Bone Pain in Cancer Patients: Mechanisms and Current Treatment



Abstract
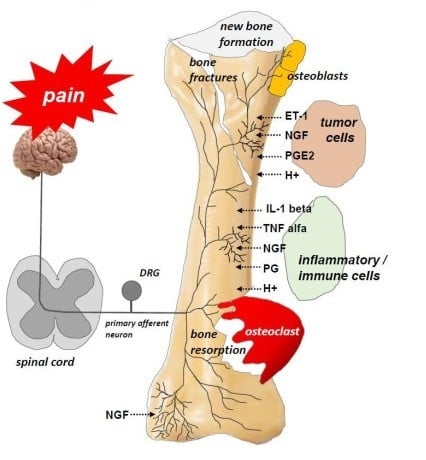
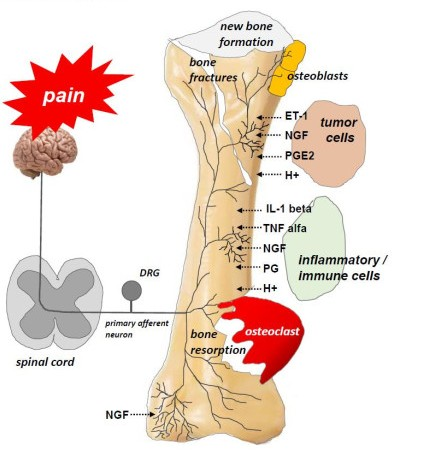{kind=link}
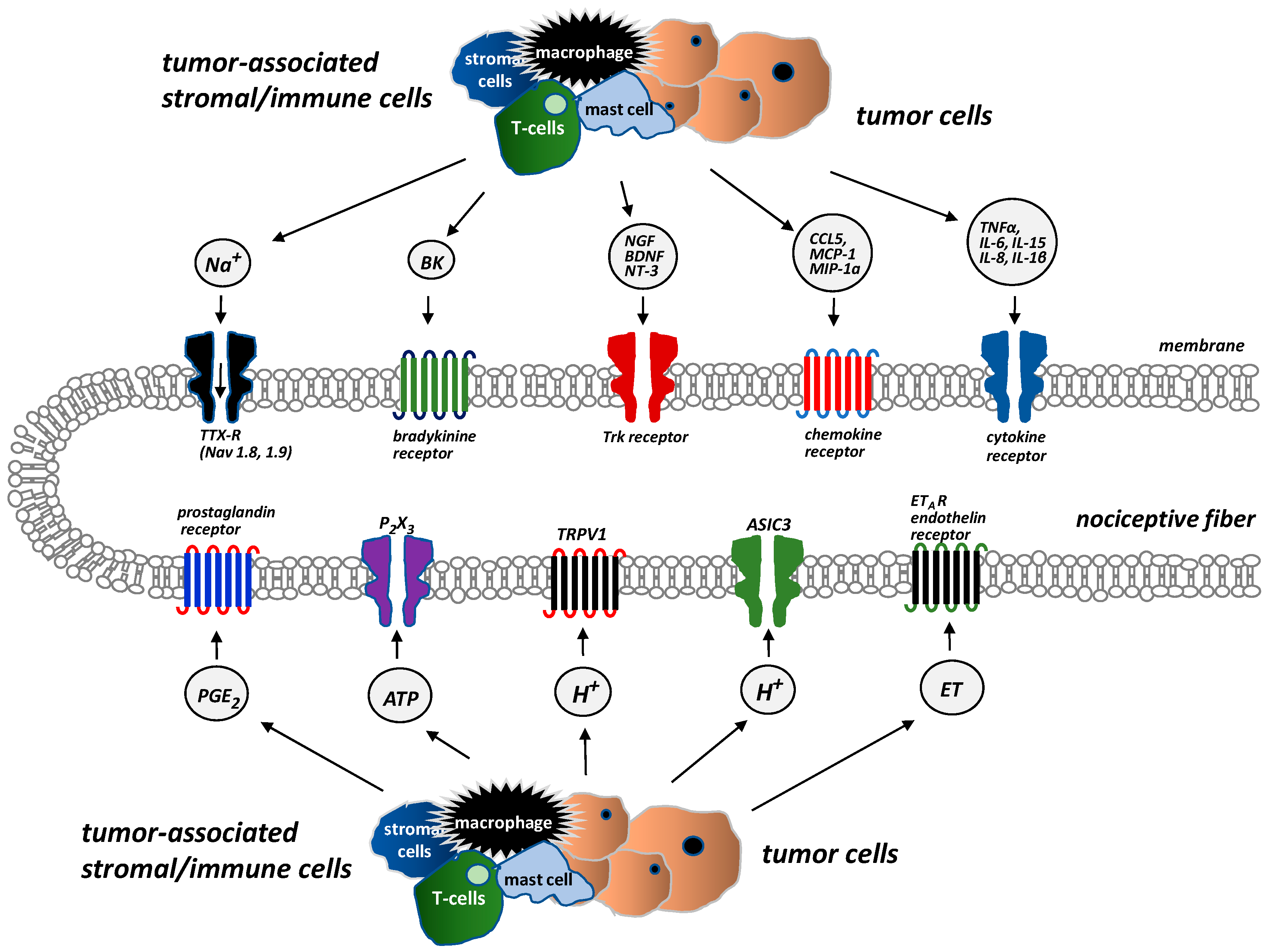{kind=link}
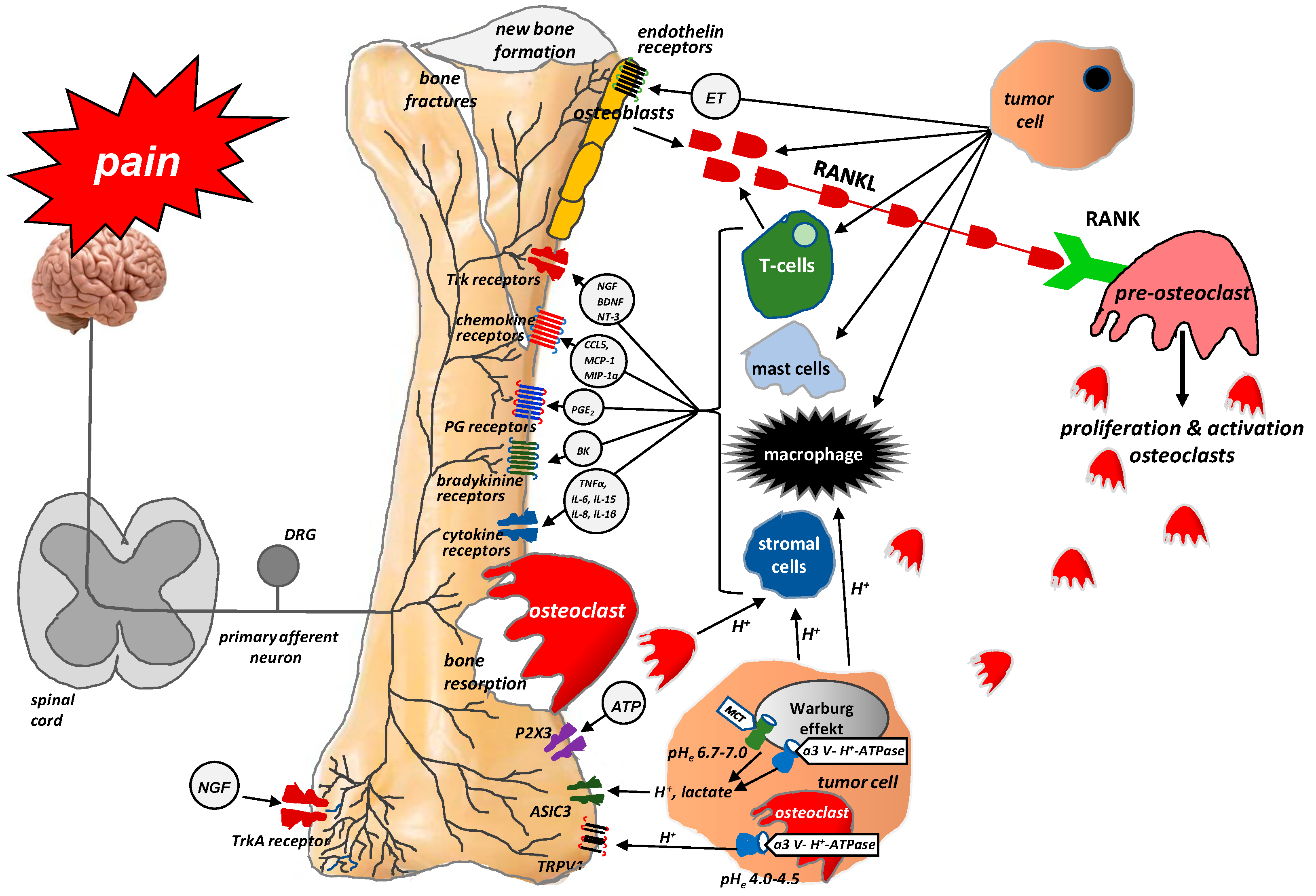{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Clinical Characteristics of Bone Pain in Cancer Patients
3. Mechanisms of Bone Pain in Cancer Patients
4. Treatment of Cancer-Induced Bone Pain
- ✓
- Anticancer treatment comprises local surgery and/or radiotherapy (RT) and systemic treatment modalities (chemotherapy, hormone