An Ancient Mutation in the TPH1 Gene is Consistent with the Changes in Mammalian Reproductive Rhythm

 ,
,
Abstract
1. Introduction
2. Results
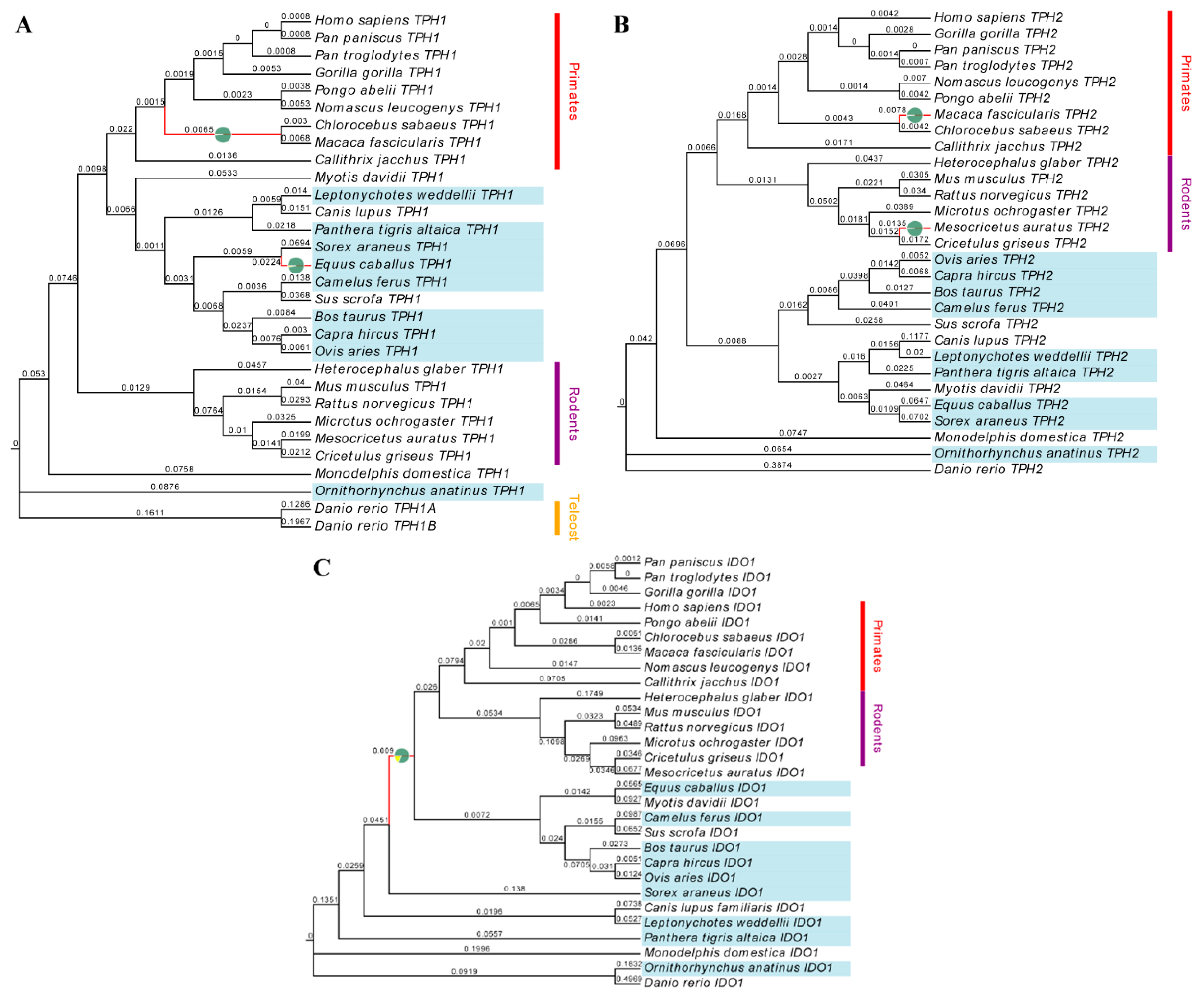
2.1. Evolution of Melatonin-Related Genes
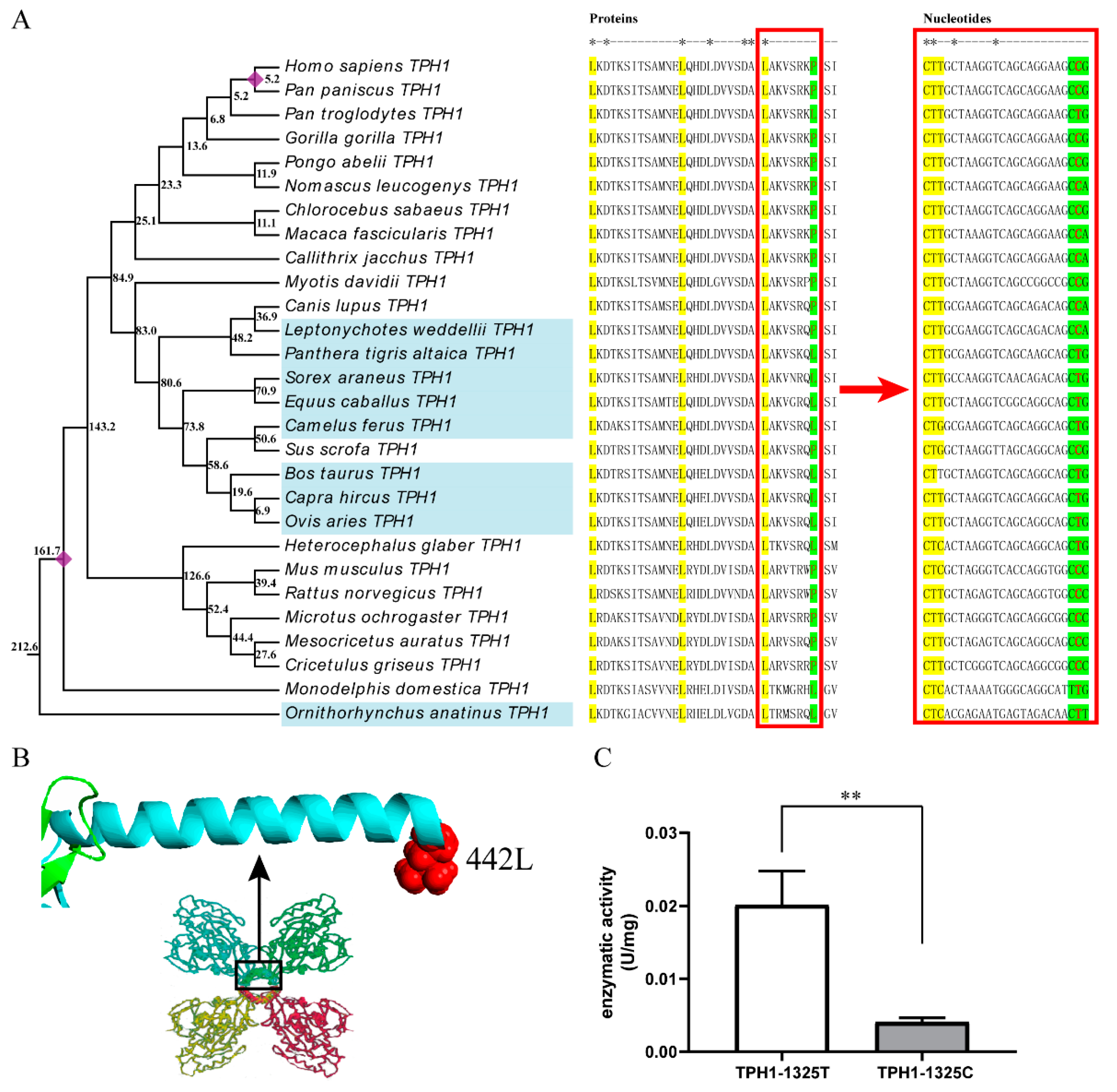
2.2. Functional Divergence of Melatonin-Related Proteins
2.3. The T/C Polymorphism at the 1325th Site in TPH1 Gene Change Enzyme Activity
3. Discussion
3.1. TPH1, TPH2 and IDO1 Genes are Positively Selected in Mammals
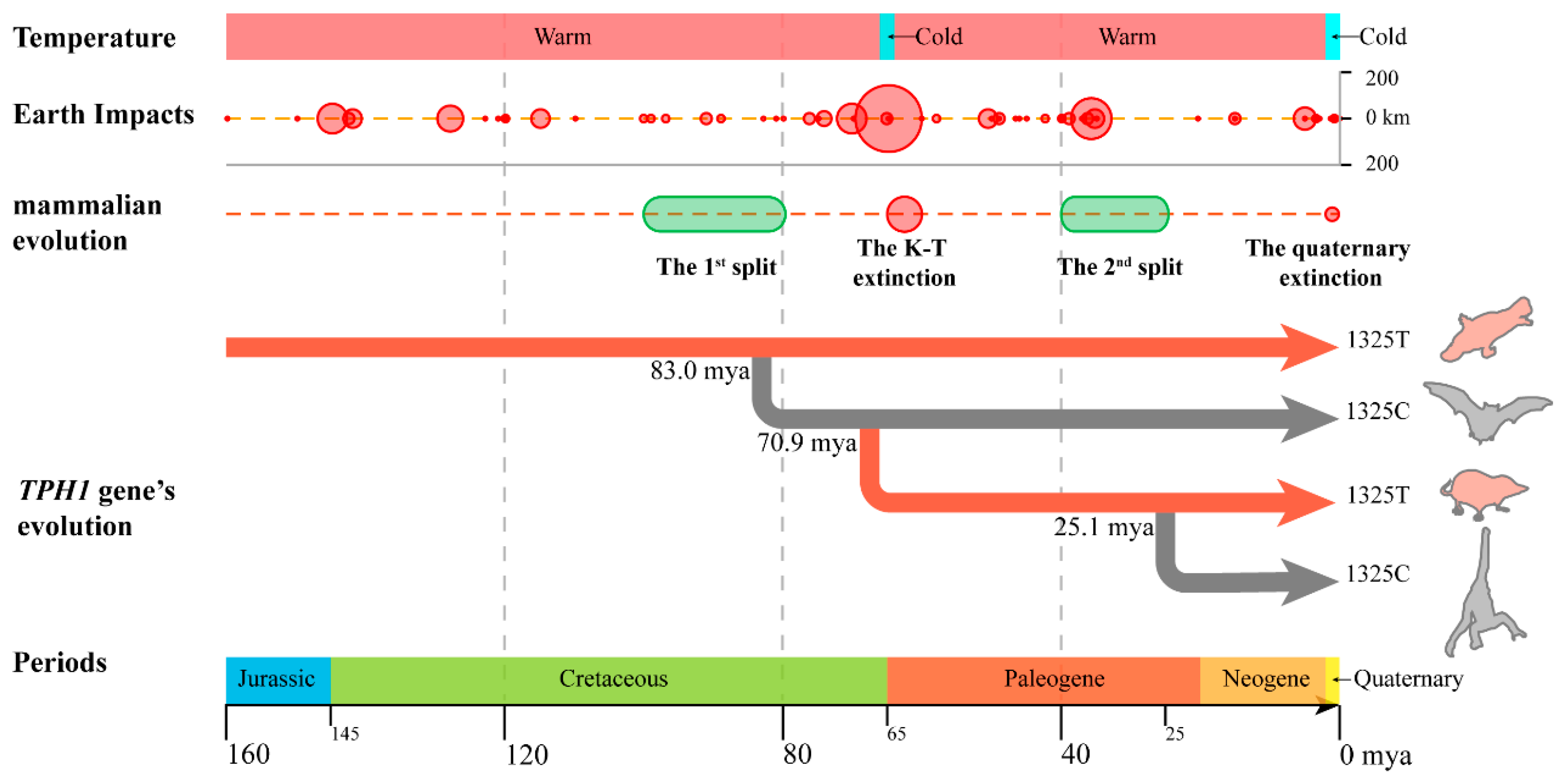
3.2. Effect of T1325C Polymorphism of the TPH1 Gene on Seasonal Reproduction in Mammals
3.3. Seasonal Reproduction Improved the Adaptability of Mammals to Cretaceous–Tertiary Extinction Events
4. Materials and Methods
4.1. Data Collection
4.2. Phylogenetic Trees and Divergence Time
4.3. Positive Selection Assessment
4.4. Functional Diversification Analysis
4.5. Structural Analysis and Homology Modeling
4.6. Cell Expression and Enzyme Activity Determinations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lewis, P.D. A review of lighting for broiler breeders. Br. Poult. Sci. 2006, 47, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, N.E.; Sack, D.A.; Jacobsen, F.M.; James, S.P.; Parry, B.L.; Arendt, J.; Tamarkin, L.; Wehr, T.A. Melatonin in seasonal affective disorder and phototherapy. J. Neural Transm. Suppl. 1986, 21, 257–267. [Google Scholar] [PubMed]
- Kvetnoy, I. Extrapineal melatonin in pathology: New perspectives for diagnosis, prognosis and treatment of illness. Neuro. Endocrinol. Lett. 2002, 23, 92–96. [Google Scholar] [PubMed]
- ViviD, D.; Bentley, G. Seasonal reproduction in vertebrates: Melatonin synthesis, binding, and functionality using Tinbergen’s four questions. Molecules 2018, 23, 652. [Google Scholar] [CrossRef] [PubMed]
- Marchesani, R.B. Metaphorical thinking on art, education and therapy: Late night conversations with bernard brenner. Psychother. Patient 2003, 12, 17–44. [Google Scholar] [CrossRef]
