poggi, A.; Tosetti, F.; Ferrari, N. The ErbB family and androgen receptor signaling are targets of Celecoxib in prostate cancer. Cancer Lett. 2017, 400, 9–17. [Google Scholar] [CrossRef]
- Ferrari, N.; Granata, I.; Capaia, M.; Piccirillo, M.; Guarracino, M.R.; Vene, R.; Brizzolara, A.; Petretto, A.; Inglese, E.; Morini, M.; et al. Adaptive phenotype drives resistance to androgen deprivation therapy in prostate cancer. Cell Commun. Signal. 2017, 15, 51. [Google Scholar] [CrossRef]
- Hsu, A.L.; Ching, T.T.; Wang, D.S.; Song, X.; Rangnekar, V.M.; Chen, C.S. The cyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking Akt activation in human prostate cancer cells independently of Bcl-2. J. Biol. Chem. 2000, 275, 11397–11403. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 162, 454. [Google Scholar] [CrossRef] [PubMed]
- Katsogiannou, M.; Ziouziou, H.; Karaki, S.; Andrieu, C.; de Villeneuve, M.H.; Rocchi, P. The hallmarks of castration-resistant prostate cancers. Cancer Treat. Rev. 2015, 41, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Pignon, J.C.; Koopmansch, B.; Nolens, G.; Delacroix, L.; Waltregny, D.; Winkler, R. Androgen receptor controls EGFR and ERBB2 gene expression at different levels in prostate cancer cell lines. Cancer Res. 2009, 69, 2941–2949. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Vadlamudi, R.; Nguyen, D.; Wang, R.A.; Costa, L.; Bagheri-Yarmand, R.; Mendelsohn, J.; Kumar, R. Growth factors regulate heterogeneous nuclear ribonucleoprotein K expression and function. J. Biol. Chem. 2001, 276, 9699–9704. [Google Scholar] [CrossRef]
- Yeap, B.B.; Wilce, J.A.; Leedman, P.J. The androgen receptor mRNA. Bioessays 2004, 26, 672–682. [Google Scholar] [CrossRef]
- Shin, C.; Kim, H. Functional roles of heterogeneous nuclear ribonucleoprotein K in post-transcriptional gene regulation. Precis. Future Med. 2018, 2, 158–166. [Google Scholar] [CrossRef]
- Barboro, P.; Salvi, S.; Rubagotti, A.; Boccardo, S.; Spina, B.; Truini, M.; Carmignani, G.; Introini, C.; Ferrari, N.; Boccardo, F.; et al. Prostate cancer: Prognostic significance of the association of heterogeneous nuclear ribonucleoprotein K and androgen receptor expression. Int. J. Oncol. 2014, 44, 1589–1598. [Google Scholar] [CrossRef]
- Ciarlo, M.; Benelli, R.; Barbieri, O.; Minghelli, S.; Barboro, P.; Balbi, C.; Ferrari, N. Regulation of neuroendocrine differentiation by AKT/hnRNPK/AR/beta-catenin signaling in prostate cancer cells. Int. J. Cancer 2012, 131, 582–590. [Google Scholar] [CrossRef]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.L.; Kamata, H.; Karin, M. IKK/NF-kappaB signaling: Balancing life and death-a new approach to cancer therapy. J. Clin. Investig. 2005, 115, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Merkhofer, E.C.; Cogswell, P.; Baldwin, A.S. Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha. Oncogene 2010, 29, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.A.; Lessard, L.; Mes-Masson, A.M.; Saad, F. Expression of NF-kappaB in prostate cancer lymph node metastases. Prostate 2004, 58, 308–313. [Google Scholar] [CrossRef]
- Jeong, J.H.; Park, S.J.; Dickinson, S.I.; Luo, J.L. A Constitutive Intrinsic Inflammatory Signaling Circuit Composed of miR-196b, Meis2, PPP3CC, and p65 Drives Prostate Cancer Castration Resistance. Mol. Cell 2017, 65, 154–167. [Google Scholar] [CrossRef]