therapy, immunotherapy, and molecular treatment). Causal treatment reduces the tumor mass and local tissue infiltration and thus decreases the pain intensity; hence, anticancer treatment may be regarded as symptomatic treatment, as well.
- ✓
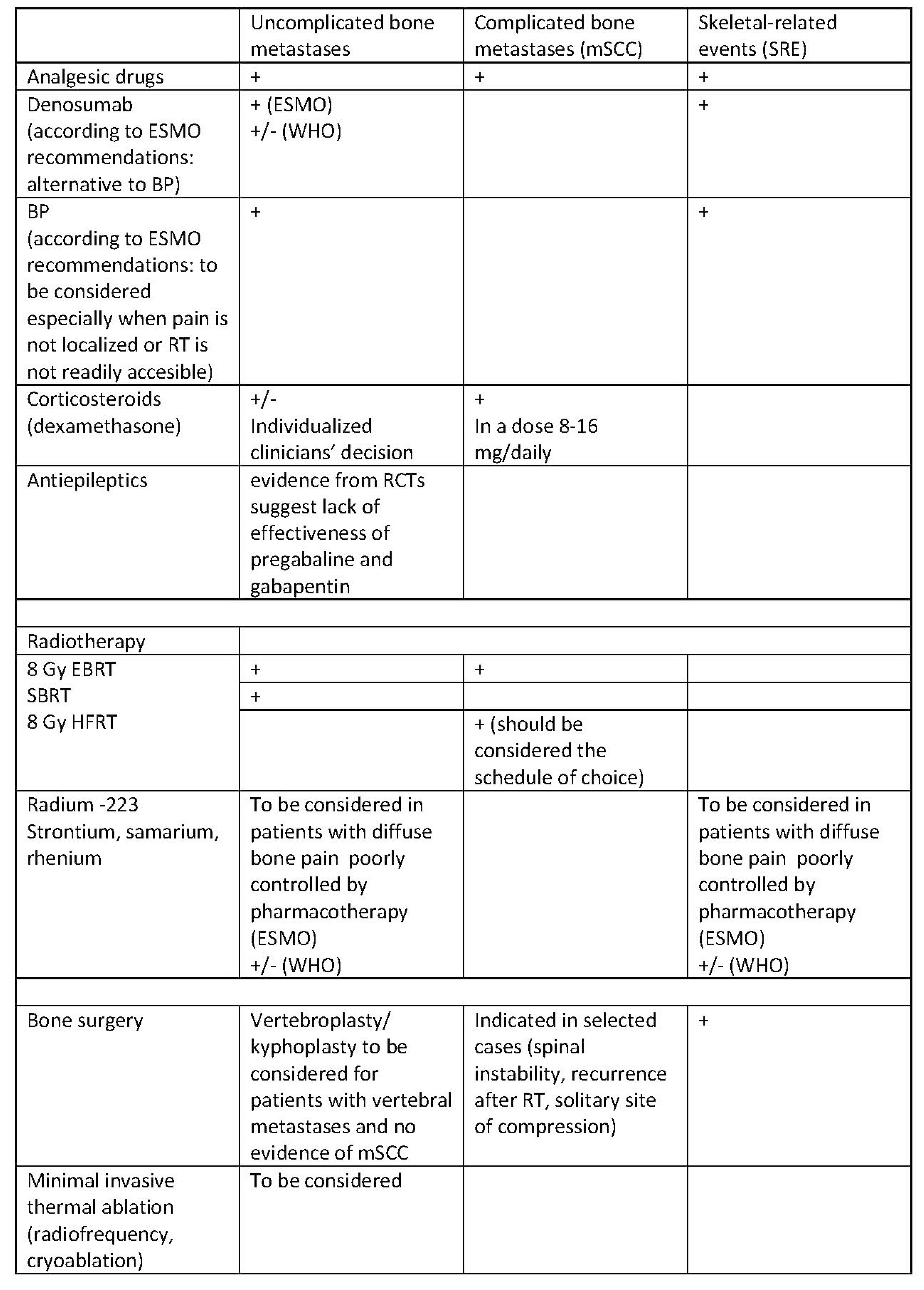
- Analgesic treatment includes pain management with analgesics (non-opioid and opioid analgesics according to the World Health Organization (WHO) analgesic ladder), bone-targeted therapies (NGF inhibitors; osteoclast inhibitors, such as bisphosphonates and denosumab), and adjuvants (corticosteroids, anticonvulsants).