- McKinney, J.; Knappskog, P.M.; Haavik, J. Different properties of the central and peripheral forms of human tryptophan hydroxylase. J. Neurochem. 2005, 92, 311–320. [Google Scholar] [CrossRef]
- Zill, P.; Büttner, A.; Eisenmenger, W.; Bondy, B.; Ackenheil, M. Regional mRNA expression of a second tryptophan hydroxylase isoform in postmortem tissue samples of two human brains. Eur. Neuropsychopharmacol. 2004, 14, 282–284. [Google Scholar] [CrossRef]
- Patel, P.D.; Pontrello, C.; Burke, S. Robust and tissue-specific expression of TPH2 versus TPH1 in rat raphe and pineal gland. Biol. Psychiatry. 2004, 55, 428–433. [Google Scholar] [CrossRef]
- Dores, R.M. Observations on the evolution of the melanocortin receptor gene family: Distinctive features of the melanocortin-2 receptor. Front. Neurosci. 2013, 7, 28. [Google Scholar] [CrossRef]
- Deprés-Brummer, P.; Metzger, G.; Morin, D.; Urien, S.; Touitou, Y.; Tillement, J.P.; Claustrat, B.; Lévi, F. Pharmacokinetically guided melatonin scheduling in rats with circadian system suppression. Eur. J. Pharmacol. 1996, 312, 171–178. [Google Scholar] [CrossRef]
- Li, C.; Li, G.; Tan, D.X.; Li, F.; Ma, X. A novel enzyme-dependent melatonin metabolite in humans. J. Pineal Res. 2012, 54, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Idle, J.R.; Krausz, K.W.; Gonzalez, F.J. Metabolism of melatonin by human cytochromes p450. Drug Metab. Dispos. 2005, 33, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Dun-Xian, T.; Lucien, M.; Eduardo, E.Z.; Zhou, Z.; Russel, R. Melatonin as a potent and inducible endogenous antioxidant: Synthesis and metabolism. Molecules 2015, 20, 18886–18906. [Google Scholar]