- Benelli, R.; Vene, R.; Minghelli, S.; Carlone, S.; Gatteschi, B.; Ferrari, N. Celecoxib induces proliferation and Amphiregulin production in colon subepithelial myofibroblasts, activating erk1-2 signaling in synergy with EGFR. Cancer Lett. 2013, 328, 73–82. [Google Scholar] [CrossRef]
- Davids, J.S.; Carothers, A.M.; Damas, B.C.; Bertagnolli, M.M. Chronic cyclooxygenase-2 inhibition promotes myofibroblast-associated intestinal fibrosis. Cancer Prev. Res. 2010, 3, 348–358. [Google Scholar] [CrossRef]
- Rothermund, C.A.; Gopalakrishnan, V.K.; Eudy, J.D.; Vishwanatha, J.K. Casodex treatment induces hypoxia-related gene expression in the LNCaP prostate cancer progression model. BMC Urol. 2005, 5, 5. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Mo, F.; Wang, K.; McConeghy, B.; Brahmbhatt, S.; Jong, L.; Mitchell, D.M.; Johnston, R.L.; Haegert, A.; Li, E.; et al. Heterogeneity in the inter-tumor transcriptome of high risk prostate cancer. Genome Biol. 2014, 15, 426. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Emmons, M.F.; Faiao-Flores, F.; Smalley, K.S.M. The role of phenotypic plasticity in the escape of cancer cells from targeted therapy. Biochem. Pharmacol. 2016, 122, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R. The cellular origins of drug resistance in cancer. Nat. Med. 2016, 22, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Gravis, G.; Fizazi, K.; Joly, F.; Oudard, S.; Priou, F.; Esterni, B.; Latorzeff, I.; Delva, R.; Krakowski, I.; Laguerre, B.; et al. Androgen-deprivation therapy alone or with docetaxel in non-castrate metastatic prostate cancer (GETUG-AFU 15): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 149–158. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Chen, Y.H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Parmar, M.K. STAMPEDE trial and patients with non-metastatic prostate cancer-Authors’ reply. Lancet 2016, 388, 235–236. [Google Scholar] [CrossRef]
- Davies, A.; Conteduca, V.; Zoubeidi, A.; Beltran, H. Biological Evolution of Castration-resistant Prostate Cancer. Eur. Urol. Focus 2019, 5, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Ponguta, L.A.; Gregory, C.W.; French, F.S.; Wilson, E.M. Site-specific androgen receptor serine phosphorylation linked to epidermal growth factor-dependent growth of castration-recurrent prostate cancer. J. Biol. Chem. 2008, 283, 20989–21001. [Google Scholar] [CrossRef]
- Di Lorenzo, G.; Tortora, G.; D’Armiento, F.P.; De Rosa, G.; Staibano, S.; Autorino, R.; D’Armiento, M.; De Laurentiis, M.; De Placido, S.; Catalano, G.; et al. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin. Cancer Res. 2002, 8, 3438–3444. [Google Scholar]
- Jathal, M.K.; Chen, L.; Mudryj, M.; Ghosh, P.M. Targeting ErbB3: The New RTK(id) on the Prostate Cancer Block. Immunol. Endocr. Metab. Agents Med. Chem. 2011, 11, 131–149. [Google Scholar] [CrossRef]
- Ghosh, P.M.; Malik, S.; Bedolla, R.; Kreisberg, J.I. Akt in prostate cancer: Possible role in androgen-independence. Curr. Drug Metab. 2003, 4, 487–496. [Google Scholar] [CrossRef]
- Wang, Y.; Kreisberg, J.I.; Ghosh, P.M. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr. Cancer Drug Targets 2007, 7, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Koul, H.K.; Pal, M.; Koul, S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Nakamura, M.; Ishida, E.; Kishi, M.; Konishi, N. Roles of p38- and c-jun NH2-terminal kinase-mediated pathways in 2-methoxyestradiol-induced p53 induction and apoptosis. Carcinogenesis 2003, 24, 1067–1075. [Google Scholar] [CrossRef]