4.1. Radiotherapy
4.2. Interventional Methods
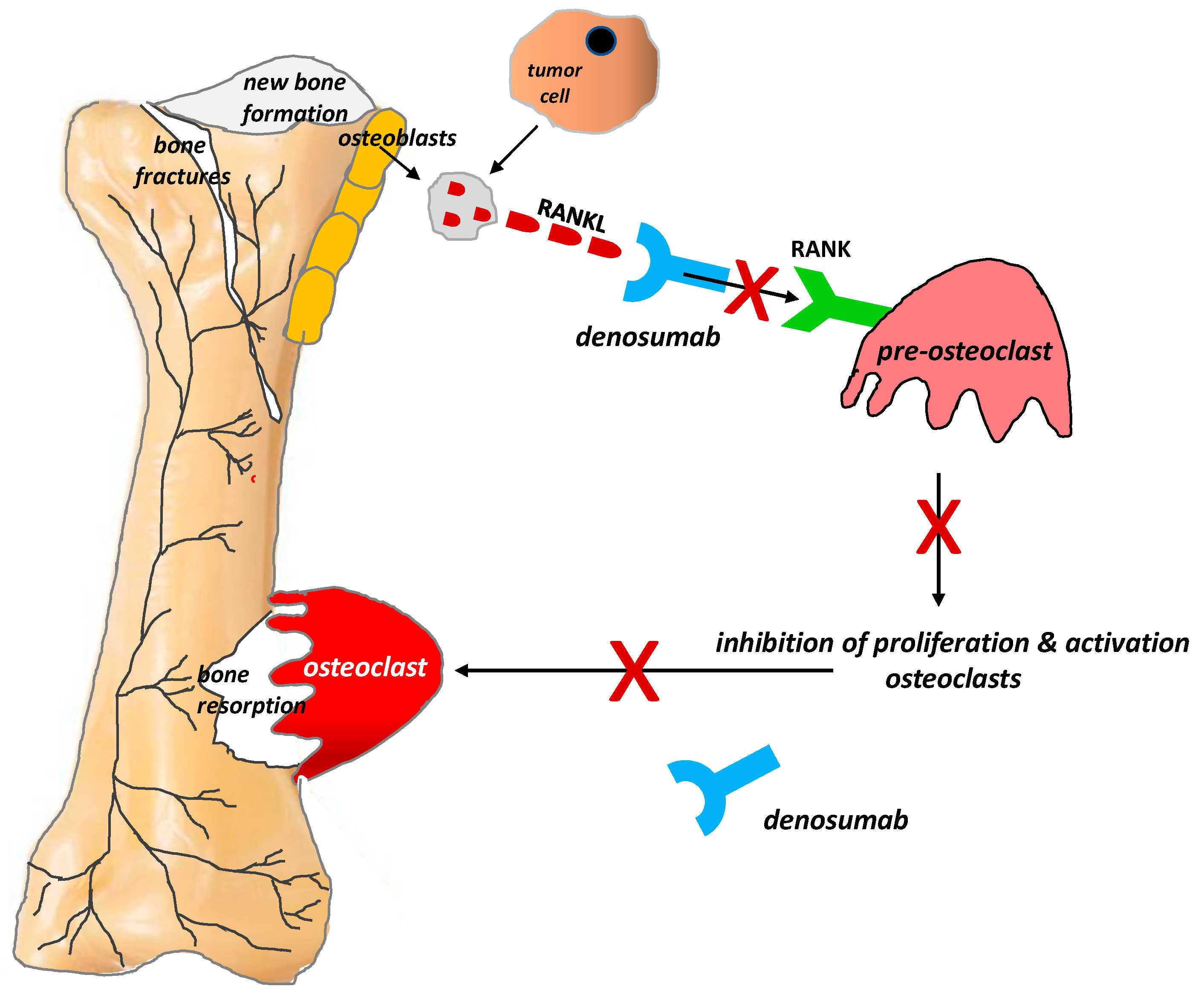
4.3. Bone-Targeting Therapies
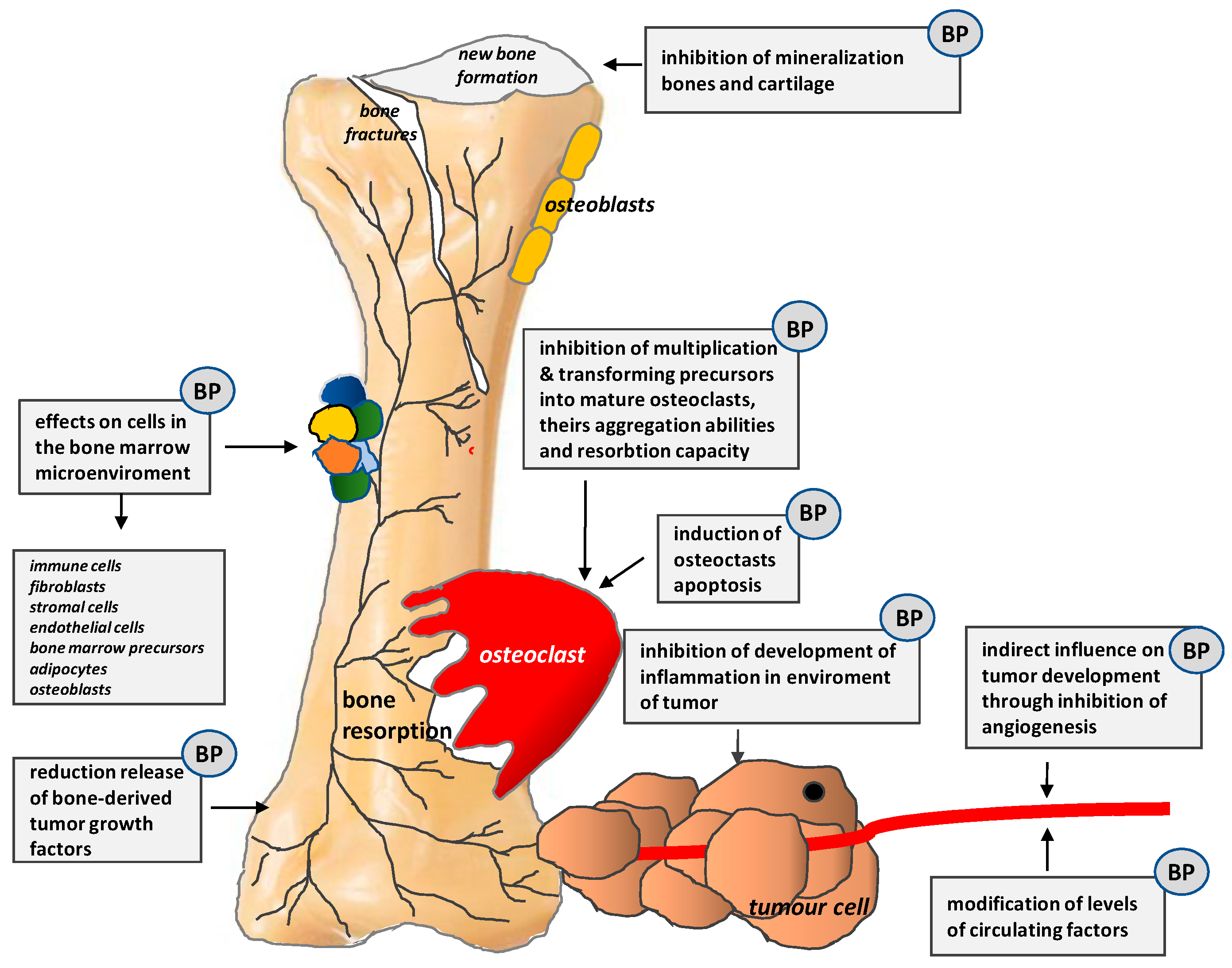
4.3.1. Bisphosphonates
4.3.2. Monoclonal Antibodies
4.3.3. Analgesics According to the WHO Analgesic Ladder
4.4. Adjuvant Analgesics
4.4.1. Corticosteroids
4.4.2. Anticonvulsants
5. Emerging Experimental Studies in Cancer-Induced Bone Pain
6. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Zhu, X.C.; Zhang, J.L.; Ge, C.T.; Yu, Y.Y.; Wang, P.; Yuan, T.F.; Fu, C.Y. Advances in cancer pain from bone metastasis. Drug Des. Dev. Ther. 2015, 9, 4239–4245. [Google Scholar]
- Mercadante, S. Malignant bone pain: Pathophysiology and treatment. Pain 1997, 69, 1–18. [Google Scholar] [CrossRef]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Davila, D.; Antoniou, A.; Chaudhry, M.A. Evaluation of Osseous Metastasis in Bone Scintigraphy. Semin. Nucl. Med. 2015, 45, 3–15. [Google Scholar] [CrossRef]
- Li, B.T.; Wong, M.H.; Pavlakis, N. Treatment and prevention of bone metastases from breast cancer: A comprehensive review of evidence for clinical practice. J. Clin. Med. 2014, 3, 1–24. [Google Scholar] [CrossRef]
- Greenberg, A.J.; Rajkumar, S.V.; Themeau, T.M.; Singh, P.P.; Dispenzieri, A.; Kumar, S.K. Relationship between initial clinical presentation and the molecular cytogenetic classification of myeloma. Leukemia 2014, 28, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.S. Painful bone metastases. Ann. Pall. Med. 2012, 1, 14–31. [Google Scholar]
- Pae, F.M.; Serafini, A.N. Systemic metabolic radiopharmaceutical therapy in the treatment of metastatic bone pain. Semin. Nucl. Med. 2010, 40, 89–104. [Google Scholar]
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin. Cancer Res. 2006, 12, 6243–6249. [Google Scholar] [CrossRef]
- Porter, A.; David, M. Palliative care for bone, spinal cord, brain and liver metastases. In Clinical Radiation Oncology; Gunderson, L.L., Tepper, J.E., Eds.; Elsevier: Philadelphia, PA, USA, 2007; pp. 437–457. [Google Scholar]
- Yoneda, T.; Hiasa, M.; Nagata, Y.; Okui, T.; White, F.A. Acidic microenvironment and bone pain in cancer-colonized bone. BoneKEy Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Figura, N.; Smith, J.; Yu, H. Mechanisms of, and Adjuvants for, Bone Pain. Hematol. Oncol. Clin. N. Am. 2018, 32, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Mantyh, P. Bone cancer pain: Causes, consequences, and therapeutic opportunities. Pain 2013, 154, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Mercadante, S.V.P.; Ferrrera, P.; Casuccio, A. Optimization of opoid therapy for preventing incident pain associated with bone metastases. J. Pain Symptom Manag. 2004, 28, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Delaney, A.; Fleetwood–Walker, S.M.; Colvin, L.A.; Fallon, M. Translational medicine: Cancer pain mechanisms and management. Br. J. Anaesth. 2008, 101, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Middlemiss, T.; Laird, B.J.A.; Fallon, M.T. Mechanisms of cancer–induced bone pain. Clin. Oncol. 2011, 23, 387–392. [Google Scholar] [CrossRef]