- Maiwald, S.; Wehner, R.; Schmitz, M.; Bornhäuser, M.; Loeb, S.; Wassmuth, R. Ido1 and ido2 gene expression analysis by quantitative polymerase chain reaction. Tissue Antigens 2011, 77, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.M.; Peet, E.; Manuck, S.B.; Williamson, D.E.; Dahl, R.E.; Ferrell, R.E.; Hariri, A.R. A regulatory variant of the human tryptophan hydroxylase-2 gene biases amygdala reactivity. Mol. Psychiatry 2005, 10, 884–888. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rotondo, A.; Schuebel, K.; Bergen, A.; Aragon, R.; Virkkunen, M.; Linnoila, M.; Goldman, D.; Nielsen, D. Identification of four variants in the tryptophan hydroxylase promoter and association to behavior. Mol. Psychiatry 1999, 4, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Chattun, M.R.; Yan, R.; Geng, J.; Zhu, R.; Shao, J.; Lu, Q.; Yao, Z. TPH-2 gene polymorphism in major depressive disorder patients with early-wakening symptom. Front. Neurosci. 2018, 12, 827. [Google Scholar] [CrossRef]
- Mura, M.C.; Luridiana, S.; Pulinas, L.; Di Stefano, M.V.; Carcangiu, V. Reproductive response to male joining with ewes with different allelic variants of the mtnr1a gene. Anim. Reprod. Sci. 2019, 200, 67–74. [Google Scholar] [CrossRef]
- Suzuki, Y. Statistical methods for detecting natural selection from genomic data. Genes Genet. Syst. 2010, 85, 359–376. [Google Scholar] [CrossRef]
- Twyman, H.; Andersson, S.; Mundy, N.I. Evolution of CYP2J19, a gene involved in colour vision and red coloration in birds: Positive selection in the face of conservation and pleiotropy. BMC Evol. Biol. 2018, 1, 22. [Google Scholar] [CrossRef]
- Berlin, S.; Smith, N.G. Testing for adaptive evolution of the female reproductive protein ZPC in mammals, birds and fishes reveals problems with the M7-M8 likelihood ratio test. BMC Evol. Biol. 2005, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less is more: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Boularand, S.; Darmon, M.C.; Ravassard, P.; Mallet, J. Characterization of the human tryptophan hydroxylase gene promoter transcriptional regulation by camp requires a new motif distinct from the camp-responsive element. J. Biol. Chem. 1995, 27, 3757–3764. [Google Scholar] [CrossRef] [PubMed]
