Cytokines in Inflammatory Disease


Abstract
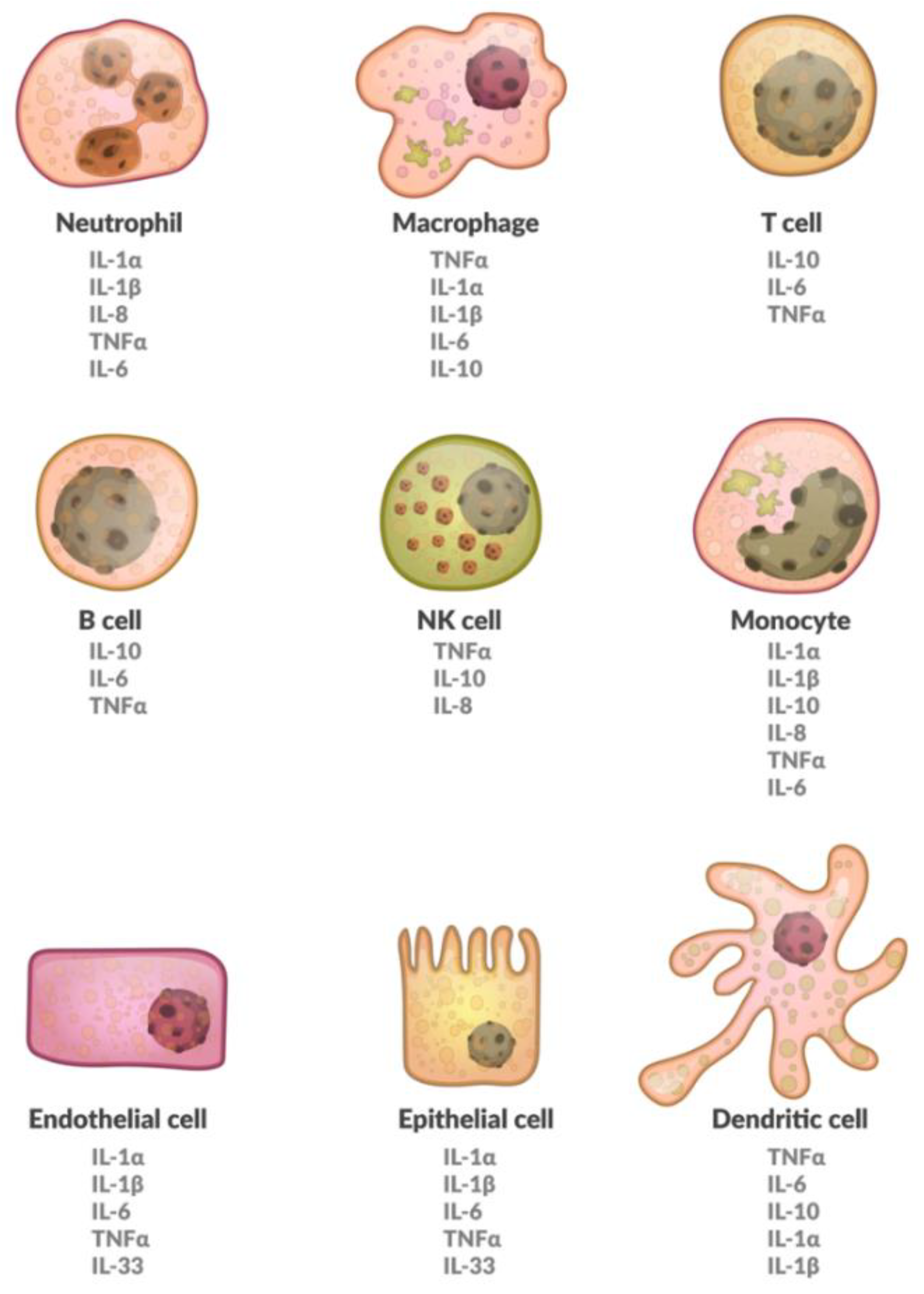
1. Introduction
2. Cytokines
2.1. Interleukin-6
2.2. Interleukin 1 Family
2.3. Tumor Necrosis Factor-Alpha
2.4. Interleukin-10
2.5. Interleukin-8
2.6. Limitations
3. Materials and Methods
4. Conclusions

{kind=link}
| Species | Study | Cell Type/Organ | Major Finding | Reference |
|---|---|---|---|---|
| IL (Interleukin)-6 | ||||
| human | in vivo | bone | IL-6 is a predictor of postmenopausal bone loss | [240] |
| meta-analysis | bone | GG genotype of IL-6-634C/G polymorphism seems to play a role in reducing bone mineral density | [47] | |
| meta-analysis | bone | IL-6-634C/G and IL-6-174G/C polymorphisms lead to modest effects on bone mineral density | [48] | |
| meta-analysis | bone | CC genotype of IL-6 G-174C polymorphism may be associated with high bone mineral density at femoral neck and distal radius and decreased risk of osteoporosis in the Caucasian population | [49] | |
| in vivo | bone | IL-6 G-174C promoter polymorphism may be a genetic marker for bone loss and wrist fracture among older women | [241] | |
| meta-analysis | bone | IL6-174 G/C gene polymorphism positively correlated with osteoporosis risk | [242,243] | |
| in vivo | bone | Variation within the low levels of IL-6 predicts bone loss and resorption | [244] | |
| in vitro | whole blood cells | Increased IL-6 production by whole blood cells from postmenopausal women with osteoporosis compared to controls | [245] | |
| in vivo | bone | IL-6 is upregulated in postmenopausal women with low bone mineral density compared to postmenopausal women with normal bone mineral density | [181] | |
| Chronic obstructive pulmonary disease (COPD) patients | bone | RANKL (Receptor activator of NF-κB ligand)-expressing neutrophils correlate negatively with bone marrow density. Plasma levels of IL-6 are increased in COPD patients and correlate with RANKL expression by neutrophils | [237] | |
| mouse | in vivo | osteoclasts | IL-6 mediates stimulation of osteoclastogenesis after estrogen loss | [246] |
| IL-1 | ||||
| human | in vitro | whole blood cells | Increased IL-1 beta production by whole blood cells from postmenopausal women with osteoporosis compared to controls | [245] |
| COPD patients | bone | RANKL-expressing neutrophils correlate negatively with bone marrow density. Plasma levels of IL-1 beta are increased in COPD patients and correlate with RANKL expression by neutrophils | [237] | |
| in vivo | bone | IL-1β (-511C/T) polymorphism is associated with pathogenesis of osteoporosis in postmenopausal women | [247] | |
| in vivo | bone | Serum IL-1β significantly higher in women with osteoporosis than controls | [248] | |
| in vivo | bone | Serum IL-1 is significantly reduced after treatment of postmenopausal with calcitriol | [249] | |
| in vitro | osteoblasts | IL-1beta and Tumor necrosis factor-alpha (TNF-α) regulate osteoblast cell number by up-regulating the Fas-mediated apoptosis of osteoblasts | [250] | |
| rat | in vivo | bone | Interleukin-1 receptor antagonist decreases bone loss and bone resorption in a rat model of postmenopausal osteoporosis | [251] |
| IL-33 | ||||
| human | in vivo | bone | IL-33 levels in postmenopausal women significantly lower compared to healthy controls, positively correlated with serum parathyroid hormone and inverse correlated with C-terminal telopeptide of type 1 collagen | [160] |
| TNF-α | ||||
| human | in vitro | whole blood cells | Increased TNF-α production by whole blood cells from postmenopausal women with osteoporosis compared to controls | [245] |
| in vivo | bone | TNF-α is upregulated in postmenopausal women with low bone mineral density compared to postmenopausal women with normal bone mineral density | [181] | |
| in vivo + in vitro | osteoclasts | Estrogen deficiency → TNF-α and RANKL ↑ → osteoclast formation and number of osteoclast precursors ↑ | [252] | |
| in vivo | bone | Association between TNF-α-308G>A polymorphism and postmenopausal osteoporosis | [253] | |
| in vivo | bone | Serum TNF-α is significantly reduced after treatment of postmenopausal with calcitriol | [249] | |
| in vivo in vitro | TNF-α increased in postmenopausal women with osteoporosis and highly correlated with the RANK and estrogen levels TNF-α synergistically promotes RANKL-induced osteoclasts formation through activation of Phosphoinositide 3-kinases (PI3K)/Akt signaling | [254] | ||
| in vitro | mesenchymal stem cells (MSC) | TNF-α suppresses osteogenic differentiation of MSCs by accelerating P2Y2 receptor in estrogen-deficiency induced osteoporosis | [255] | |
| in vitro | osteoblasts | IL-1beta and TNF-α regulate osteoblast cell number by up-regulating the Fas-mediated apoptosis of osteoblasts | [250] | |
| mouse | in vivo | bone marrow-derived mesenchymal stem cells (BMMSCs) | TNF-α inhibits Forkhead box protein O1 (FoxO1) and thereby aggravates oxidative damage in BMMSCs during osteoporosis | [256] |
| rat | in vitro | bone cultures of fetal rat calvariae | TNF-α causes osteoclastic bone resorption and inhibits bone collagen synthesis | [257] |
| IL-10 | ||||
| human | patients with systemic lupus erythematosus | bone | IL-10 level is elevated in systemic lupus erythematosus patients with osteoporosis | [217] |
| in vivo | bone | IL-10 gene-597 C>A polymorphism is associated with higher risk for osteoporosis | [258] | |
| in vivo | bone | Lower levels of IL-10 in postmenopausal women with osteoporosis | [259,260] | |
| in vivo | bone | Association between IL10-1082G>A polymorphism and postmenopausal osteoporosis | [253] | |