- Nieder, C.; Pawinski, A.; Dalhaug, A. Continuous controversy about radiation oncologists’ choice of treatment regimens for bone metastases: Should we blame doctors, cancer-related features, or design of previous clinical studies? Radiat. Oncol. 2013, 8, 85. [Google Scholar] [CrossRef]
- Carrafiello, G.; Lagana, D.; Pellegrino, C.; Mangini, M.; Fontana, F.; Piacentino, F.; Recaldini, C.; Rovera, F.; Dionigi, G.; Boni, L.; et al. Ablation of painful metastatic bone tumors: A systematic review. Int. J. Surg. 2008, 6, 47–52. [Google Scholar] [CrossRef]
- Falk, S.; Dickenson, A.H. Pain and nociception: Mechanisms of cancer–induced bone pain. J. Clin. Oncol. 2014, 32, 1647–1654. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.R.; Camacho, D.F.; Shiozawa, Y.; Pienta, K.J.; Taichman, R.S. Mechanisms of cancer cell metastasis to the bone: A multistep process. Future Oncol. 2011, 7, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Gdowski, A.S.; Ranjan, A.; Vishwanatha, J.K. Current concepts in bone metastasis, contemporary therapeutic strategies and ongoing clinical trials. J. Exp. Clin. Cancer Res. 2017, 36, 108. [Google Scholar] [CrossRef]
- Douma, S.; Van Laar, T.; Zevenhoven, J.; Meuwissen, R.; Van Garderen, E.; Peeper, D.S. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature 2004, 430, 1034–1039. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.; Volkmer, J.P.; Levin, A.M.; Volkmer, A.K.; Ozkan, E.; Fernhoff, N.B.; van de Rijn, M.; Weissman, I.L.; et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef]
- Sun, Y.X.; Fang, M.; Wang, J.; Cooper, C.R.; Pienta, K.J.; Taichman, R.S. Expression and activation of alpha v beta 3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells. Prostate 2007, 67, 61–73. [Google Scholar] [CrossRef]
- Dushyanthen, S.; Cossigny, D.A.F.; Quan, G.M.Y. The Osteoblastic and Osteoclastic Interactions in Spinal Metastases Secondary to Prostate Cancer. Cancer Growth Metast. 2013, 6, 61–80. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Eber, M.R.; Berry, J.E.; Taichman, R.S. Bone marrow as a metastatic niche for disseminated tumor cells from solid tumors. Bonekey Rep. 2015, 4, 689. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhou, H.; Dunstan, C.R.; Sutherland, R.L.; Seibel, M.J. The role of the bone microenvironment in skeletal metastasis. J. Bone Oncol. 2013, 2, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Eber, M.R.; Widner, D.B.; Shiozawa, Y. Role of the Bone Microenvironment in the Development of Painful Complications of Skeletal Metastases. Cancers 2018, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- Buijs, J.T.; Stayrook, K.R.; Guise, T.A. The role of TGF-β in bone metastasis: Novel therapeutic perspectives. BoneKEy Rep. 2012. [Google Scholar] [CrossRef] [PubMed]