- Walther, D.J.; Peter, J.U.; Bashammakh, S.; Hörtnagl, H.; Voits, M.; Fink, H.; Bader, M. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 2003, 299, 76. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.M.; Hernandez, L.L.; Horseman, N.D. Serotonin and serotonin transport in the regulation of lactation. J. Mammary Gland Biol. Neoplasia 2014, 19, 139–146. [Google Scholar] [CrossRef]
- Bernard, M.; Guerlotté, J.; Grève, P.; Gréchez-Cassiau, A.; Iuvone, M.P.; Zatz, M.; Chong, N.W.; Klein, D.C.; Voisin, P. Melatonin synthesis pathway: Circadian regulation of the genes encoding the key enzymes in the chicken pineal gland and retina. Reprod. Nutr. Dev. 1999, 39, 325–334. [Google Scholar] [CrossRef]
- Weems, P.W.; Goodman, R.L.; Lehman, M.N. Neural mechanisms controlling seasonal reproduction: Principles derived from the sheep model and its comparison with hamsters. Front. Neuroendocrinol. 2015, 37, 43–51. [Google Scholar] [CrossRef]
- Qian, S.W.; Yang, X.Z.; Li, H.Y.; Li, H.; Zhang, S.S.; Lu, Y.; Wang, J.P.; Zhang, Z.X.; Xu, J. Analysis on time queue data of circadian rhythms of 5-hydroxytryptamine. CJTCMP 2016, 31, 93–95. [Google Scholar]
- Keller, G.; Stinnesbeck, W.; Adatte, T.; Stüben, D. Multiple impacts across the Cretaceous–Tertiary boundary. Earth-Sci. Rev. 2003, 62, 327–363. [Google Scholar] [CrossRef]
- Bininda-Emonds, O.R.; Cardillo, M.; Jones, K.E.; MacPhee, R.D.; Beck, R.M.; Grenyer, R.; Price, S.A.; Vos, R.A.; Gittleman, J.L.; Purvis, A. The delayed rise of present-day mammals. Nature 2007, 446, 507–512. [Google Scholar] [CrossRef]
- MacLeod, N.; Rawson, P.F.; Forey, P.L.; Banner, F.T.; Boudagher-Fadel, M.K.; Bown, P.R.; Burnett, J.A.; Chambers, P.; Culver, S.; Evans, S.E.; et al. The Cretaceous-tertiary biotic transition. J. Geol. Soc. 1997, 154, 265–292. [Google Scholar] [CrossRef]