| mouse | in vitro | RAW264.7 monocytes | IL-10 directly inhibits osteoclastogenesis is by suppressing Nuclear factor of activated T-cells, cytoplasmic 1 activity | [261] |
| in vivo | bone | IL-10 is important for promoting osteoblast maturation and reducing bone loss during early stages of type-1 diabetes | [216] | |
| in vivo | bone | IL-10−/− mice develop the reduced bone mass, increased mechanical fragility, and suppressed bone formation (hallmarks of osteoporosis) | [262] | |
| IL-8 | ||||
| human | in vivo | bone | IL-8 significantly increase in post-menopausal women with osteoporosis and bone loss | [235] |
| COPD patients | bone | RANKL-expressing neutrophils correlate negatively with bone marrow density, plasma levels of IL-8 are increased in COPD patients and correlate with RANKL expression by neutrophils | [237] | |
| in vitro | osteoblasts | IL-8 may contribute to osteoporosis in rheumatoid arthritis by enhanced osteoblast-mediated osteoclastogenesis (partly via IL-6 production) | [263] | |
| rats | in vivo | bone | IL-8 and bone loss reduction after atorvastatin treatment of rats with glucocorticoid-induced osteoporosis | [236] |
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADAM | A disintegrin and metalloproteinase |
| AP | Activator protein |
| APC | Antigen presenting cells |
| ARDS | Acute respiratory distress syndrome |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| ATP | Adenosine triphosphate |
| CARD | Caspase activation and recruitment domain |
| CD | Cluster of differentiation |
| COPD | Chronic obstructive pulmonary disease |
| COX | Cyclooxygenase |
| CRP | C-reactive protein |
| CSIF | Cytokine synthesis inhibitory factor |
| DAMP | Damage-associated molecular patterns |
| DNA | Deoxyribonucleic acid |
| fMLP | Formyl-methionyl-leucyl-phenylalanine |
| FoxO1 | Forkhead box protein O1 |
| gp | Glycoprotein |
| HMGB1 | High-mobility group box 1 |
| HSP | Heat shock proteins |
| ICAM | Intercellular cell adhesion molecules |
| IFN | Interferon |
| IL | Interleukin |
| ILC | Innate lymphoid cells |
| IL-1R | Interleukin-1 receptor |
| iNOS | Inducible nitric oxide synthase |
| kDa | Kilo Dalton |
| LPS | Lipopolysaccharide |
| LRR | Leucine-rich repeat |
| MAP | Mitogen activated protein |
| mbIL6R | membrane-bound IL-6 receptor |
| MBL | Mannose-binding lectin |
| MCP | Monocyte chemotactic protein |
| M-CSF | Macrophage-colony stimulating factor |
| MHC | Major histocompatibility complex |
| MODS | Multi organ dysfunction syndrome |
| MyD88 | Myeloid differentiation primary response 88 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-HEV | Nuclear factor from high endothelial venules |
| NF-κB | nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells |
| NLR | NOD-Like Receptors |
| NLRP | NOD-, LRR- and pyrin domaine-containing protein |
| NO | Nitric oxide |
| NOD | Nucleotide-binding oligomerization domain |
| NPC | Nasopharyngeal carcinoma |
| PAMP | Pathogen-associated molecular patterns |
| PBMC | Peripheral blood mononuclear cells |
| PG | Prostaglandin |
| PGN | Peptidoglycan |
| PI3K | Phosphoinositide 3-kinases |
| PLA | Phospholipase A |
| PMNL | Polymorphonuclear leukocytes |
| PRR | Pattern recognition receptors |
| R, r | Receptor |
| RANKL | Receptor activator of NF-κB ligand |
| ROS | Reactive oxygen species |
| s | Soluble |
| sIL6R | IL-6 receptor |
| STAT | Signal transducer and activator of transcription |
| TACE | TNF converting enzyme |
| TBI | Traumatic brain injury |
| Th | T helper cells |
| TLR | Toll-like receptors |
| TNF-α | Tumor necrosis factor-alpha |
| VCAM | Vascular cell adhesion molecule |
| Wnt | Wingless-related integration site |
References
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, Danger, and the Extended Family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Janeway, C.A. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 1–13. [Google Scholar] [CrossRef]
- Häcker, G.; Redecke, V.; Häcker, H. Activation of the immune system by bacterial CpG-DNA. Immunology 2002, 105, 245–251. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Seong, S.Y.; Matzinger, P. Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 2004, 4, 469–478. [Google Scholar] [CrossRef]
- Frank, M.G.; Adhikary, S.; Sobesky, J.L.; Weber, M.D.; Watkins, L.R.; Maier, S.F. The danger-associated molecular pattern HMGB1 mediates the neuroinflammatory effects of methamphetamine. Brain Behav. Immun. 2016, 51, 99–108. [Google Scholar] [CrossRef]
- Zong, M.; Bruton, J.D.; Grundtman, C.; Yang, H.; Li, J.H.; Alexanderson, H.; Palmblad, K.; Andersson, U.; Harris, H.E.; Lundberg, I.E.; et al. TLR4 as receptor for HMGB1 induced muscle dysfunction in myositis. Ann. Rheum. Dis. 2013, 72, 1390–1399. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 1–18. [Google Scholar] [CrossRef]
- Gay, N.J.; Packman, L.C.; Weldon, M.A.; Barna, J.C. A leucine-rich repeat peptide derived from the Drosophila Toll receptor forms extended filaments with a beta-sheet structure. FEBS Lett. 1991, 291, 87–91. [Google Scholar] [CrossRef]
- Creagh, E.M.; O’Neill, L.A.J. TLRs, NLRs and RLRs: A trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006, 27, 352–357. [Google Scholar] [CrossRef]
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef]
- Tang, D.; Shi, Y.; Kang, R.; Li, T.; Xiao, W.; Wang, H.; Xiao, X. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J. Leukoc. Biol. 2007, 81, 741–747. [Google Scholar] [CrossRef]