- Zaporowska-Stachowiak, I.; Łuczak, J.; Hoffmann, K.; Stachowiak, K.; Bryl, W.; Sopata, M. Managing metastatic bone pain: New perspectives, different solutions. Biomed. Pharmacother. 2017, 93, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Clohisy, D.R.; Mantyh, P.W. Bone cancer pain and the role of RANKL/OPG. J. Musculoskelet. Neuronal Interact 2004, 4, 293–300. [Google Scholar]
- Mantyh, P.W. Bone cancer: From mechanism to therapy. Curr. Opin. Support Palliat. Care 2014, 8, 83–90. [Google Scholar] [CrossRef]
- Stopeck, A.T.; Lipton, A.; Body, J.J.; Steger, G.G.; Tonkin, K.; de Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: A randomized, double-blind study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef]
- Jimenez-Andrade, J.M.; Bloom, A.P.; Stake, J.I.; Mantyh, W.G.; Taylor, R.N.; Freeman, K.T.; Ghilardi, J.R.; Kuskowski, M.A.; Mantyh, P.W. Pathological sprouting of adult nociceptors in chronic prostate cancer–induced bone pain. J. Neurosci. 2010, 30, 14649–14656. [Google Scholar] [CrossRef]
- Bloom, A.P.; Jimenez-Andrade, J.M.; Taylor, R.N.; Castañeda-Corral, G.; Kaczmarska, M.J.; Freeman, K.T.; Coughlin, K.A.; Ghilardi, J.R.; Kuskowski, M.A.; Mantyh, P.W. Breast cancer–induced bone remodeling, skeletal pain and sprouting of sensory nerve fibers. J. Pain 2011, 12, 698–711. [Google Scholar] [CrossRef]
- Petruska, J.C.; Mendell, L.M. The many functions of nerve growth factor: Multiple actions on nociceptors. Neurosci. Lett. 2004, 361, 168–171. [Google Scholar] [CrossRef]
- Lozano-Ondoua, A.N.; Symons-Liguori, A.M.; Vanderah, T.W. Mechanisms of cancer-induced bone pain. Neurosci. Lett. 2013, 17, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Mantyh, W.G.; Jimenez-Andrade, J.M.; Stake, J.I.; Bloom, A.P.; Kaczmarska, M.J.; Taylor, R.N.; Freeman, K.T.; Ghilardi, J.R.; Kuskowski, M.A.; Mantyh, P.W. Blockade of nerve sprouting and neuroma formation markedly attenuates the development of late stage cancer pain. Neuroscience 2010, 171, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Fallon, M.; Giusti, R.; Aielli, F.; Hoskin, P.; Rolke, R.; Sharma, M.; Ripamonti, C.I. ESMO Guidelines Committee. Management of cancer pain in adult patients: ESMO Clinical Practice Guidelines. Ann. Oncol. 2018, 29, 166–191. [Google Scholar] [CrossRef] [PubMed]
- WHO Guidelines for the Pharmacological and Radiotherapeutic Management of Cancer Pain in Adults and Adolescents; World Health Organization: Geneva, Switzerland, 2018; ISBN 978-92-4-155039-0.
