PP2Ac Modulates AMPK-Mediated Induction of Autophagy in Mycobacterium bovis-Infected Macrophages

 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. PP2Ac Expression in Mycobacterium Bovis (M. bovis)-Infected Macrophages
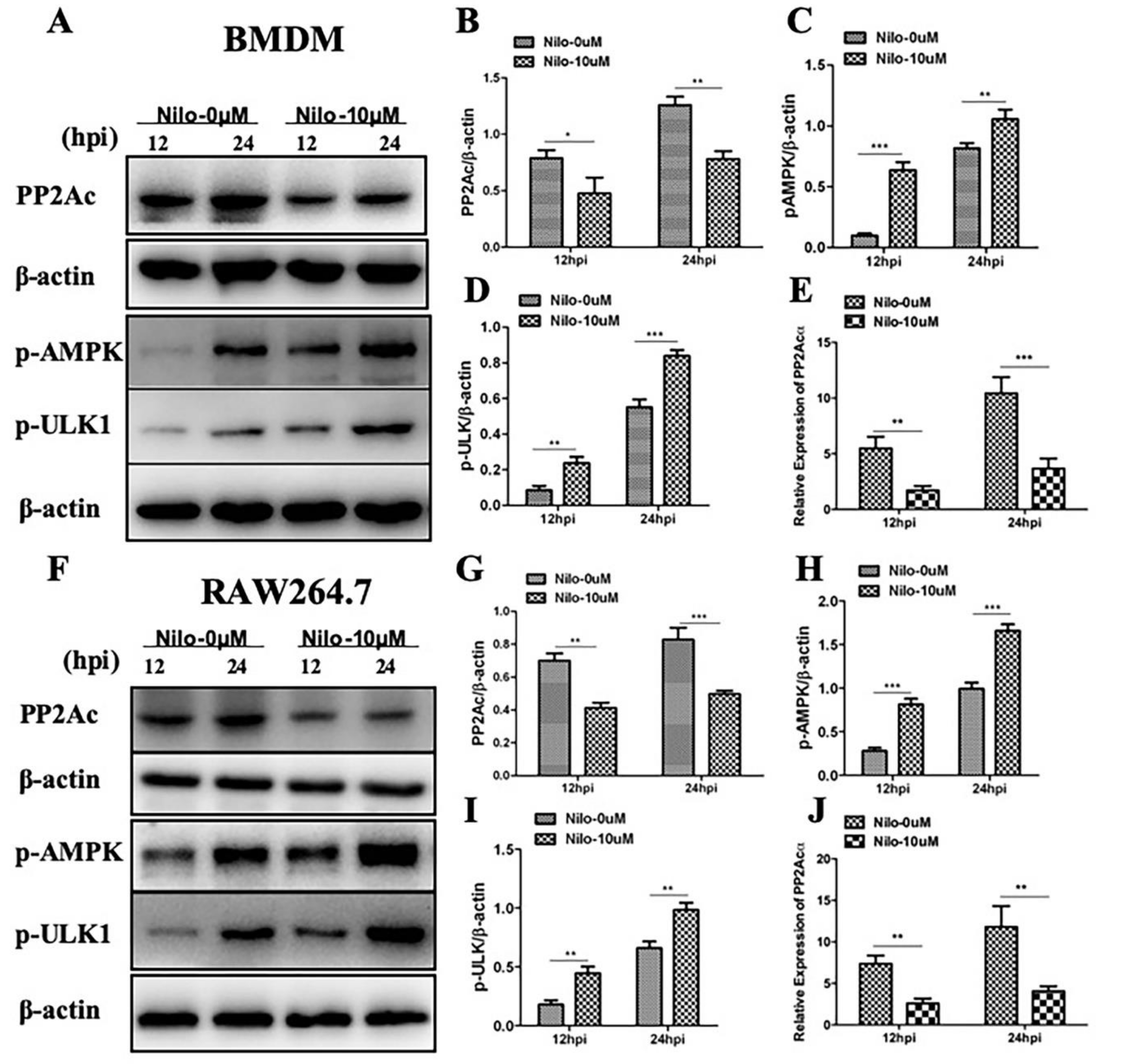
2.2. Targeting PP2Ac via TKI-Nilotinib in M. bovis-Infected Macrophages
2.3. The Effects of PP2Ac Expression on AMP-Activated Protein Kinase (AMPK) Signaling in M. bovis-Infected Macrophages
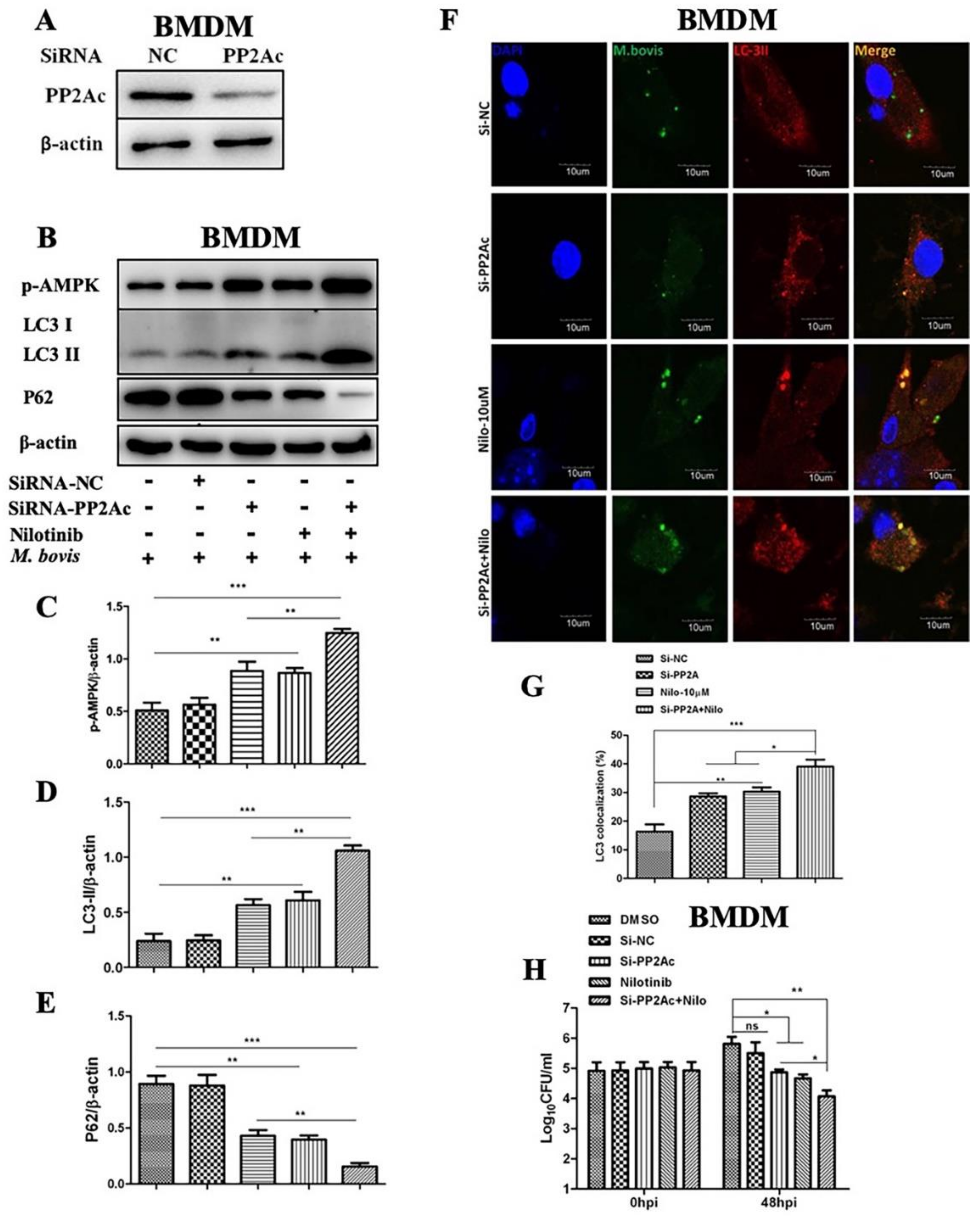
2.4. AMPK Signaling Modulates Autophagy in M. bovis-Infected Macrophages
2.5. PP2Ac Promotes Intracellular Survival of M. bovis in Murine Macrophages
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Antibodies and Reagents
4.3. Bacterial Culture Preparation
4.4. Preparation of Macrophages for In Vitro Experiments
4.5. Cell Infection and Treatment
4.6. Western Blot Analysis
4.7. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.8. Enumeration of Viable Bacteria
4.9. Immunofluorescence Analysis
4.10. Cell Viability and Phagocytic Ability Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| M. bovis | Mycobacterium bovis |
| M. tb | Mycobacterium tuberculosis |
| MTBC | Mycobacterium tuberculosis complex |
| TKI | Tyrosine Kinase Inhibitor |
| PP2Ac | catalytic subunit of protein phosphatase 2A |
| AMPK | AMP-activated protein kinase |
| BMDM | Bone marrow derived macrophages |
| ELISA | Enzyme linked immunosorbent assay |
| WB | Western bloat |
| CFU | Colony-forming unit |
References
- Ashford, D.A.; Whitney, E.; Raghunathan, P.; Cosivi, O. Epidemiology of selected mycobacteria that infect humans and other animals. Rev. Sci. Tech. 2001, 20, 325–337. [Google Scholar] [CrossRef] [PubMed]
- De la Rua-Domenech, R. Human Mycobacterium bovis infection in the United Kingdom: Incidence, risks, control measures and review of the zoonotic aspects of bovine tuberculosis. Tuberculosis (Edinb) 2006, 86, 77–109. [Google Scholar] [CrossRef] [PubMed]
- Grange, J.M. Mycobacterium bovis infection in human beings. Tuberculosis 2001, 81, 71–77. [Google Scholar] [CrossRef] [PubMed]