- Schaper, F.; Rose-John, S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015, 26, 475–487. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef]
- Geginat, J.; Larghi, P.; Paroni, M.; Nizzoli, G.; Penatti, A.; Pagani, M.; Gagliani, N.; Meroni, P.; Abrignani, S.; Flavell, R.A. The light and the dark sides of Interleukin-10 in immune-mediated diseases and cancer. Cytokine Growth Factor Rev. 2016, 30, 87–93. [Google Scholar] [CrossRef]
- Charo, I.F.; Ransohoff, R.M. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, J.N.; Jéru, I.; Lecron, J.C.; Medlej-Hashim, M. Cytokine signatures in hereditary fever syndromes (HFS). Cytokine Growth Factor Rev. 2017, 33, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Diehl, S.; Rincón, M. The two faces of IL-6 on Th1/Th2 differentiation. Mol. Immunol. 2002, 39, 531–536. [Google Scholar] [CrossRef]
- Hirano, T.; Yasukawa, K.; Harada, H.; Taga, T.; Watanabe, Y.; Matsuda, T.; Kashiwamura, S.I.; Nakajima, K.; Koyama, K.; Iwamatsu, A.; et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 1986, 324, 73–76. [Google Scholar] [CrossRef]
- Hibi, M.; Murakami, M.; Saito, M.; Hirano, T.; Taga, T.; Kishimoto, T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell 1990, 63, 1149–1157. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Radtke, S.; Wüller, S.; Yang, X.P.; Lippok, B.E.; Mütze, B.; Mais, C.; de Leur, H.S.V.; Bode, J.G.; Gaestel, M.; Heinrich, P.C.; et al. Cross-regulation of cytokine signalling: Pro-inflammatory cytokines restrict IL-6 signalling through receptor internalisation and degradation. J. Cell Sci. 2010, 123, 947–959. [Google Scholar] [CrossRef]
- Boulanger, M.J.; Chow, D.C.; Brevnova, E.E.; Garcia, K.C. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science (N.Y.) 2003, 300, 2101–2104. [Google Scholar] [CrossRef]
- Franchimont, N.; Lambert, C.; Huynen, P.; Ribbens, C.; Relic, B.; Chariot, A.; Bours, V.; Piette, J.; Merville, M.P.; Malaise, M. Interleukin-6 receptor shedding is enhanced by interleukin-1beta and tumor necrosis factor alpha and is partially mediated by tumor necrosis factor alpha-converting enzyme in osteoblast-like cells. Arthritis Rheum. 2005, 52, 84–93. [Google Scholar] [CrossRef]
- Walev, I.; Vollmer, P.; Palmer, M.; Bhakdi, S.; Rose-John, S. Pore-forming toxins trigger shedding of receptors for interleukin 6 and lipopolysaccharide. Proc. Natl. Acad. Sci. USA 1996, 93, 7882–7887. [Google Scholar] [CrossRef] [PubMed]
- Lust, J.A.; Donovan, K.A.; Kline, M.P.; Greipp, P.R.; Kyle, R.A.; Maihle, N.J. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 1992, 4, 96–100. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.M.; Moon, Y.P.; Liu, K.M.; Spitalnik, S.; Paik, M.C.; Cheung, K.; Sacco, R.L.; Elkind, M.S.V. High-sensitivity C-reactive protein and interleukin-6-dominant inflammation and ischemic stroke risk: The northern Manhattan study. Stroke 2014, 45, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Gryko, M.; Kędra, B.; Szmitkowski, M. Diagnostic usefulness of serum interleukin 6 (IL-6) and C-reactive protein (CRP) in the differentiation between pancreatic cancer and chronic pancreatitis. J. Clin. Lab. Anal. 2010, 24, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Panichi, V.; Maggiore, U.; Taccola, D.; Migliori, M.; Rizza, G.M.; Consani, C.; Bertini, A.; Sposini, S.; Perez-Garcia, R.; Rindi, P.; et al. Interleukin-6 is a stronger predictor of total and cardiovascular mortality than C-reactive protein in haemodialysis patients. Nephrol. Dial. Transplant. 2004, 19, 1154–1160. [Google Scholar] [CrossRef]
- Hurst, S.M.; Wilkinson, T.S.; McLoughlin, R.M.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef]
- Chen, Q.; Fisher, D.T.; Clancy, K.A.; Gauguet, J.M.M.; Wang, W.C.; Unger, E.; Rose-John, S.; von Andrian, U.H.; Baumann, H.; Evans, S.S. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat. Immunol. 2006, 7, 1299–1308. [Google Scholar] [CrossRef]
- Jenkins, B.J.; Grail, D.; Inglese, M.; Quilici, C.; Bozinovski, S.; Wong, P.; Ernst, M. Imbalanced gp130-dependent signaling in macrophages alters macrophage colony-stimulating factor responsiveness via regulation of c-fms expression. Mol. Cell. Biol. 2004, 24, 1453–1463. [Google Scholar] [CrossRef]
- McLoughlin, R.M.; Jenkins, B.J.; Grail, D.; Williams, A.S.; Fielding, C.A.; Parker, C.R.; Ernst, M.; Topley, N.; Jones, S.A. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 9589–9594. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 Are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Luig, M.; Kluger, M.A.; Goerke, B.; Meyer, M.; Nosko, A.; Yan, I.; Scheller, J.; Mittrucker, H.W.; Rose-John, S.; Stahl, R.A.K.; et al. Inflammation-Induced IL-6 Functions as a Natural Brake on Macrophages and Limits GN. J. Am. Soc. Nephrol. 2015, 26, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Neuhöfer, P.; Song, L.; Rabe, B.; Lesina, M.; Kurkowski, M.U.; Treiber, M.; Wartmann, T.; Regnér, S.; Thorlacius, H.; et al. IL-6 trans-signaling promotes pancreatitis-associated lung injury and lethality. J. Clin. Investig. 2013, 123, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, M.; Bataille, R.; Seguin, J.; Zhang, X.G.; Chaptal, P.A.; Klein, B. Constitutive production of interleukin-6 and immunologic features in cardiac myxomas. Arthritis Rheum. 1990, 33, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.A.; Taylor, J.M.G.; Terrell, J.E.; Islam, M.; Li, Y.; Fowler, K.E.; Wolf, G.T.; Teknos, T.N. Interleukin-6 predicts recurrence and survival among head and neck cancer patients. Cancer 2008, 113, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Tsang, C.M.; Deng, W.; Yip, Y.L.; Lui, V.W.Y.; Wong, S.C.C.; Cheung, A.L.M.; Hau, P.M.; Zeng, M.; Lung, M.L.; et al. Enhanced IL-6/IL-6R Signaling Promotes Growth and Malignant Properties in EBV-Infected Premalignant and Cancerous Nasopharyngeal Epithelial Cells. PLoS ONE 2013, 8, e62284. [Google Scholar] [CrossRef]