- Sleep, N.H.; Lowe, D.R. Physics of crustal fracturing and chert dike formation triggered by asteroid impact, ~3.26 ga, barberton greenstone belt, south africa. Geochem. Geophys. Geosyst. 2014, 15, 1054–1070. [Google Scholar] [CrossRef]
- Keller, G. The Cretaceous–Tertiary mass extinction, Chicxulub impact, and Deccan volcanism. In Earth and Life; Springer Netherlands: Dordrecht, The Netherlands, 2012; pp. 759–793. [Google Scholar]
- Westerhold, T.; Röhl, U.; Raffi, I.; Fornaciari, E.; Monechi, S.; Reale, V.; Bowles, J.; Evans, H.F. Astronomical calibration of the Paleocene time. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2008, 257, 377–403. [Google Scholar] [CrossRef]
- Sahney, S.; Benton, M.J.; Ferry, P.A. Links between global taxonomic diversity, ecological diversity and the expansion of vertebrates on land. Biol. Lett. 2010, 6, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2002. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.M.; Jiang, X.P. Positive selection drives lactoferrin evolution in mammals. Genetica 2010, 138, 757–762. [Google Scholar] [CrossRef]
- Xu, S.; Chen, Y.; Cheng, Y.; Yang, D.; Zhou, X.; Xu, J.; Zhou, K.; Yang, G. Positive selection at the ASPM gene coincides with brain size enlargements in cetaceans. Proc. Biol. Sci. 2012, 279, 4433–4440. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.H. EvolView, an online tool for visualizing, annotating and managing phylogenetic trees. Nucleic Acids Res. 2012, 40, W569–W572. [Google Scholar] [CrossRef]
- Mika, K.M.; Lynch, V.J. An ancient fecundability-associated polymorphism switches a repressor into an enhancer of endometrial tap2 expression. Am. J. Hum. Genet. 2016, 99, 1059–1071. [Google Scholar] [CrossRef]
- Wang, Y.; Gu, X. Evolutionary patterns of gene families generated in the early stage of vertebrates. J. Mol. Evol. 2000, 51, 88–96. [Google Scholar] [CrossRef]