- Lutz, S.; Berk, L.; Chang, E.; Chow, E.; Hahn, C.; Hoskin, P.; Howell, D.; Konski, A.; Kachnic, L.; Lo, S.; et al. Palliative radiotherapy for bone metastases: An ASTRO evidence-based guideline. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Chow, R.; Hoskin, P.; Hollenberg, D.; Lam, M.; Dennis, K.; Lutz, S.; Lam, H.; Mesci, A.; DeAngelis, C.; Chan, S.; et al. Efficacy of single fraction conventional radiation therapy for painful uncomplicated bonemetastases: A systematic review and meta-analysis. Ann. Palliat Med. 2017, 6, 125–142. [Google Scholar] [CrossRef]
- Coleman, R.; Body, J.J.; Aapro, M.; Hadji, P.; Herrstedt, J. Bone health in cancer patients: ESMO Clinical Practice Guidelines. Ann. Oncol. 2014, 25, 124–137. [Google Scholar] [CrossRef]
- Roque, I.; Figuls, M.; Martnez-Zapata, M.J.; Scott-Brown, M.; Alonso-Coello, P. Withdrawn: Radioisotopes for metastatic bone pain. Cochrane Database Syst. Rev. 2017, 3, CD003347. [Google Scholar]
- Sartor, O.; Coleman, R.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; et al. Effect of radium-223 dichloride on symptomatic skeletal events in patients with castration-resistant prostate cancer and bone metastases: Results from a phase 3, double blind, randomized trial. Lancet Oncol 2014, 15, 738–746. [Google Scholar] [CrossRef]
- Altaf, F.; Weber, M.; Dea, N.; Boriani, S.; Ames, C.; Williams, R.; Verlaan, J.J.; Laufer, I.; Fisher, C.G. Evidence-Based review and survey of expert opinion of reconstruction of metastatic spine tumors. Spine 2016, 41, S254–S261. [Google Scholar] [CrossRef]
- National Institute for Care and Health Excellence. Percutaneous Vertebroplasty. NICE Interventional Procedure Guidance [IPG12]. 2008. Available online: https://www.nice.org.uk/guidance/ipg12 (accessed on 20 November 2019).
- Kyriakou, C.; Molloy, S.; Vrionis, F.; Bastian, L.; Zonder, J.A.; Giralt, S.; Raje, N.; Kyle, R.A.; Roodman, D.G.D.; Dimopoulos, M.A.; et al. The role of cement augmentation with percutaneous vertebroplasty and balloon kyphoplasty for the treatment of vertebral compression fractures in multiple myeloma: A consensus statement from the International Myeloma Working Group (IMWG). Blood Cancer J. 2019, 9, 27. [Google Scholar] [CrossRef]
- Ringe, K.I.; Panzica, M.; von Falck, C. Thermoablation of Bone Tumors. Fortschr. Röntgenstr. 2016, 188, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Van Poznak, C.H.; Temin, S.; Yee, G.C.; Janjan, N.A.; Barlow, E.; Biermann, J.S.; Bosserman, L.D.; Geoghegan, C.; Hillner, B.E.; Theriault, R.L.; et al. American Society of Clinical Oncology American Society of Clinical Oncology executive summary of the clinical practice guideline update on the role of bone modifying agents in metastatic breast cancer. J. Clin. Oncol. 2011, 29, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Van Poznak, C.; Somerfield, M.R.; Barlow, W.E.; Biermann, J.S.; Bosserman, L.D.; Clemons, M.J.; Dhesy-Thind, S.K.; Dillmon, M.S.; Eisen, A.; Frank, E.S.; et al. Role of Bone-Modifying Agents in Metastatic Breast Cancer: An American Society of Clinical Oncology-Cancer Care Ontario Focused Guideline Update. J. Clin. Oncol. 2017, 35, 3978–3986. [Google Scholar] [CrossRef]
- Lipton, A.; Jun, S. RANKL inhibition in the treatment of bone metastases. Curr. Opin. Support Palliat. Care 2008, 2, 197–203. [Google Scholar] [CrossRef] [PubMed]
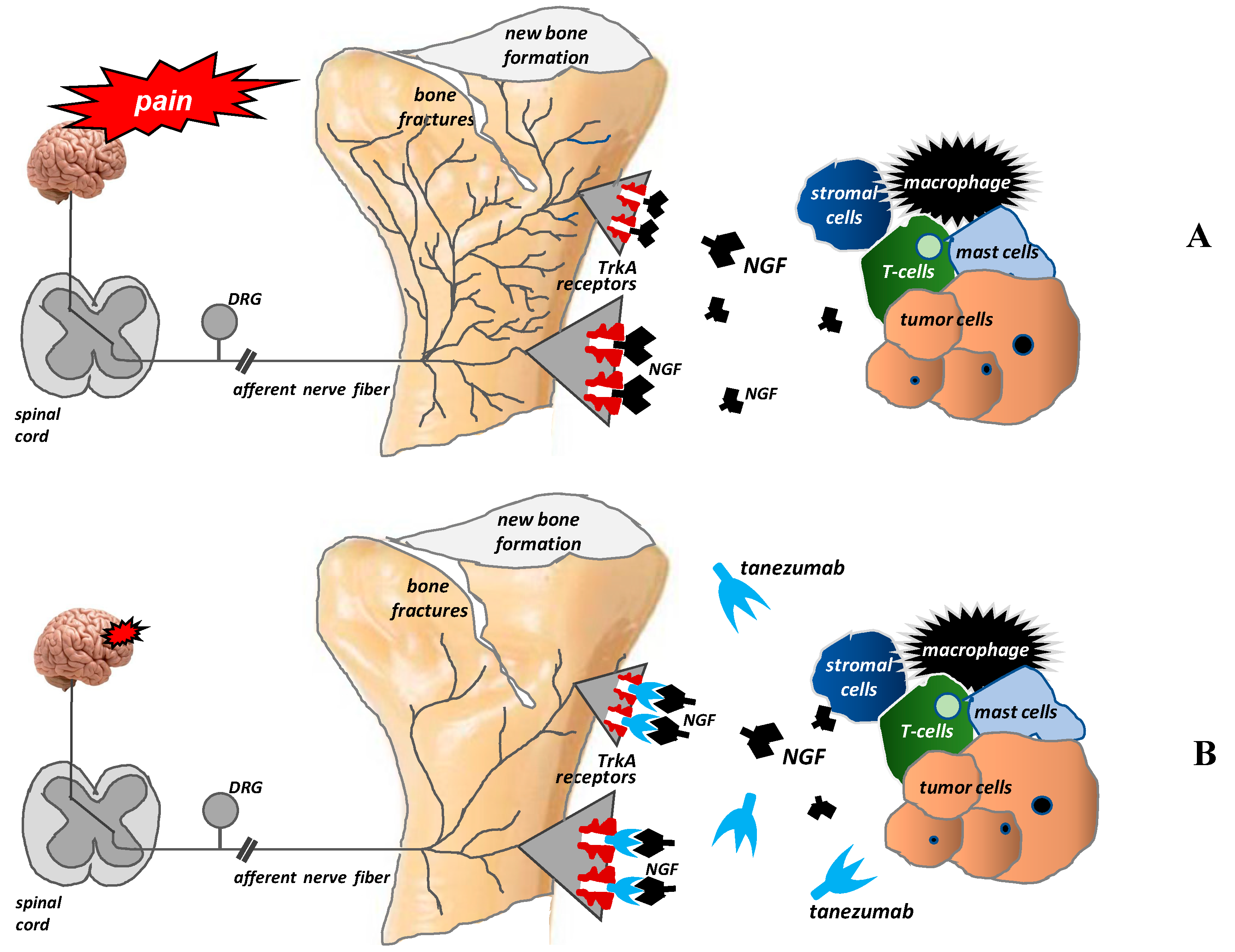
- Pezet, S.; McMahon, S.B. Neurotrophins: Mediators and modulators of pain. Annu. Rev. Neurosci. 2006, 29, 507–538. [Google Scholar] [CrossRef]
- Vega, J.A.; García-Suárez, O.; Hannestad, J.; Pérez-Pérez, M.; Germanà, A. Neurotrophins and the immune system. J. Anat. 2003, 203, 1–19. [Google Scholar] [CrossRef]
- Patel, M.K.; Kaye, A.D.; Urman, R.D. Tanezumab: Therapy targeting nerve growth factor in pain pathogenesis. J. Anaesthesiol. Clin. Pharmacol. 2018, 34, 111–116. [Google Scholar] [CrossRef]
