Integrated Methylome and Transcriptome Analysis between the CMS-D2 Line ZBA and Its Maintainer Line ZB in Upland Cotton

 , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Methylation Landscapes of ZB and ZBA
2.2. Differential Methylome Analysis between ZB and ZBA
2.3. Correlation Between Altered DNA Methylation Patterns and Differential Gene Expression during Anther Development
2.4. GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) Enrichment Analysis of DMEGs
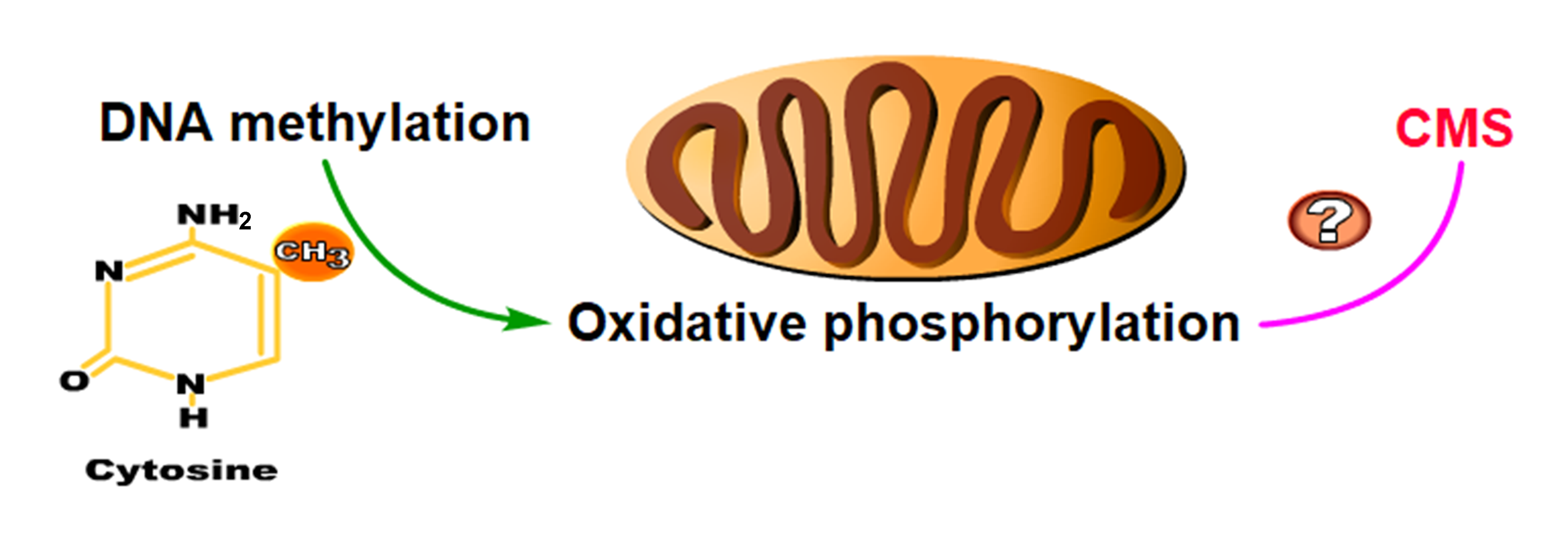
2.5. DNA Hypermethylation Maintains Oxidative Phosphorylation Homeostasis to Ensure the Normal Development of Cotton Anthers
3. Discussion
3.1. Overview of DNA Methylome in Cotton CMS Line Decoded by WGBS
3.2. Widespread Dynamic DNA Methylation and Its Association with Differential Gene Expression during Anther Development
3.3. Possible Regulatory Role of DNA Methylation in Cotton CMS
4. Materials and Methods
4.1. Plant Materials
4.2. DNA and RNA Extraction, Quantification, and Qualification
4.3. Bisulfite Treatment and Library Construction
4.4. DNA Methylation Data Analysis
4.5. RNA Sequencing and Data Analysis
4.6. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis
4.7. ROS and ATP Quantification
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CMS | Cytoplasmic male sterility |
| mCs | Methylated cytosine sites |
| MLs | Methylation levels |
| DMRs | Differentially methylated regions |
| DMGs | Differentially methylated genes |
| DEGs | Differentially expressed genes |
| DMEGs | DEGs associated with DMRs |
| ETC | Electron transport chain |
| ATP | Adenosine triphosphate |
| ROS | Reactive oxygen species |
| PCD | Programmed cell death |
| Rf | Restorer of fertility |
| CMS-WA | Wild-abortive CMS |
| TCA | Tricarboxylic acid |
| PPR | Pentatricopeptide repeat |
| MYB | V-myb avian myeloblastosis vira I oncogene homolog |
| GMS | Genic male sterility |
| NGS | Next-generation sequencing |
| WGBS | Whole-genome bisulfite sequencing |
| TTC | 2,3,5-Triphenyltetrazolium chloride |
| DMCs | Differentially methylated cytosines |
| FPKM | Fragments per kilobase of exons per million fragments mapped |
| GO | Gene ontology |
| BP | Biological process |
| CC | Cellular component |
| MF | Molecular function |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| HT | High-temperature |
| COX11 | CYTOCHROME C OXIDASE 11 |
| ICR-CAAS | Institute of Cotton Research of Chinese Academy of Agricultural Sciences |
| CTAB | Cetyltriethylammnonium bromide |
| NCBI | National Center for Biotechnology Information |
| SRA | Sequence Read Archive |
| FDR | False discovery rate |
| cDNA | Complementary DNA |
| SD | Standard deviation |
References
- Shahzad, K.; Qi, T.; Guo, L.; Tang, H.; Zhang, X.; Wang, H.; Qiao, X.; Zhang, M.; Zhang, B.; Feng, J.; et al. Adaptability and Stability Comparisons of Inbred and Hybrid Cotton in Yield and Fiber Quality Traits. Agronomy 2019, 9, 516. [Google Scholar] [CrossRef]
- Kubo, T.; Kitazaki, K.; Matsunaga, M.; Kagami, H.; Mikami, T. Male sterility-inducing mitochondrial genomes: How do they differ? Crit. Rev. Plant Sci. 2011, 30, 378–400. [Google Scholar] [CrossRef]