- Wang, Y.; Gu, X. Functional divergence in the caspase gene family and altered functional constraints: Statistical analysis and prediction. Genetics 2001, 158, 1311–1320. [Google Scholar] [PubMed]
- Koda, Y.; Tachida, H.; Soejima, M.; Takenaka, O.; Kimura, H. Ancient origin of the null allele se 428 of the human ABO-secretor locus (FUT2). J. Mol. Evol. 2000, 50, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.E.; Freitas, L.B.; Salzano, F.M. Molecular evolution and functional divergence of alcohol dehydrogenases in animals, fungi and plants. Genet. Mol. Biol. 2018, 41, 341–354. [Google Scholar] [CrossRef]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Nielsen, R.; Bustamante, C.; Clark, A.G.; Glanowski, S.; Sackton, T.B.; Hubisz, M.J.; Fledel-Alon, A.; Tanenbaum, D.M.; Civello, D.; White, T.J.; et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005, 3, e170. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Sternberg, M.J. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Model a | ln L | Estimates of Parameters a | LRT b | Positive Sites c |
|---|---|---|---|---|---|
| TPH1 | M2a | −6828.237 | p1 = 0.953, p2 = 0.047, p3 = 0.000 | 0.000 | 97, 442 |
| ω1 = 0.075, ω2 = 1.000, ω3 = 9.527 | |||||
| M1a | −6828.237 | p1 = 0.953, p2 = 0.047 | |||
| ω1 = 0.075, ω2 = 1.000 | |||||
| M8 | −6817.136 | p0 = 0.982, p = 0.472, q = 4.095 | 36.742 * | ||
| (p1 = 0.018), ω = 1.391 | |||||
| M7 | −6835.507 | P = 0.348, q =2.304 | |||
| TPH2 | M2a | −8015.897 | p1 = 0.949, p2 = 0.043, p3 = 0.008 | 0.000 | 47, 66 |
| ω1 = 0.034, ω2 = 1.000, ω3 = 1.000 | |||||
| M1a | −8015.897 | p1 = 0.949, p2 = 0.051 | |||
| ω1 = 0.034 ω2 = 1.000 | |||||
| M8 | −7982.384 | p0 = 0.983, p = 0.044, q = 0.337 | 47.952 * | ||
| (p1 = 0.017), ω = 1.228 | |||||
| M7 | −8006.360 | p = 0.109, q = 0.929 | |||
| IDO1 | M2a | −8015.897 | p1 = 0.666, p2 = 0.293, p3 = 0.004 | 23.524 * | 194, 197, 205, 219, 229, 286, 315, 336, 400, 404 |