- McClean, C.M.; Tobin, D.M. Macrophage form, function, and phenotype in mycobacterial infection: Lessons from tuberculosis and other diseases. Pathog. Dis. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Ji, J.H.; Choi, Y.C.; Ryu, C.J.; Ko, S.Y. Regulation of Polo-like kinase 1 by DNA damage in mitosis. Inhibition of mitotic PLK-1 by protein phosphatase 2A. J. Biol. Chem. 2007, 282, 2473–2482. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gu, P.; Jiang, X.; Ruan, H.B.; Li, C.; Gao, X. Protein phosphatase 2A catalytic subunit alpha (PP2Acalpha) maintains survival of committed erythroid cells in fetal liver erythropoiesis through the STAT5 pathway. Am. J. Pathol. 2011, 178, 2333–2343. [Google Scholar] [CrossRef]
- Lechward, K.; Awotunde, O.S.; Swiatek, W.; Muszynska, G. Protein phosphatase 2A: Variety of forms and diversity of functions. Acta. Biochim. Pol. 2001, 48, 921–933. [Google Scholar]
- Xie, L.; Liu, C.; Wang, L.; Gunawardena, H.P.; Yu, Y.; Du, R.; Taxman, D.J.; Dai, P.; Yan, Z.; Yu, J.; et al. Protein phosphatase 2A catalytic subunit alpha plays a MyD88-dependent, central role in the gene-specific regulation of endotoxin tolerance. Cell Rep. 2013, 3, 678–688. [Google Scholar] [CrossRef][Green Version]
- Wu, Y.; Song, P.; Xu, J.; Zhang, M.; Zou, M.H. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 9777–9788. [Google Scholar] [CrossRef]
- Guo, S.G.; Chen, C.Y.; Ji, F.; Mao, L.; Xie, Y. PP2A catalytic subunit silence by microRNA-429 activates AMPK and protects osteoblastic cells from dexamethasone. Biochem. Biophys. Res. Commun. 2017, 487, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Kim, J.J.; Lee, H.M.; Jin, H.S.; Lee, S.H.; Park, J.H.; Kim, S.J.; Kim, J.M.; Han, Y.M.; Lee, M.S.; et al. The AMPK-PPARGC1A pathway is required for antimicrobial host defense through activation of autophagy. Autophagy 2014, 10, 785–802. [Google Scholar] [CrossRef] [PubMed]
- O’Garra, A.; Redford, P.S.; McNab, F.W.; Bloom, C.I.; Wilkinson, R.J.; Berry, M.P.R. The Immune Response in Tuberculosis. Ann. Rev. Immunol. 2013, 31, 475–527. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.W.; Sun, J.X.; Wang, Y.L.; He, W.G.; Wang, L.X.; Zheng, Y.J.; Wu, J.; Zhang, Y.; Jiang, X. Antimycobacterial and Anti-inflammatory Mechanisms of Baicalin via Induced Autophagy in Macrophages Infected with Mycobacterium tuberculosis. Front. Microbiol. 2017, 8, 2142. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef]