- Li, X.; Shahzad, K.; Guo, L.; Qi, T.; Zhang, X.; Wang, H.; Tang, H.; Qiao, X.; Zhang, J.; Wu, J.; et al. Using yield quantitative trait locus targeted SSR markers to study the relationship between genetic distance and yield heterosis in upland cotton (Gossypium hirsutum). Plant Breed. 2019, 138, 105–113. [Google Scholar] [CrossRef]
- Shahzad, K.; Li, X.; Qi, T.; Guo, L.; Tang, H.; Zhang, X.; Wang, H.; Zhang, M.; Zhang, B.; Qiao, X.; et al. Genetic analysis of yield and fiber quality traits in upland cotton (Gossypium hirsutum L.) cultivated in different ecological regions of China. J. Cotton Res. 2019, 2, 14. [Google Scholar] [CrossRef]
- Havey, M.J. The Use of Cytoplasmic Male Sterility for Hybrid Seed Production. In Molecular Biology and Biotechnology of Plant Organelles; Springer: Berlin/Heidelberg, Germany, 2004; pp. 623–634. [Google Scholar]
- Budar, F.; Pelletier, G. Male sterility in plants: Occurrence, determinism, significance and use. Comptes Rendus Acad. Sci. III 2001, 324, 543–550. [Google Scholar] [CrossRef]
- Wu, J.; Gong, Y.; Cui, M.; Qi, T.; Guo, L.; Zhang, J.; Xing, C. Molecular characterization of cytoplasmic male sterility conditioned by Gossypium harknessii cytoplasm (CMS-D2) in upland cotton. Euphytica 2011, 181, 17–29. [Google Scholar] [CrossRef]
- Wu, J.; Cao, X.; Guo, L.; Qi, T.; Wang, H.; Tang, H.; Zhang, J.; Xing, C. Development of a candidate gene marker for Rf1 based on a PPR gene in cytoplasmic male sterile CMS-D2 Upland cotton. Mol. Breed. 2014, 34, 231–240. [Google Scholar] [CrossRef]
- Yang, L.; Wu, Y.; Zhang, M.; Zhang, J.; Stewart, J.M.; Xing, C.; Wu, J.; Jin, S. Transcriptome, cytological and biochemical analysis of cytoplasmic male sterility and maintainer line in CMS-D8 cotton. Plant Mol. Biol. 2018, 97, 537–551. [Google Scholar] [CrossRef]
- Zhang, X.; Meng, Z.; Zhou, T.; Sun, G.; Shi, J.; Yu, Y.; Zhang, R.; Guo, S. Mitochondrial SCAR and SSR markers for distinguishing cytoplasmic male sterile lines from their isogenic maintainer lines in cotton. Plant Breed. 2012, 131, 563–570. [Google Scholar] [CrossRef]
- Kong, X.; Liu, D.; Liao, X.; Zheng, J.; Diao, Y.; Liu, Y.; Zhou, R. Comparative Analysis of the Cytology and Transcriptomes of the Cytoplasmic Male Sterility Line H276A and Its Maintainer Line H276B of Cotton (Gossypium barbadense L.). Int. J. Mol. Sci. 2017, 18, 2240. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, M.; Zhang, X.; Guo, L.; Qi, T.; Wang, H.; Tang, H.; Zhang, J.; Xing, C. Development of InDel markers for the restorer gene Rf1 and assessment of their utility for marker-assisted selection in cotton. Euphytica 2017, 213, 251. [Google Scholar] [CrossRef]
- Budar, F.; Touzet, P.; De Paepe, R. The nucleo-mitochondrial conflict in cytoplasmic male sterilities revisited. Genetica 2003, 117, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Huang, W.; Huang, Q.; Qin, X.; Yu, C.; Wang, L.; Li, S.; Zhu, R.; Zhu, Y. Mitochondria and cytoplasmic male sterility in plants. Mitochondrion 2014, 19, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Horn, R.; Gupta, K.J.; Colombo, N. Mitochondrion role in molecular basis of cytoplasmic male sterility. Mitochondrion 2014, 19, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Xu, H.; Liu, Z.; Guo, J.; Li, H.; Chen, L.; Fang, C.; Zhang, Q.; Bai, M.; Yao, N.; et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 2013, 45, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Wise, R.P.; Schnable, P.S. The rf2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science 1996, 272, 1334–1336. [Google Scholar] [CrossRef]