| ω1 = 0.125, ω2 = 1.000, ω3 = 2.447 | |||||
| M1a | −8015.897 | p1 = 0.673, p2 = 0.327 | |||
| ω1 = 0.120, ω2 = 1.000 | |||||
| M8 | −7982.384 | p0 = 0.750, p = 0.816, q = 3.690 | 52.290 * | ||
| (p1 = 0.250), ω = 1.204 | |||||
| M7 | −8006.360 | P = 0.078, q = 0.098 |
| Comparison (Cluster 1/Cluster 2) | θa | SE b | LRT c | N (0.9) d |
|---|---|---|---|---|
| TPH1/TPH2 | 0.513 | 0.061 | 42.837 * | 5 |
| IDO1/IDO2 | 0.560 | 0.080 | 91.682 * | 16 |
| MTNR1A/MTNR1B | 0.554 | 0.070 | 45.038 * | 5 |
| CYP1A1/CYP1A2 | −0.019 | 0.093 | 0.176 | 0 |
| CYP1A1/CYP1B1 | 0.802 | 0.102 | 60.455 * | 16 |
| CYP1A1/CYP2C19 | 0.660 | 0.116 | 34.390 * | 4 |
| CYP1A2/CYP1B1 | 0.766 | 0.100 | 62.899 * | 13 |
| CYP1A2/CYP2C19 | 0.892 | 0.118 | 57.428 * | 22 |
| CYP1B1/CYP2C19 | 0.839 | 0.100 | 77.931 * | 51 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Jiang, X.; Liu, G.; Wassie, T.; Girmay, S. An Ancient Mutation in the TPH1 Gene is Consistent with the Changes in Mammalian Reproductive Rhythm. Int. J. Mol. Sci. 2019, 20, 6065. https://doi.org/10.3390/ijms20236065
Liu C, Jiang X, Liu G, Wassie T, Girmay S. An Ancient Mutation in the TPH1 Gene is Consistent with the Changes in Mammalian Reproductive Rhythm. International Journal of Molecular Sciences. 2019; 20(23):6065. https://doi.org/10.3390/ijms20236065
Chicago/Turabian StyleLiu, Chenhui, Xunping Jiang, Guiqiong Liu, Teketay Wassie, and Shishay Girmay. 2019. "An Ancient Mutation in the TPH1 Gene is Consistent with the Changes in Mammalian Reproductive Rhythm" International Journal of Molecular Sciences 20, no. 23: 6065. https://doi.org/10.3390/ijms20236065
APA StyleLiu, C., Jiang, X., Liu, G., Wassie, T., & Girmay, S. (2019). An Ancient Mutation in the TPH1 Gene is Consistent with the Changes in Mammalian Reproductive Rhythm. International Journal of Molecular Sciences, 20(23), 6065. https://doi.org/10.3390/ijms20236065