- Yan, L.; Hu, R.; Tu, S.; Cheng, W.J.; Zheng, Q.; Wang, J.W.; Kan, W.S.; Ren, Y.J. Meta-analysis of association between IL-6 -634C/G polymorphism and osteoporosis. Genet. Mol. Res. 2015, 14, 19225–19232. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Y.; He, M.; Wang, R.; Ma, J.; Zhang, Y.; Zhao, L.; Yu, K. Association between interleukin-6 gene polymorphisms and bone mineral density: A meta-analysis. Genet. Test. Mol. Biomark. 2013, 17, 898–909. [Google Scholar] [CrossRef]
- Ni, Y.; Li, H.; Zhang, Y.; Zhang, H.; Pan, Y.; Ma, J.; Wang, L. Association of IL-6 G-174C polymorphism with bone mineral density. J. Bone Min. Metab. 2014, 32, 167–173. [Google Scholar] [CrossRef]
- Reinhart, K.; Meisner, M.; Brunkhorst, F.M. Markers for Sepsis Diagnosis: What is Useful? Crit. Care Clin. 2006, 22, 503–519. [Google Scholar] [CrossRef]
- Cruickshank, A.M.; Fraser, W.D.; Burns, H.J.G.; Van Damme, J.; Shenkin, A. Response of Serum Interleukin-6 in Patients Undergoing Elective Surgery of Varying Severity. Clin. Sci. 1990, 79, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Gentile, L.F.; Cuenca, A.G.; Vanzant, E.L.; Efron, P.A.; McKinley, B.; Moore, F.; Moldawer, L.L. Is there value in plasma cytokine measurements in patients with severe trauma and sepsis? Methods 2013, 61, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Kitanovski, L.; Jazbec, J.; Hojker, S.; Gubina, M.; Derganc, M. Diagnostic accuracy of procalcitonin and interleukin-6 values for predicting bacteremia and clinical sepsis in febrile neutropenic children with cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2006, 25, 413–415. [Google Scholar] [CrossRef]
- Haasper, C.; Kalmbach, M.; Dikos, G.D.; Meller, R.; Müller, C.; Krettek, C.; Hildebrand, F.; Frink, M. Prognostic value of procalcitonin (PCT) and/or interleukin-6 (IL-6) plasma levels after multiple trauma for the development of multi organ dysfunction syndrome (MODS) or sepsis. Technol. Health Care 2010, 18, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Pallás Beneyto, L.A.; Rodríguez Luis, O.; Saiz Sánchez, C.; Cotell Simón, O.; Bautista Rentero, D.; Miguel Bayarri, V. Valor pronóstico de la interleucina 6 en la mortalidad de pacientes con sepsis. Med. Clínica 2016, 147, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Goldin, N.P.; Wolff, S.M. Demonstration and characterization of two distinct human leukocytic pyrogens. J. Exp. Med. 1974, 139, 1369–1381. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef]
- Di Paolo, N.C.; Shayakhmetov, D.M. Interleukin 1α and the inflammatory process. Nat. Immunol. 2016, 17, 906–913. [Google Scholar] [CrossRef]
- Netea, M.G.; Van de Veerdonk, F.L.; Van der Meer, J.W.M.; Dinarello, C.A.; Joosten, L.A.B. Inflammasome-Independent Regulation of IL-1-Family Cytokines. Annu. Rev. Immunol. 2015, 33, 49–77. [Google Scholar] [CrossRef]
- Bersudsky, M.; Luski, L.; Fishman, D.; White, R.M.; Ziv-Sokolovskaya, N.; Dotan, S.; Rider, P.; Kaplanov, I.; Aychek, T.; Dinarello, C.A.; et al. Non-redundant properties of IL-1α and IL-1β during acute colon inflammation in mice. Gut 2014, 63, 598–609. [Google Scholar] [CrossRef]
- McCarthy, D.A.; Ranganathan, A.; Subbaram, S.; Flaherty, N.L.; Patel, N.; Trebak, M.; Hempel, N.; Melendez, J.A. Redox-control of the alarmin, Interleukin-1α. Redox Biol. 2013, 1, 218–225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, B.; Lee, Y.; Kim, E.; Kwak, A.; Ryoo, S.; Bae, S.H.; Azam, T.; Kim, S.; Dinarello, C.A. The Interleukin-1α Precursor is Biologically Active and is Likely a Key Alarmin in the IL-1 Family of Cytokines. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Inukai, Y.; Takii, T.; Furutani, Y.; Shibata, Y.; Hayashi, H.; Sakurada, S.; Okamoto, T.; Inoue, J.; Oomoto, Y.; et al. Molecular analysis of constitutive IL-1alpha gene expression in human melanoma cells: Autocrine stimulation through NF-kappaB activation by endogenous IL-1alpha. Cytokine 1998, 10, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Groß, O.; Yazdi, A.S.; Thomas, C.J.; Masin, M.; Heinz, L.X.; Guarda, G.; Quadroni, M.; Drexler, S.K.; Tschopp, J. Inflammasome Activators Induce Interleukin-1α Secretion via Distinct Pathways with Differential Requirement for the Protease Function of Caspase-1. Immunity 2012, 36, 388–400. [Google Scholar] [CrossRef]
- Kuida, K.; Lippke, J.A.; Ku, G.; Harding, M.W.; Livingston, D.J.; Su, M.S.; Flavell, R.A. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science (N. Y.) 1995, 267, 2000–2003. [Google Scholar] [CrossRef]
- Colotta, F.; Re, F.; Muzio, M.; Bertini, R.; Polentarutti, N.; Sironi, M.; Giri, J.G.; Dower, S.K.; Sims, J.E.; Mantovani, A. Interleukin-1 type II receptor: A decoy target for IL-1 that is regulated by IL-4. Science (N. Y.) 1993, 261, 472–475. [Google Scholar] [CrossRef]