- Kim, I.; He, Y.Y. Targeting the AMP-Activated Protein Kinase for Cancer Prevention and Therapy. Front. Oncol. 2013, 3, 175. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Jie, L.; Kumar, P.; Hong, G.S.; Leow, M.K.S.; Paleja, B.; Tsenova, L.; Kurepina, N.; Chen, J.M.; Zolezzi, F.; et al. Metformin as adjunct antituberculosis therapy. Sci. Transl. Med. 2014, 6, 263ra159. [Google Scholar] [CrossRef]
- Chandra, P.; Rajmani, R.S.; Verma, G.; Bhavesh, N.S.; Kumar, D. Targeting Drug-Sensitive and -Resistant Strains of Mycobacterium tuberculosis by Inhibition of Src Family Kinases Lowers Disease Burden and Pathology. Msphere 2016, 1, 1–13. [Google Scholar] [CrossRef]
- Singh, P.; Subbian, S. Harnessing the mTOR Pathway for Tuberculosis Treatment. Front. Microbiol. 2018, 9, 70. [Google Scholar] [CrossRef]
- Yu, H.C.; Lin, C.S.; Tai, W.T.; Liu, C.Y.; Shiau, C.W.; Chen, K.F. Nilotinib Induces Autophagy in Hepatocellular Carcinoma through AMPK Activation. J. Biol. Chem. 2013, 288, 18249–18259. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Yu, M.C.; Chien, C.C.; Wu, M.S.; Lee, Y.C.; Chen, Y.C. Nilotinib reduced the viability of human ovarian cancer cells via mitochondria-dependent apoptosis, independent of JNK activation. Toxicol. In Vitro 2016, 31, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Schachter, J.B.; Moussa, C.E. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. (Berl.) 2014, 92, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Zhao, D.; Shah, S.Z.A.; Sabir, N.; Wang, J.; Liao, Y.; Song, Y.; Dong, H.; Hussain Mangi, M.; Ni, J.; et al. Nilotinib: A Tyrosine Kinase Inhibitor Mediates Resistance to Intracellular Mycobacterium Via Regulating Autophagy. Cells 2019, 8, 506. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, M.; Calin, G.A.; Perrotti, D. From the Biology of PP2A to the PADs for Therapy of Hematologic Malignancies. Front. Oncol. 