- Balk, J.; Leaver, C.J. The PET1-CMS mitochondrial mutation in sunflower is associated with premature programmed cell death and cytochrome c release. Plant Cell 2001, 13, 1803–1818. [Google Scholar] [CrossRef]
- Ji, J.; Huang, W.; Yin, C.; Gong, Z. Mitochondrial cytochrome c oxidase and F1Fo-ATPase dysfunction in peppers (Capsicum annuum L.) with cytoplasmic male sterility and its association with orf507 and Psiatp6-2 genes. Int. J. Mol. Sci. 2013, 14, 1050–1068. [Google Scholar] [CrossRef]
- Wan, C.; Li, S.; Wen, L.; Kong, J.; Wang, K.; Zhu, Y. Damage of oxidative stress on mitochondria during microspores development in Honglian CMS line of rice. Plant Cell Rep. 2007, 26, 373–382. [Google Scholar] [CrossRef]
- Yang, P.; Han, J.; Huang, J. Transcriptome sequencing and de novo analysis of cytoplasmic male sterility and maintenance in JA-CMS cotton. PLoS ONE 2014, 9, e112320. [Google Scholar] [CrossRef]
- Li, J.; Han, S.; Ding, X.; He, T.; Dai, J.; Yang, S.; Gai, J. Comparative Transcriptome Analysis between the Cytoplasmic Male Sterile Line NJCMS1A and Its Maintainer NJCMS1B in Soybean (Glycine max (L.) Merr.). PLoS ONE 2015, 10, e0126771. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Tian, H.; Huang, Y.Q.; Hu, J.; Ji, Y.X.; Li, S.Q.; Feng, Y.Q.; Guo, L.; Zhu, Y.G. Alterations of mitochondrial protein assembly and jasmonic acid biosynthesis pathway in Honglian (HL)-type cytoplasmic male sterility rice. J. Biol. Chem. 2012, 287, 40051–40060. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qin, T.; Wei, C.; Sun, J.; Dong, T.; Zhou, R.; Chen, Q.; Wang, Q. Using Transcriptome Analysis to Screen for Key Genes and Pathways Related to Cytoplasmic Male Sterility in Cotton (Gossypium hirsutum L.). Int. J. Mol. Sci. 2019, 20, 5120. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Cao, D.; Li, S.; Su, A.; Geng, J.; Grover, C.E.; Hu, S.; Hua, J. The complete mitochondrial genome of Gossypium hirsutum and evolutionary analysis of higher plant mitochondrial genomes. PLoS ONE 2013, 8, e69476. [Google Scholar] [CrossRef]
- Li, S.; Chen, Z.; Zhao, N.; Wang, Y.; Nie, H.; Hua, J. The comparison of four mitochondrial genomes reveals cytoplasmic male sterility candidate genes in cotton. BMC Genomics 2018, 19, 775. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Saze, H.; Kakutani, T. Differentiation of epigenetic modifications between transposons and genes. Curr. Opin. Plant. Biol. 2011, 14, 81–87. [Google Scholar] [CrossRef]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef]
- Zhu, J.K. Active DNA demethylation mediated by DNA glycosylases. Annu. Rev. Genet. 2009, 43, 143. [Google Scholar] [CrossRef]
- He, X.J.; Chen, T.; Zhu, J.K. Regulation and function of DNA methylation in plants and animals. Cell Res. 2011, 21, 442. [Google Scholar] [CrossRef]