- Sopata, M.; Katz, N.; Carey, W.; Smith, M.D.; Keller, D.; Verburg, K.M.; West, C.R.; Wolfram, G.; Brown, M.T. Efficacy and safety of tanezumab in the treatment of pain from bone metastases. Pain 2015, 156, 1703–1713. [Google Scholar] [CrossRef]
- Urch, C. The pathophysiology of cancer-induced bone pain: Current understanding. Palliat. Med. 2004, 18, 267–274. [Google Scholar] [CrossRef]
- Isono, M.; Suzuki, T.; Hosono, K.; Hayashi, I.; Sakagami, H.; Uematsu, S.; Akira, S.; DeClerck, Y.A.; Okamoto, H.; Majima, M. Microsomal prostaglandin E synthase-1 enhances bone cancer growth and bone cancer-related pain behaviors in mice. Life Sci. 2011, 88, 693–700. [Google Scholar] [CrossRef]
- Derry, S.; Wiffen, P.J.; Moore, R.A.; McNicol, E.D.; Bell, R.F.; Carr, D.B.; McIntyre, M.; Wee, B. Oral nonsteroidal anti-inflammatory drugs (NSAIDs) for cancer pain in adults. Cochrane Database Syst. Rev. 2017. [Google Scholar] [CrossRef]
- Caraceni, A.; Hanks, G.; Kaasa, S.; Bennett, M.I.; Brunelli, C.; Cherny, N.; Dale, O.; De Conno, F.; Fallon, M.; Hanna, M.; et al. Use of opioid analgesics in the treatment of cancer pain: Evidence-based recommendations from the EAPC. Lancet Oncol. 2012, 13, 58–68. [Google Scholar] [CrossRef]
- Gupta, K. Iatrogenic angiogenesis. In Morphine and Metastasis; Parat, M., Ed.; Springer Science+Business Media: Dordrecht, The Netherlands, 2013; pp. 63–78. [Google Scholar]
- Zajączkowska, R.; Leppert, W.; Mika, J.; Kocot-Kępska, M.; Woroń, J.; Wrzosek, A.; Wordliczek, J. Perioperative immunosupression and risk of cancer progression: The impact of opioids on pain management. Pain Res. Manag. 2018, 9293704. [Google Scholar] [CrossRef]
- Edwards, K.A.; Havelin, J.J.; Mcintosh, M.I.; Ciccone, H.A.; Pangilinan, K.; Imbert, I.; Largent-Milnes, T.M.; King, T.; Vanderah, T.W.; Streicher, J.M. A Kappa Opioid Receptor Agonist Blocks Bone Cancer Pain without Altering Bone Loss, Tumor Size, or Cancer Cell Proliferation in a Mouse Model of Cancer-Induced Bone Pain. J. Pain 2018, 19, 612–625. [Google Scholar] [CrossRef]
- Bruera, E.; Watanabe, S. Corticosteroids as adjuvant analgesics. J. Pain Symptom Manag. 1994, 9, 442–445. [Google Scholar] [CrossRef]
- Lussier, D.; Huskey, A.; Portenoy, R.K. Adjuvant analgesics in cancer pain management. Oncologist 2004, 9, 571–591. [Google Scholar] [CrossRef]
- Mensah-Nyagan, A.G.; Meyer, L.; Schaeffer, V.; Kibaly, C.; Patte-Mensah, C. Evidence for a key role of steroids in the modulation of pain. Psychoneuroendocrinology 2009, 34, 169–177. [Google Scholar] [CrossRef]
- Haywood, A.; Good, P.; Khan, S.; Leupp, A.; Jenkins-Marsh, S.; Rickett, K.; Hardy, J.R. Corticosteroids for the management of cancer-related pain in adults. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef]
- Bruera, E.; Roca, E.; Cedaro, L.; Carraro, S.; Chacon, R. Action of oral methylprednisolone in terminal cancer patients: A prospective randomized double-blind study. Cancer Treat Rep. 1985, 69, 751–754. [Google Scholar]
- Leppert, W.; Buss, T. The role of corticosteroids in the treatment of pain in cancer patients. Curr. Pain Headache Rep. 2012, 16, 307–313. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef]
- Donovan-Rodriguez, T.; Dickenson, A.H.; Urch, C.E. Gabapentin normalizes spinal neuronal responses that correlate with behavior in a rat model of cancer-induced bone pain. Anesthesiology 2005, 102, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Caraceni, A.; Zecca, E.; Martini, C.; Pigni, A.; Bracchi, P. Gabapentin for breakthrough pain due to bone metastases. Palliat. Med. 2008, 22, 392–393. [Google Scholar] [CrossRef] [PubMed]
- Sjölund, K.F.; Yang, R.; Lee, K.H.; Resnick, M. Randomized study of pregabalin in patients with cancer–induced bone pain. Pain Ther. 2013, 2, 37–48. [Google Scholar] [CrossRef]