- Staal, J.; Beyaert, R. Inflammation and NF-kappaB Signaling in Prostate Cancer: Mechanisms and Clinical Implications. Cells 2018, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.P.; Li, J.; Yadav, S.S.; Tewari, A.K. Recent insights into NF-kappaB signalling pathways and the link between inflammation and prostate cancer. BJU Int. 2014, 114, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yamashita, H.; Yu, X.; Wang, J.; Franco, O.E.; Wang, Y.; Hayward, S.W.; Matusik, R.J. Inhibition of NF-kappa B signaling restores responsiveness of castrate-resistant prostate cancer cells to anti-androgen treatment by decreasing androgen receptor-variant expression. Oncogene 2015, 34, 3700–3710. [Google Scholar] [CrossRef] [PubMed]
- Barboro, P.; Borzi, L.; Repaci, E.; Ferrari, N.; Balbi, C. Androgen receptor activity is affected by both nuclear matrix localization and the phosphorylation status of the heterogeneous nuclear ribonucleoprotein K in anti-androgen-treated LNCaP cells. PLoS ONE 2013, 8, e79212. [Google Scholar] [CrossRef]
- Capaia, M.; Granata, I.; Guarracino, M.; Petretto, A.; Inglese, E.; Cattrini, C.; Ferrari, N.; Boccardo, F.; Barboro, P. A hnRNP K(-)AR-Related Signature Reflects Progression toward Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2018, 19, 1920. [Google Scholar] [CrossRef]
- Harris, R.E.; Beebe-Donk, J.; Alshafie, G.A. Cancer chemoprevention by cyclooxygenase 2 (COX-2) blockade: Results of case control studies. Subcell. Biochem. 2007, 42, 193–212. [Google Scholar]
- Smith, M.R.; Manola, J.; Kaufman, D.S.; Oh, W.K.; Bubley, G.J.; Kantoff, P.W. Celecoxib versus placebo for men with prostate cancer and a rising serum prostate-specific antigen after radical prostatectomy and/or radiation therapy. J. Clin. Oncol. 2006, 24, 2723–2728. [Google Scholar] [CrossRef]
- Harris, R.E. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology 2009, 17, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Benelli, R.; Monteghirfo, S.; Balbi, C.; Barboro, P.; Ferrari, N. Novel antivascular efficacy of metronomic docetaxel therapy in prostate cancer: hnRNP K as a player. Int. J. Cancer 2009, 124, 2989–2996. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benelli, R.; Barboro, P.; Costa, D.; Astigiano, S.; Barbieri, O.; Capaia, M.; Poggi, A.; Ferrari, N. Multifocal Signal Modulation Therapy by Celecoxib: A Strategy for Managing Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 6091. https://doi.org/10.3390/ijms20236091
Benelli R, Barboro P, Costa D, Astigiano S, Barbieri O, Capaia M, Poggi A, Ferrari N. Multifocal Signal Modulation Therapy by Celecoxib: A Strategy for Managing Castration-Resistant Prostate Cancer. International Journal of Molecular Sciences. 2019; 20(23):6091. https://doi.org/10.3390/ijms20236091
Chicago/Turabian StyleBenelli, Roberto, Paola Barboro, Delfina Costa, Simonetta Astigiano, Ottavia Barbieri, Matteo Capaia, Alessandro Poggi, and Nicoletta Ferrari. 2019. "Multifocal Signal Modulation Therapy by Celecoxib: A Strategy for Managing Castration-Resistant Prostate Cancer" International Journal of Molecular Sciences 20, no. 23: 6091. https://doi.org/10.3390/ijms20236091
APA StyleBenelli, R., Barboro, P., Costa, D., Astigiano, S., Barbieri, O., Capaia, M., Poggi, A., & Ferrari, N. (2019). Multifocal Signal Modulation Therapy by Celecoxib: A Strategy for Managing Castration-Resistant Prostate Cancer. International Journal of Molecular Sciences, 20(23), 6091. https://doi.org/10.3390/ijms20236091