- Chan, S.W.; Henderson, I.R.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tian, M.; Ci, D.; Zhang, D. Methylation of microRNA genes regulates gene expression in bisexual flower development in andromonoecious poplar. J. Exp. Bot. 2015, 66, 1891–1905. [Google Scholar] [CrossRef] [PubMed]
- Grafi, G. Epigenetics in plant development and response to stress. Biochim. Biophys. Acta 2011, 1809, 351–352. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Min, L.; Wang, M.; Wang, C.; Zhao, Y.; Li, Y.; Fang, Q.; Wu, Y.; Xie, S.; Ding, Y.; et al. Disrupted Genome Methylation in Response to High Temperature Has Distinct Affects on Microspore Abortion and Anther Indehiscence. Plant Cell 2018, 30, 1387–1403. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdzic, M.; Becker, J.D.; Feijo, J.A.; Martienssen, R.A. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 2009, 136, 461–472. [Google Scholar] [CrossRef]
- Xu, P.; Yan, W.; He, J.; Li, Y.; Zhang, H.; Peng, H.; Wu, X. DNA methylation affected by male sterile cytoplasm in rice (Oryza sativa L.). Mol. Breed. 2013, 31, 719–727. [Google Scholar] [CrossRef]
- Ba, Q.; Zhang, G.; Wang, J.; Niu, N.; Ma, S.; Wang, J. Gene expression and DNA methylation alterations in chemically induced male sterility anthers in wheat (Triticum aestivum L.). Acta Physiol. Plant. 2014, 36, 503–512. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, Y.; Lu, Y.; Wang, J.; Zhang, S.; Lan, H.; Rong, T.; Cao, M. DNA methylation analysis of sterile and fertile CMS-C hybrids and their parents in maize. J. Plant Biochem. Biotechnol. 2016, 25, 3–11. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, X.; Guo, L.; Qi, T.; Liu, G.; Feng, J.; Shahzad, K.; Zhang, B.; Li, X.; Wang, H.; et al. Single-base resolution methylomes of cotton CMS system reveal epigenomic changes in response to high-temperature stress during anther development. J. Exp. Bot 2019. [Google Scholar] [CrossRef]
- Han, F.; Zhang, X.; Liu, X.; Su, H.; Kong, C.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Liu, Y.; et al. Comparative analysis of genome wide DNA methylation profiles for the genic male sterile cabbage line 01-20S and its maintainer line. Genes 2017, 8, 159. [Google Scholar] [CrossRef]
- Omidvar, V.; Fellner, M. DNA methylation and transcriptomic changes in response to different lights and stresses in 7B-1 male-sterile tomato. PLoS ONE 2015, 10, e0121864. [Google Scholar] [CrossRef] [PubMed]
- Mutz, K.O.; Heilkenbrinker, A.; Lonne, M.; Walter, J.G.; Stahl, F. Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tontifilippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Wang, T.; Coarfa, C.; Nagarajan, R.P.; Hong, C.; Downey, S.L.; Johnson, B.E.; Fouse, S.D.; Delaney, A.; Zhao, Y.; et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010, 28, 1097–1105. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, X.; Chen, X.; Shu, N.; Wang, J.; Wang, D.; Wang, S.; Fan, W.; Guo, L.; Guo, X.; et al. Single-base resolution methylomes of upland cotton (Gossypium hirsutum L.) reveal epigenome modifications in response to drought stress. BMC Genomics 2017, 18, 297. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef]
- Ausin, I.; Greenberg, M.V.; Simanshu, D.K.; Hale, C.J.; Vashisht, A.A.; Simon, S.A.; Lee, T.F.; Feng, S.; Espanola, S.D.; Meyers, B.C.; et al. Involved in de novo 2-containing complex involved in RNA-directed DNA methylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 8374–8381. [Google Scholar] [CrossRef]