- Fallon, M.; Hoskin, P.; Colvin, L.A.; Fleetwood-Walker, S.M.; Adamson, D.; Byrne, A.; Murray, G.D.; Laird, B.J. Randomized Double-Blind Trial of Pregabalin Versus Placebo in Conjunction with Palliative Radiotherapy for Cancer-Induced Bone Pain. J. Clin. Oncol. 2016, 34, 550–556. [Google Scholar] [CrossRef]
- Kerba, M.; Wu, J.S.; Duan, Q.; Hagen, N.A.; Bennett, M.I. Neuropathic pain features in patients with bone metastases referred for palliative radiotherapy. J. Clin. Oncol. 2010, 28, 4892–4897. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; DeAngelis, C.; Bruera, E.; Chow, R.; Lechner, B.; Chow, E. Does Pregabalin Still Have a Role in Treating Cancer-Induced Bone Pain? J. Clin. Oncol. 2016, 34, 524–526. [Google Scholar] [CrossRef]
- Miller, S. Effectiveness of gabapentin and pregabalin for cancer-induced bone pain: A systematic review. BMJ Support. Palliat. Care 2017, 7, 1–54. [Google Scholar] [CrossRef]
- Tian, J.; Song, T.; Wang, H.; Wang, W.; Zhang, Z.; Yan, R. Thalidomide alleviates bone cancer pain by down-regulating expressions of NF-κB and GFAP in spinal astrocytes in a mouse model. Int. J. Neurosci. 2019, 129, 896–903. [Google Scholar] [CrossRef]
- Grenald, S.A.; Doyle, T.M.; Zhang, H.; Slosky, L.M.; Chen, Z.; Largent-Milnes, T.M.; Spiegel, S.; Vanderah, T.W.; Salvemini, D. Targeting the S1P/S1PR1 axis mitigates cancer-induced bone pain and neuroinflammation. Pain 2017, 158, 1733–1742. [Google Scholar] [CrossRef]
- Falk, S. Carbenoxolone as a novel therapy for attenuation of cancer-induced bone pain. Pain 2018, 159, 1127–1136. [Google Scholar] [CrossRef]
- Slosky, L.M.; BassiriRad, N.M.; Symons, A.M.; Thompson, M.; Doyle, T.; Forte, B.L.; Staatz, W.D.; Bui, L.; Neumann, W.L.; Mantyh, P.W.; et al. The cystine/glutamate antiporter system xc− drives breast tumor cell glutamate release and cancer-induced bone pain. Pain 2016, 157, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Ungard, R.G.; Seidlitz, E.P.; Singh, G. Inhibition of breast cancer-cell glutamate release with sulfasalazine limits cancer-induced bone pain. Pain 2014, 155, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, J.; Balenko, M.; Zacal, N.; Singh, G. Identification of capsazepine as a novel inhibitor of system xc- and cancer-induced bone pain. J. Pain Res. 2017, 10, 915–925. [Google Scholar] [CrossRef]
- Ebbinghaus, M.; Segond von Banchet, G.; Massier, J.; Gajda, M.; Brauer, R.; Kress, M.; Schaible, H.-G. Interleukin-6-dependent influence of nociceptive sensory neurons on antigen-induced arthritis. Arthritis Res. Ther. 2015, 17, 1–13. [Google Scholar] [CrossRef]
- Kiguchi, N.; Maeda, T.; Kobayashi, Y.; Kondo, T.; Ozaki, M.; Kishioka, S. The critical role of invading peripheral macrophage-derived interleukin-6 in vincristine-induced mechanical allodynia in mice. Eur. J. Pharmacol. 2008, 592, 87–92. [Google Scholar] [CrossRef]
- Remeniuk, B.; King, T.; Sukhtankar, D.; Nippert, A.; Li, N.; Li, F.; Cheng, K.; Rice, K.C.; Porreca, F. Disease modifying actions of interleukin-6 blockade in a rat model of bone cancer pain. Pain 2018, 159, 684–698. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zajączkowska, R.; Kocot-Kępska, M.; Leppert, W.; Wordliczek, J. Bone Pain in Cancer Patients: Mechanisms and Current Treatment. Int. J. Mol. Sci. 2019, 20, 6047. https://doi.org/10.3390/ijms20236047
Zajączkowska R, Kocot-Kępska M, Leppert W, Wordliczek J. Bone Pain in Cancer Patients: Mechanisms and Current Treatment. International Journal of Molecular Sciences. 2019; 20(23):6047. https://doi.org/10.3390/ijms20236047
Chicago/Turabian StyleZajączkowska, Renata, Magdalena Kocot-Kępska, Wojciech Leppert, and Jerzy Wordliczek. 2019. "Bone Pain in Cancer Patients: Mechanisms and Current Treatment" International Journal of Molecular Sciences 20, no. 23: 6047. https://doi.org/10.3390/ijms20236047
APA StyleZajączkowska, R., Kocot-Kępska, M., Leppert, W., & Wordliczek, J. (2019). Bone Pain in Cancer Patients: Mechanisms and Current Treatment. International Journal of Molecular Sciences, 20(23), 6047. https://doi.org/10.3390/ijms20236047