- Anquetil, F.; Sabouri, S.; Thivolet, C.; Rodriguez-Calvo, T.; Zapardiel-Gonzalo, J.; Amirian, N.; Schneider, D.; Castillo, E.; Lajevardi, Y.; von Herrath, M.G. Alpha cells, the main source of IL-1β in human pancreas. J. Autoimmun. 2017, 81, 68–73. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: NF- B Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Qu, Y.; Franchi, L.; Nunez, G.; Dubyak, G.R. Nonclassical IL-1 Secretion Stimulated by P2X7 Receptors Is Dependent on Inflammasome Activation and Correlated with Exosome Release in Murine Macrophages. J. Immunol. 2007, 179, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Gattorno, M.; Tassi, S.; Carta, S.; Delfino, L.; Ferlito, F.; Pelagatti, M.A.; D’Osualdo, A.; Buoncompagni, A.; Alpigiani, M.G.; Alessio, M.; et al. Pattern of interleukin-1β secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients withCIAS1 mutations. Arthritis Rheum. 2007, 56, 3138–3148. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Zhen, J.; Zhang, L.; Pan, J.; Ma, S.; Yu, X.; Li, X.; Chen, S.; Du, W. AIM2 Mediates Inflammation-Associated Renal Damage in Hepatitis B Virus-Associated Glomerulonephritis by Regulating Caspase-1, IL-1 β, and IL-18. Mediat. Inflamm. 2014, 2014, 190860. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef]
- Feldmann, J.; Prieur, A.M.; Quartier, P.; Berquin, P.; Certain, S.; Cortis, E.; Teillac-Hamel, D.; Fischer, A.; de Saint Basile, G. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am. J. Hum. Genet. 2002, 71, 198–203. [Google Scholar] [CrossRef]
- Aksentijevich, I.; Nowak, M.; Mallah, M.; Chae, J.J.; Watford, W.T.; Hofmann, S.R.; Stein, L.; Russo, R.; Goldsmith, D.; Dent, P.; et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): A new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002, 46, 3340–3348. [Google Scholar] [CrossRef]
- Cuisset, L.; Jeru, I.; Dumont, B.; Fabre, A.; Cochet, E.; Le Bozec, J.; Delpech, M.; Amselem, S.; Touitou, I.; French, C.S.G. Mutations in the autoinflammatory cryopyrin-associated periodic syndrome gene: Epidemiological study and lessons from eight years of genetic analysis in France. Ann. Rheum. Dis. 2011, 70, 495–499. [Google Scholar] [CrossRef]
- ter Haar, N.M.; Oswald, M.; Jeyaratnam, J.; Anton, J.; Barron, K.S.; Brogan, P.A.; Cantarini, L.; Galeotti, C.; Grateau, G.; Hentgen, V.; et al. Recommendations for the management of autoinflammatory diseases. Ann. Rheum. Dis. 2015, 74, 1636–1644. [Google Scholar] [CrossRef]
- Landmann, E.C.; Walker, U.A. Pharmacological treatment options for cryopyrin-associated periodic syndromes. Expert Rev. Clin. Pharm. 2017, 10, 855–864. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arter. Thromb/Vasc/Biol/ 2003, 23, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Jeurissen, M.L.; van Gorp, P.J.; Gijbels, M.J.; Walenbergh, S.M.; Houben, T.; van Gorp, R.; Pottgens, C.C.; Stienstra, R.; Netea, M.G.; et al. Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr(-/-) mice. FEBS J. 2015, 282, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Xing, S.; Gong, Z.; Mu, W.; Xing, Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediat. Inflamm. 2014, 2014, 507208. [Google Scholar] [CrossRef]
- Menu, P.; Pellegrin, M.; Aubert, J.F.; Bouzourene, K.; Tardivel, A.; Mazzolai, L.; Tschopp, J. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011, 2, e137. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, M.; Huang, K.; Zhang, Z.; Shao, N.; Zhang, Y.; Wang, W.; Wang, S. Oxidized low-density lipoprotein induces secretion of interleukin-1beta by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2012, 425, 121–126. [Google Scholar] [CrossRef]
- Son, S.J.; Rhee, K.J.; Lim, J.; Kim, T.U.; Kim, T.J.; Kim, Y.S. Triglyceride-induced macrophage cell death is triggered by caspase-1. Biol. Pharm. Bull. 2013, 36, 108–113. [Google Scholar] [CrossRef][Green Version]
- Guo, C.; Fu, R.; Wang, S.; Huang, Y.; Li, X.; Zhou, M.; Zhao, J.; Yang, N. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin. Exp. Immunol. 2018, 194, 231–243. [Google Scholar] [CrossRef]
- Ippagunta, S.K.; Brand, D.D.; Luo, J.; Boyd, K.L.; Calabrese, C.; Stienstra, R.; Van de Veerdonk, F.L.; Netea, M.G.; Joosten, L.A.; Lamkanfi, M.; et al. Inflammasome-independent role of apoptosis-associated speck-like protein containing a CARD (ASC) in T cell priming is critical for collagen-induced arthritis. J. Biol. Chem. 2010, 285, 12454–12462. [Google Scholar] [CrossRef]
- Joosten, L.A.; Netea, M.G.; Fantuzzi, G.; Koenders, M.I.; Helsen, M.M.; Sparrer, H.; Pham, C.T.; van der Meer, J.W.; Dinarello, C.A.; van den Berg, W.B. Inflammatory arthritis in caspase 1 gene-deficient mice: Contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum. 2009, 60, 3651–3662. [Google Scholar] [CrossRef]
- Vande Walle, L.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.D.; Van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Choulaki, C.; Papadaki, G.; Repa, A.; Kampouraki, E.; Kambas, K.; Ritis, K.; Bertsias, G.; Boumpas, D.T.; Sidiropoulos, P. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res. 2015, 17, 257. [Google Scholar] [CrossRef] [PubMed]
- Ruscitti, P.; Cipriani, P.; Di Benedetto, P.; Liakouli, V.; Berardicurti, O.; Carubbi, F.; Ciccia, F.; Alvaro, S.; Triolo, G.; Giacomelli, R. Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)-1beta via the nucleotide-binding domain and leucine-rich repeat containing family pyrin 3(NLRP3)-inflammasome activation: A possible implication for therapeutic decision in these patients. Clin. Exp. Immunol. 2015, 182, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Shin, T.H.; Kim, H.S.; Kang, T.W.; Lee, B.C.; Lee, H.Y.; Kim, Y.J.; Shin, J.H.; Seo, Y.; Won Choi, S.; Lee, S.; et al. Human umbilical cord blood-stem cells direct macrophage polarization and block inflammasome activation to alleviate rheumatoid arthritis. Cell Death Dis. 2016, 7, e2524. [Google Scholar] [CrossRef]