- Li, J.; Zhu, L.; Hull, J.J.; Liang, S.; Daniell, H.; Jin, S.; Zhang, X. Transcriptome analysis reveals a comprehensive insect resistance response mechanism in cotton to infestation by the phloem feeding insect Bemisia tabaci (whitefly). Plant Biotechnol. J. 2016, 14, 1956–1975. [Google Scholar] [CrossRef]
- Tu, L.; Zhang, X.; Liu, D.; Jin, S.; Cao, J.; Zhu, L.; Deng, F.; Tan, J.; Zhang, C. Suitable internal control genes for qRT-PCR normalization in cotton fiber development and somatic embryogenesis. Chin. Sci. Bull. 2007, 52, 3110–3117. [Google Scholar] [CrossRef]
- Artico, S.; Nardeli, S.M.; Brilhante, O.; Grossi-de-Sa, M.F.; Alves-Ferreira, M. Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC Plant Biol. 2010, 10, 49. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Q.; Zhang, B. Evaluation and selection of reliable reference genes for gene expression under abiotic stress in cotton (Gossypium hirsutum L.). Gene 2013, 530, 44–50. [Google Scholar] [CrossRef]
- Chen, C.; Xia, R.; Chen, H.; He, Y. TBtools, a Toolkit for Biologists integrating various HTS-data handling tools with a user-friendly interface. bioRxiv 2018, 289660. [Google Scholar] [CrossRef]
- Hu, L.; Liang, W.; Yin, C.; Cui, X.; Zong, J.; Wang, X.; Hu, J.; Zhang, D. Rice MADS3 regulates ROS homeostasis during late anther development. Plant Cell 2011, 23, 515–533. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.T.; Wan, Z.Y.; Li, S.; Zhang, Y. Spatiotemporal Production of Reactive Oxygen Species by NADPH Oxidase Is Critical for Tapetal Programmed Cell Death and Pollen Development in Arabidopsis. Plant Cell 2014, 26, 2007–2023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kimatu, J.N.; Xu, K.; Liu, B. DNA cytosine methylation in plant development. J. Genet. Genom. 2010, 37, 1–12. [Google Scholar] [CrossRef]
- Zhong, S.; Fei, Z.; Chen, Y.R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J.; et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J.K.; Lang, Z. Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. USA 2019, 116, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef]
- Feng, S.J.; Liu, X.S.; Tao, H.; Tan, S.K.; Chu, S.S.; Oono, Y.; Zhang, X.D.; Chen, J.; Yang, Z.M. Variation of DNA methylation patterns associated with gene expression in rice (Oryza sativa) exposed to cadmium. Plant. Cell Environ. 2016, 39, 2629–2649. [Google Scholar] [CrossRef]
- Ding, J.; Shen, J.; Mao, H.; Xie, W.; Li, X.; Zhang, Q. RNA-directed DNA methylation is involved in regulating photoperiod-sensitive male sterility in rice. Mol. Plant. 2012, 5, 1210–1216. [Google Scholar] [CrossRef]
- Chen, X.; Hu, J.; Zhang, H.; Ding, Y. DNA methylation changes in photoperiod-thermo-sensitive male sterile rice PA64S under two different conditions. Gene 2014, 537, 143–148. [Google Scholar] [CrossRef]
- Hu, J.; Chen, X.; Zhang, H.; Ding, Y. Genome-wide analysis of DNA methylation in photoperiod- and thermo-sensitive male sterile rice Peiai 64S. BMC Genom. 2015, 16, 102. [Google Scholar] [CrossRef]