2015, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.M.; Hardigan, A.; Geraghty, P.; Salim, S.; Gaffney, A.; Thankachen, J.; Arellanos, L.; D’Armiento, J.M.; Foronjy, R.F. Protein phosphatase 2A regulates innate immune and proteolytic responses to cigarette smoke exposure in the lung. Toxicol. Sci. 2012, 126, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.Z.A.; Zhao, D.M.; Hussain, T.; Yang, L.F. Role of the AMPK pathway in promoting autophagic flux via modulating mitochondrial dynamics in neurodegenerative diseases: Insight into prion diseases. Ageing Res. Rev. 2017, 40, 51–63. [Google Scholar] [CrossRef]
- Wang, T.; Yu, Q.; Chen, J.; Deng, B.; Qian, L.; Le, Y. PP2A mediated AMPK inhibition promotes HSP70 expression in heat shock response. PLoS ONE 2010. [Google Scholar] [CrossRef]
- Young, A.R.J.; Chan, E.Y.W.; Hu, X.W.; Koch, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef]
- Larsen, K.B.; Lamark, T.; Overvatn, A.; Harneshaug, I.; Johansen, T.; Bjorkoey, G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy 2010, 6, 784–793. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease REPLY. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.C.; Yao, Z.Y.; Klionsky, D.J. How to control self-digestion: Transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015, 25, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Steiger, J.; Stephan, A.; Inkeles, M.S.; Realegeno, S.; Bruns, H.; Kroll, P.; de Castro Kroner, J.; Sommer, A.; Batinica, M.; Pitzler, L.; et al. Imatinib Triggers Phagolysosome Acidification and Antimicrobial Activity against Mycobacterium bovis Bacille Calmette-Guerin in Glucocorticoid-Treated Human Macrophages. J. Immunol. 2016, 197, 222–232. [Google Scholar] [CrossRef]
- Napier, R.J.; Rafi, W.; Cheruvu, M.; Powell, K.R.; Zaunbrecher, M.A.; Bornmann, W.; Salgame, P.; Shinnick, T.M.; Kalman, D. Imatinib-sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe 2011, 10, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Bruns, H.; Stegelmann, F.; Fabri, M.; Dohner, K.; van Zandbergen, G.; Wagner, M.; Skinner, M.; Modlin, R.L.; Stenger, S. Abelson Tyrosine Kinase Controls Phagosomal Acidification Required for Killing of Mycobacterium tuberculosis in Human Macrophages. J. Immunol. 2012, 189, 4069–4078. [Google Scholar] [CrossRef]