- Sui, J.; Li, H.; Fang, Y.; Liu, Y.; Li, M.; Zhong, B.; Yang, F.; Zou, Q.; Wu, Y. NLRP1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in han chinese. Arthritis Rheum. 2012, 64, 647–654. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, Y.; Zou, F.; Wu, M.; Fan, C.; Ding, Y. 11beta-Hydroxysteroid dehydrogenase 1 inhibition attenuates collagen-induced arthritis. Int. Immunopharmacol. 2013, 17, 489–494. [Google Scholar] [CrossRef]
- Li, F.; Guo, N.; Ma, Y.; Ning, B.; Wang, Y.; Kou, L. Inhibition of P2X4 suppresses joint inflammation and damage in collagen-induced arthritis. Inflammation 2014, 37, 146–153. [Google Scholar] [CrossRef]
- Grandemange, S.; Sanchez, E.; Louis-Plence, P.; Tran Mau-Them, F.; Bessis, D.; Coubes, C.; Frouin, E.; Seyger, M.; Girard, M.; Puechberty, J.; et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann Rheum. Dis. 2017, 76, 1191–1198. [Google Scholar] [CrossRef]
- Mansoori, M.N.; Shukla, P.; Kakaji, M.; Tyagi, A.M.; Srivastava, K.; Shukla, M.; Dixit, M.; Kureel, J.; Gupta, S.; Singh, D. IL-18BP is decreased in osteoporotic women: Prevents Inflammasome mediated IL-18 activation and reduces Th17 differentiation. Sci. Rep. 2016, 6, 33680. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, L.; Wang, Z.; Li, C.; Li, S.; Li, L.; Fan, Q.; Zheng, L. Melatonin Suppresses Estrogen Deficiency-Induced Osteoporosis and Promotes Osteoblastogenesis by Inactivating the NLRP3 Inflammasome. Calcif. Tissue Int. 2018, 103, 400–410. [Google Scholar] [CrossRef]
- Snouwaert, J.N.; Nguyen, M.; Repenning, P.W.; Dye, R.; Livingston, E.W.; Kovarova, M.; Moy, S.S.; Brigman, B.E.; Bateman, T.A.; Ting, J.P.; et al. An NLRP3 Mutation Causes Arthropathy and Osteoporosis in Humanized Mice. Cell Rep. 2016, 17, 3077–3088. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kim, K.S.; Kim, M.J.; Kim, H.S.; Lee, K.Y.; Kang, K.W. Auranofin Inhibits RANKL-Induced Osteoclastogenesis by Suppressing Inhibitors of kappaB Kinase and Inflammasome-Mediated Interleukin-1beta Secretion. Oxid. Med. Cell Longev. 2019, 2019, 3503912. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, K.; Wan, X.; Wang, F.; Guo, Z.; Mo, Z. NLRP3 inflammasome activation in mesenchymal stem cells inhibits osteogenic differentiation and enhances adipogenic differentiation. Biochem. Biophys. Res. Commun. 2017, 484, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Chen, H.; Li, F.; Zhu, Y.; Yin, W.; Zhuo, Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-kappaB pathway in acute glaucoma. J. Neuroinflamm. 2015, 12, 137. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 11181–11186. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Doyle, S.L. An eye on the future of inflammasomes and drug development in AMD. J. Mol. Med. (Berl.) 2013, 91, 1059–1070. [Google Scholar] [CrossRef]
- Doyle, S.L.; Campbell, M.; Ozaki, E.; Salomon, R.G.; Mori, A.; Kenna, P.F.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Lavelle, E.C.; et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 2012, 18, 791–798. [Google Scholar] [CrossRef]
- Ildefonso, C.J.; Biswal, M.R.; Ahmed, C.M.; Lewin, A.S. The NLRP3 Inflammasome and its Role in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2016, 854, 59–65. [Google Scholar] [CrossRef]
- Yerramothu, P.; Vijay, A.K.; Willcox, M.D.P. Inflammasomes, the eye and anti-inflammasome therapy. Eye (Lond.) 2018, 32, 491–505. [Google Scholar] [CrossRef]
- Relja, B.; Horstmann, J.P.; Kontradowitz, K.; Jurida, K.; Schaible, A.; Neunaber, C.; Oppermann, E.; Marzi, I. Nlrp1 inflammasome is downregulated in trauma patients. J. Mol. Med. (Berl) 2015, 93, 1391–1400. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.P.; Lotocki, G.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D.; Keane, R.W. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J. Cereb. Blood Flow. Metab. 2009, 29, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Kovarova, M.; Hesker, P.R.; Jania, L.; Nguyen, M.; Snouwaert, J.N.; Xiang, Z.; Lommatzsch, S.E.; Huang, M.T.; Ting, J.P.; Koller, B.H. NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J. Immunol. 2012, 189, 2006–2016. [Google Scholar] [CrossRef] [PubMed]
- Kerr, N.; Lee, S.W.; Perez-Barcena, J.; Crespi, C.; Ibanez, J.; Bullock, M.R.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. Inflammasome proteins as biomarkers of traumatic brain injury. PLoS ONE 2018, 13, e0210128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Q.; Zeng, L.; Gu, W.; Zhang, L.Y.; Zhou, J.; Jiang, D.P.; Du, D.Y.; Hu, P.; Yang, C.; Yan, J.; et al. Clinical relevance of single nucleotide polymorphisms within the entire NLRP3 gene in patients with major blunt trauma. Crit. Care 2011, 15, R280. [Google Scholar] [CrossRef]
- Hosseinian, N.; Cho, Y.; Lockey, R.F.; Kolliputi, N. The role of the NLRP3 inflammasome in pulmonary diseases. Adv Respir. Dis. 2015, 9, 188–197. [Google Scholar] [CrossRef]
- Jones, H.D.; Crother, T.R.; Gonzalez-Villalobos, R.A.; Jupelli, M.; Chen, S.; Dagvadorj, J.; Arditi, M.; Shimada, K. The NLRP3 inflammasome is required for the development of hypoxemia in LPS/mechanical ventilation acute lung injury. Am. J. Respir. Cell Mol. Biol. 2014, 50, 270–280. [Google Scholar] [CrossRef]
- Kuipers, M.T.; Aslami, H.; Janczy, J.R.; van der Sluijs, K.F.; Vlaar, A.P.; Wolthuis, E.K.; Choi, G.; Roelofs, J.J.; Flavell, R.A.; Sutterwala, F.S.; et al. Ventilator-induced lung injury is mediated by the NLRP3 inflammasome. Anesthesiology 2012, 116, 1104–1115. [Google Scholar] [CrossRef]
- Kerr, N.A.; De Rivero Vaccari, J.P.; Abbassi, S.; Kaur, H.; Zambrano, R.; Wu, S.; Dietrich, W.D.; Keane, R.W. Traumatic Brain Injury-Induced Acute Lung Injury: Evidence for Activation and Inhibition of a Neural-Respiratory-Inflammasome Axis. J. Neurotrauma 2018, 35, 2067–2076. [Google Scholar] [CrossRef]