- Chodavarapu, R.K.; Feng, S.; Ding, B.; Simon, S.A.; Lopez, D.; Jia, Y.; Wang, G.L.; Meyers, B.C.; Jacobsen, S.E.; Pellegrini, M. Transcriptome and methylome interactions in rice hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 12040–12045. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.; Xia, E.H.; Yao, Q.Y.; Liu, X.D.; Gao, L.Z. Autotetraploid rice methylome analysis reveals methylation variation of transposable elements and their effects on gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, E7022–E7029. [Google Scholar] [CrossRef] [PubMed]
- Gent, J.I.; Ellis, N.A.; Guo, L.; Harkess, A.E.; Yao, Y.; Zhang, X.; Dawe, R.K. CHH islands: De novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013, 23, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Eichten, S.R.; Hermanson, P.J.; Zaunbrecher, V.M.; Song, J.; Wendt, J.; Rosenbaum, H.; Madzima, T.F.; Sloan, A.E.; Huang, J.; et al. Genetic perturbation of the maize methylome. Plant Cell 2014, 26, 4602–4616. [Google Scholar] [CrossRef]
- Li, Q.; Song, J.; West, P.T.; Zynda, G.; Eichten, S.R.; Vaughn, M.W.; Springer, N.M. Examining the Causes and Consequences of Context-Specific Differential DNA Methylation in Maize. Plant Physiol 2015, 168, 1262–1274. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, T.; Stelly, D.M.; Chen, Z.J. Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol. 2017, 18, 99. [Google Scholar] [CrossRef]
- Song, Q.X.; Lu, X.; Li, Q.T.; Chen, H.; Hu, X.Y.; Ma, B.; Zhang, W.K.; Chen, S.Y.; Zhang, J.S. Genome-wide analysis of DNA methylation in soybean. Mol. Plant 2013, 6, 1961–1974. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, J.; Liu, Y.; Liu, S.; Liu, Z.; Duan, Z.; Wang, Z.; Zhu, B.; Guo, Y.L.; Tian, Z. DNA methylation footprints during soybean domestication and improvement. Genome Biol. 2018, 19, 128. [Google Scholar] [CrossRef]
- Xu, W.; Yang, T.; Dong, X.; Li, D.Z.; Liu, A. Genomic DNA Methylation Analyses Reveal the Distinct Profiles in Castor Bean Seeds with Persistent Endosperms. Plant Physiol. 2016, 171, 1242–1258. [Google Scholar] [CrossRef]
- Parkin, I.A.; Koh, C.; Tang, H.; Robinson, S.J.; Kagale, S.; Clarke, W.E.; Town, C.D.; Nixon, J.; Krishnakumar, V.; Bidwell, S.L.; et al. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 2014, 15, R77. [Google Scholar] [CrossRef]
- Millar, A.H.; Whelan, J.; Soole, K.L.; Day, D.A. Organization and regulation of mitochondrial respiration in plants. Annu. Rev. Plant. Biol. 2011, 62, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Djanaguiraman, M.; Perumal, R.; Jagadish, S.V.K.; Ciampitti, I.A.; Welti, R.; Prasad, P.V.V. Sensitivity of sorghum pollen and pistil to high-temperature stress. Plant. Cell Environ. 2017, 41, 1065–1082. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dai, X.; Li, L.; Jiao, Z.; Huang, Q. Metabolism of reactive oxygen species in cytoplasmic male sterility of rice by marking upmost pulvinus interval. Appl. Biochem. Biotechnol. 2015, 175, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Zhang, X.; Zhu, Y.; Zhu, W.; Xie, H.; Wang, X. Metabolism of reactive oxygen species in cotton cytoplasmic male sterility and its restoration. Plant Cell Rep. 2007, 26, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shi, X.; Li, S.; Hu, G.; Zhang, L.; Song, X. Tapetal-Delayed Programmed Cell Death (PCD) and Oxidative Stress-Induced Male Sterility of Aegilops uniaristata Cytoplasm in Wheat. Int. J. Mol. Sci. 2018, 19, 1708. [Google Scholar] [CrossRef]