- Hou, T.; Xiao, Z.; Li, Y.; You, Y.H.; Li, H.; Liu, Y.P.; Xi, Y.Y.; Li, J.; Duan, S.B.; Liu, H.; et al. Norcantharidin inhibits renal interstitial fibrosis by downregulating PP2Ac expression. Am. J. Transl. Res. 2015, 7, 2199–2211. [Google Scholar]
- Fujiwara, N.; Usui, T.; Ohama, T.; Sato, K. Regulation of Beclin 1 Protein Phosphorylation and Autophagy by Protein Phosphatase 2A (PP2A) and Death-associated Protein Kinase 3 (DAPK3). J. Biol. Chem. 2016, 291, 10858–10866. [Google Scholar] [CrossRef]
- Boute, M.; Carreras, F.; Rossignol, C.; Doz, E.; Winter, N.; Epardaud, M. The C3HeB/FeJ mouse model recapitulates the hallmark of bovine tuberculosis lung lesions following Mycobacterium bovis aerogenous infection. Vet. Res. 2017, 48, 73. [Google Scholar] [CrossRef]
- Weaver, B.K.; Bohn, E.; Judd, B.A.; Gil, M.P.; Schreiber, R.D. ABIN-3: A molecular basis for species divergence in interleukin-10-induced anti-inflammatory actions. Mol. Cell Biol. 2007, 27, 4603–4616. [Google Scholar] [CrossRef]
- Mahadik, K.; Prakhar, P.; Rajmani, R.S.; Singh, A.; Balaji, K.N. c-Abl-TWIST1 Epigenetically Dysregulate Inflammatory Responses during Mycobacterial Infection by Co-Regulating Bone Morphogenesis Protein and miR27a. Front. Immunol. 2018, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.Z.A.; Zhao, D.M.; Taglialatela, G.; Khan, S.H.; Hussain, T.; Dong, H.D.; Lai, M.Y.; Zhou, X.M.; Yang, L.F. Early Minocycline and Late FK506 Treatment Improves Survival and Alleviates Neuroinflammation, Neurodegeneration, and Behavioral Deficits in Prion-Infected Hamsters. Neurotherapeutics 2017, 14, 463–483. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Cui, H.C.; Banerjee, S.; Tan, Z.; Salomao, R.; Fu, M.G.; Abraham, E.; Thannickal, V.J.; Liu, G. miR-27a Regulates Inflammatory Response of Macrophages by Targeting IL-10. J. Immunol. 2014, 193, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Wallner, F.K.; Hopkins, M.H.; Lindvall, T.; Olofsson, P.; Tilevik, A. Cytokine correlation analysis based on drug perturbation. Cytokine 2017, 90, 73–79. [Google Scholar] [CrossRef]