- Zou, P.; Liu, X.; Li, G.; Wang, Y. Resveratrol pretreatment attenuates traumatic brain injury in rats by suppressing NLRP3 inflammasome activation via SIRT1. Mol. Med. Rep. 2018, 17, 3212–3217. [Google Scholar] [CrossRef]
- Osuka, A.; Hanschen, M.; Stoecklein, V.; Lederer, J.A. A protective role for inflammasome activation following injury. Shock 2012, 37, 47–55. [Google Scholar] [CrossRef]
- Xu, X.; Yin, D.; Ren, H.; Gao, W.; Li, F.; Sun, D.; Wu, Y.; Zhou, S.; Lyu, L.; Yang, M.; et al. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol. Dis. 2018, 117, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Li, M.; He, F.; Zhou, S.; Zhu, L. Targeting the NLRP3 inflammasome to attenuate spinal cord injury in mice. J Neuroinflamm. 2017, 14, 207. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Yuan, Z.; Wang, G.; Wang, X.; Li, K. The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice. Int. Immunopharmacol. 2019, 70, 147–155. [Google Scholar] [CrossRef]
- Li, J.; Chen, J.; Mo, H.; Chen, J.; Qian, C.; Yan, F.; Gu, C.; Hu, Q.; Wang, L.; Chen, G. Minocycline Protects Against NLRP3 Inflammasome-Induced Inflammation and P53-Associated Apoptosis in Early Brain Injury After Subarachnoid Hemorrhage. Mol. Neurobiol. 2016, 53, 2668–2678. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Coutts, G.; Lenart, N.; Cruickshank, S.M.; Pelegrin, P.; Skinner, J.; Rothwell, N.; Allan, S.M.; Brough, D. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl. Acad. Sci. USA 2015, 112, 4050–4055. [Google Scholar] [CrossRef] [PubMed]
- Horai, R.; Asano, M.; Sudo, K.; Kanuka, H.; Suzuki, M.; Nishihara, M.; Takahashi, M.; Iwakura, Y. Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J. Exp. Med. 1998, 187, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Wu, D.; Zuo, Q.; Wang, Z.; Fan, W. Ginsenoside Rb1 prevents interleukin-1 beta induced inflammation and apoptosis in human articular chondrocytes. Int. Orthop. 2013, 37, 2065–2070. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Van der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef]
- Ren, G.; Zhao, X.; Zhang, L.; Zhang, J.; L’Huillier, A.; Ling, W.; Roberts, A.I.; Le, A.D.; Shi, S.; Shao, C.; et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J. Immunol. 2010, 184, 2321–2328. [Google Scholar] [CrossRef]
- Al-Sadi, R.M.; Ma, T.Y. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J. Immunol. 2007, 178, 4641–4649. [Google Scholar] [CrossRef]
- Carmi, Y.; Voronov, E.; Dotan, S.; Lahat, N.; Rahat, M.A.; Fogel, M.; Huszar, M.; White, M.R.; Dinarello, C.A.; Apte, R.N. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J. Immunol. 2009, 183, 4705–4714. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Oleinika, K.; Tonon, S.; Doyle, R.; Bosma, A.; Carter, N.A.; Harris, K.A.; Jones, S.A.; Klein, N.; Mauri, C. Regulatory B cells are induced by gut microbiota-driven interleukin-1β and interleukin-6 production. Nat. Med. 2014, 20, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Van Griensven, M. Zytokine als Marker bei Polytrauma. Der. Unf. 2014, 117, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Kurt, A.N.C.; Aygun, A.D.; Godekmerdan, A.; Kurt, A.; Dogan, Y.; Yilmaz, E. Serum IL-1beta, IL-6, IL-8, and TNF-alpha levels in early diagnosis and management of neonatal sepsis. Mediat. Inflamm. 2007, 2007, 31397. [Google Scholar] [CrossRef] [PubMed]
- Al-Fadhli, M.A.; Saraya, M.A.; Qasem, J.A. Evaluation of leptin, interleukin-1 beta and tumor necrosis factor alpha in serum of malaria patients as prognostic markers of treatment outcome. Asian Pac. J. Trop. Biomed. 2014, 4, 441–445. [Google Scholar] [CrossRef]
- De Pablo, R.; Monserrat, J.; Reyes, E.; Diaz-Martin, D.; Rodriguez Zapata, M.; Carballo, F.; De la Hera, A.; Prieto, A.; Alvarez-Mon, M. Mortality in Patients With Septic Shock Correlates With Anti-Inflammatory But not Proinflammatory Immunomodulatory Molecules. J. Intensive Care Med. 2011, 26, 125–132. [Google Scholar] [CrossRef]
- Maruotti, N.; Corrado, A.; Cantatore, F.P. Osteoporosis and rheumatic diseases. Reumatismo 2014, 66, 125–135. [Google Scholar] [CrossRef]
- Lacativa, P.G.; Farias, M.L. Osteoporosis and inflammation. Arq. Bras. Endocrinol. Metab. 2010, 54, 123–132. [Google Scholar] [CrossRef]
- Gu, H.F.; Gu, L.J.; Wu, Y.; Zhao, X.H.; Zhang, Q.; Xu, Z.R.; Yang, Y.M. Efficacy and Safety of Denosumab in Postmenopausal Women With Osteoporosis: A Meta-Analysis. Medicine (Baltim.) 2015, 94, e1674. [Google Scholar] [CrossRef]
- Choi, N.K.; Solomon, D.H.; Tsacogianis, T.N.; Landon, J.E.; Song, H.J.; Kim, S.C. Comparative Safety and Effectiveness of Denosumab Versus Zoledronic Acid in Patients With Osteoporosis: A Cohort Study. J. Bone Min. Res. 2017, 32, 611–617. [Google Scholar] [CrossRef]
- Kullenberg, T.; Lofqvist, M.; Leinonen, M.; Goldbach-Mansky, R.; Olivecrona, H. Long-term safety profile of anakinra in patients with severe cryopyrin-associated periodic syndromes. Rheumatology (Oxf.) 2016, 55, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.V.; Sanmarti, R.; Canete, J.D. The safety of tumor necrosis factor-alpha inhibitors in the treatment of rheumatoid arthritis. Expert Opin. Drug Saf. 2016, 15, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA. 2007, 104, 282–287. [Google Scholar] [CrossRef]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrancais, E.; Dujardin, A.; Ortega, N.; Girard, J.P. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: In situ analysis using a novel Il-33-LacZ gene trap reporter strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef]