- Peng, X.; Li, F.; Li, S.; Zhu, Y. Expression of a mitochondrial gene orfH79 from the CMS-HongLian rice inhibits Saccharomyces cerevisiae growth and causes excessive ROS accumulation and decrease in ATP. Biotechnol. Lett. 2009, 31, 409–414. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, M.; Zhang, B.; Zhang, X.; Guo, L.; Qi, T.; Wang, H.; Zhang, J.; Xing, C. Genome-wide comparative transcriptome analysis of CMS-D2 and its maintainer and restorer lines in upland cotton. BMC Genom. 2017, 18, 454. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, X.; Liu, G.; Guo, L.; Qi, T.; Zhang, M.; Li, X.; Wang, H.; Tang, H.; Qiao, X.; et al. A combined small RNA and transcriptome sequencing analysis reveal regulatory roles of miRNAs during anther development of upland cotton carrying cytoplasmic male sterile Gossypium harknessii (D2) cytoplasm. BMC Plant Biol. 2018, 18, 242. [Google Scholar] [CrossRef]
- Mehetre, S.S. Anther and pollen development in cotton haploids and their parents. Proc. Plant Sci. 1982, 91, 409–416. [Google Scholar] [CrossRef]
- Zhang, J.; Stewart, J.M. Economical and rapid method for extracting cotton genomic DNA. J. Cotton Sci. 2000, 4, 193–201. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Ghosh, S.; Chan, C.K. Analysis of RNA-Seq Data Using TopHat and Cufflinks. Methods Mol. Biol. 2016, 1374, 339–361. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Rychlik, W. OLIGO 7 primer analysis software. Methods Mol. Biol. 2007, 402, 35–60. [Google Scholar] [CrossRef]
- St John, J.B. Determination of ATP in Chlorella with the luciferin-luciferase enzyme system. Anal. Biochem. 1970, 37, 409–416. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Guo, L.; Qi, T.; Zhang, X.; Tang, H.; Wang, H.; Qiao, X.; Zhang, B.; Feng, J.; Zuo, Z.; et al. Integrated Methylome and Transcriptome Analysis between the CMS-D2 Line ZBA and Its Maintainer Line ZB in Upland Cotton. Int. J. Mol. Sci. 2019, 20, 6070. https://doi.org/10.3390/ijms20236070
Zhang M, Guo L, Qi T, Zhang X, Tang H, Wang H, Qiao X, Zhang B, Feng J, Zuo Z, et al. Integrated Methylome and Transcriptome Analysis between the CMS-D2 Line ZBA and Its Maintainer Line ZB in Upland Cotton. International Journal of Molecular Sciences. 2019; 20(23):6070. https://doi.org/10.3390/ijms20236070
Chicago/Turabian StyleZhang, Meng, Liping Guo, Tingxiang Qi, Xuexian Zhang, Huini Tang, Hailin Wang, Xiuqin Qiao, Bingbing Zhang, Juanjuan Feng, Zhidan Zuo, and et al. 2019. "Integrated Methylome and Transcriptome Analysis between the CMS-D2 Line ZBA and Its Maintainer Line ZB in Upland Cotton" International Journal of Molecular Sciences 20, no. 23: 6070. https://doi.org/10.3390/ijms20236070
APA StyleZhang, M., Guo, L., Qi, T., Zhang, X., Tang, H., Wang, H., Qiao, X., Zhang, B., Feng, J., Zuo, Z., Li, T., Shahzad, K., Wu, J., & Xing, C. (2019). Integrated Methylome and Transcriptome Analysis between the CMS-D2 Line ZBA and Its Maintainer Line ZB in Upland Cotton. International Journal of Molecular Sciences, 20(23), 6070. https://doi.org/10.3390/ijms20236070