- Wang, J.; Hussain, T.; Yue, R.; Liao, Y.; Li, Q.; Yao, J.; Song, Y.; Sun, X.; Wang, N.; Xu, L.; et al. MicroRNA-199a Inhibits Cellular Autophagy and Downregulates IFN-beta Expression by Targeting TBK1 in Mycobacterium bovis Infected Cells. Front. Cell Infect. Microbiol. 2018, 8, 238. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef]
- Hussain, T.; Zhao, D.; Shah, S.Z.A.; Wang, J.; Yue, R.; Liao, Y.; Sabir, N.; Yang, L.; Zhou, X. MicroRNA 27a-3p Regulates Antimicrobial Responses of Murine Macrophages Infected by Mycobacterium avium subspecies paratuberculosis by Targeting Interleukin-10 and TGF-beta-Activated Protein Kinase 1 Binding Protein 2. Front. Immunol. 2017, 8, 1915. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, T.; Zhao, D.; Shah, S.Z.A.; Sabir, N.; Wang, J.; Liao, Y.; Song, Y.; Hussain Mangi, M.; Yao, J.; Dong, H.; et al. PP2Ac Modulates AMPK-Mediated Induction of Autophagy in Mycobacterium bovis-Infected Macrophages. Int. J. Mol. Sci. 2019, 20, 6030. https://doi.org/10.3390/ijms20236030
Hussain T, Zhao D, Shah SZA, Sabir N, Wang J, Liao Y, Song Y, Hussain Mangi M, Yao J, Dong H, et al. PP2Ac Modulates AMPK-Mediated Induction of Autophagy in Mycobacterium bovis-Infected Macrophages. International Journal of Molecular Sciences. 2019; 20(23):6030. https://doi.org/10.3390/ijms20236030
Chicago/Turabian StyleHussain, Tariq, Deming Zhao, Syed Zahid Ali Shah, Naveed Sabir, Jie Wang, Yi Liao, Yinjuan Song, Mazhar Hussain Mangi, Jiao Yao, Haodi Dong, and et al. 2019. "PP2Ac Modulates AMPK-Mediated Induction of Autophagy in Mycobacterium bovis-Infected Macrophages" International Journal of Molecular Sciences 20, no. 23: 6030. https://doi.org/10.3390/ijms20236030
APA StyleHussain, T., Zhao, D., Shah, S. Z. A., Sabir, N., Wang, J., Liao, Y., Song, Y., Hussain Mangi, M., Yao, J., Dong, H., Yang, L., & Zhou, X. (2019). PP2Ac Modulates AMPK-Mediated Induction of Autophagy in Mycobacterium bovis-Infected Macrophages. International Journal of Molecular Sciences, 20(23), 6030. https://doi.org/10.3390/ijms20236030