- Palmer, G.; Lipsky, B.P.; Smithgall, M.D.; Meininger, D.; Siu, S.; Talabot-Ayer, D.; Gabay, C.; Smith, D.E. The IL-1 receptor accessory protein (AcP) is required for IL-33 signaling and soluble AcP enhances the ability of soluble ST2 to inhibit IL-33. Cytokine 2008, 42, 358–364. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef]
- Baekkevold, E.S.; Roussigné, M.; Yamanaka, T.; Johansen, F.E.; Jahnsen, F.L.; Amalric, F.; Brandtzaeg, P.; Erard, M.; Haraldsen, G.; Girard, J.P. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am. J. Pathol. 2003, 163, 69–79. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef]
- Kakkar, R.; Hei, H.; Dobner, S.; Lee, R.T. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J. Biol. Chem. 2012, 287, 6941–6948. [Google Scholar] [CrossRef]
- Byers, D.E.; Alexander-Brett, J.; Patel, A.C.; Agapov, E.; Dang-Vu, G.; Jin, X.; Wu, K.; You, Y.; Alevy, Y.; Girard, J.P.; et al. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J. Clin. Investig. 2013, 123, 3967–3982. [Google Scholar] [CrossRef] [PubMed]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Kearley, J.; Silver, J.S.; Sanden, C.; Liu, Z.; Berlin, A.A.; White, N.; Mori, M.; Pham, T.H.; Ward, C.K.; Criner, G.J.; et al. Cigarette Smoke Silences Innate Lymphoid Cell Function and Facilitates an Exacerbated Type I Interleukin-33-Dependent Response to Infection. Immunity 2015, 42, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Komai-Koma, M.; Xu, D.; Li, Y.; McKenzie, A.N.J.; McInnes, I.B.; Liew, F.Y. IL-33 is a chemoattractant for human Th2 cells. Eur. J. Immunol. 2007, 37, 2779–2786. [Google Scholar] [CrossRef] [PubMed]
- Rank, M.A.; Kobayashi, T.; Kozaki, H.; Bartemes, K.R.; Squillace, D.L.; Kita, H. IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol. 2009, 123, 1047–1054. [Google Scholar] [CrossRef]
- Sun, J.C.; Ma, A.; Lanier, L.L. Cutting edge: IL-15-independent NK cell response to mouse cytomegalovirus infection. J. Immunol. 2009, 183, 2911–2914. [Google Scholar] [CrossRef]
- Bourgeois, E.; Van, L.P.; Samson, M.; Diem, S.; Barra, A.; Roga, S.; Gombert, J.M.; Schneider, E.; Dy, M.; Gourdy, P.; et al. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur. J. Immunol. 2009, 39, 1046–1055. [Google Scholar] [CrossRef]
- Villarreal, D.O.; Weiner, D.B. Interleukin 33: A switch-hitting cytokine. Curr. Opin. Immunol. 2014, 28, 102–106. [Google Scholar] [CrossRef]
- Bonilla, W.V.; Frohlich, A.; Senn, K.; Kallert, S.; Fernandez, M.; Johnson, S.; Kreutzfeldt, M.; Hegazy, A.N.; Schrick, C.; Fallon, P.G.; et al. The Alarmin Interleukin-33 Drives Protective Antiviral CD8+ T Cell Responses. Science 2012, 335, 984–989. [Google Scholar] [CrossRef]
- Ginaldi, L.; De Martinis, M.; Saitta, S.; Sirufo, M.M.; Mannucci, C.; Casciaro, M.; Ciccarelli, F.; Gangemi, S. Interleukin-33 serum levels in postmenopausal women with osteoporosis. Sci. Rep. 2019, 9, 3786. [Google Scholar] [CrossRef]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Mountz, J.D.; Kimberly, R.P. Immunobiology of tumor necrosis factor receptor superfamily. Immunol. Res. 2002, 26, 323–336. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.M. Tumor necrosis factor. Cancer Lett. 2013, 328, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Grell, M. Tumor necrosis factor (TNF) receptors in cellular signaling of soluble and membrane-expressed TNF. J. Inflamm. 1995, 47, 8–17. [Google Scholar] [PubMed]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279. [Google Scholar] [CrossRef]
- Bradley, J. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, H.; Zhang, J.; Volk, A.; Zhang, S.; Wei, W.; Zhang, S.; Breslin, P.; Zhang, J. TNF-/Fas-RIP-1-induced cell death signaling separates murine hematopoietic stem cells/progenitors into 2 distinct populations. Blood 2011, 118, 6057–6067. [Google Scholar] [CrossRef][Green Version]
- Tsurumi, A.; Que, Y.A.; Ryan, C.M.; Tompkins, R.G.; Rahme, L.G. TNF-α/IL-10 Ratio Correlates with Burn Severity and May Serve as a Risk Predictor of Increased Susceptibility to Infections. Front. Public Health 2016, 4, 216. [Google Scholar] [CrossRef]
- Spielmann, S.; Kerner, T.; Ahlers, O.; Keh, D.; Gerlach, M.; Gerlach, H. Early detection of increased tumour necrosis factor alpha (TNFalpha) and soluble TNF receptor protein plasma levels after trauma reveals associations with the clinical course. Acta Anaesthesiol. Scand. 2001, 45, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Reikerås, O.; Borgen, P. Activation of markers of inflammation, coagulation and fibrinolysis in musculoskeletal trauma. PLoS ONE 2014, 9, e107881. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Fong, Y.; Hesse, D.G.; Manogue, K.R.; Lee, A.T.; Kuo, G.C.; Lowry, S.F.; Cerami, A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 1987, 330, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Mark, K.S.; Trickler, W.J.; Miller, D.W. Tumor necrosis factor-alpha induces cyclooxygenase-2 expression and prostaglandin release in brain microvessel endothelial cells. J. Pharmacol. Exp. Ther. 2001, 297, 1051–1058. [Google Scholar] [PubMed]
- Van Kampen, C.; Mallard, B.A. Regulation of bovine intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) on cultured aortic endothelial cells. Vet. Immunol. Immunopathol. 2001, 79, 129–138. [Google Scholar] [CrossRef]
- Stoecklein, V.M.; Osuka, A.; Lederer, J.A. Trauma equals danger--damage control by the immune system. J. Leukoc. Biol. 2012, 92, 539–551. [Google Scholar] [CrossRef]
- Yoshida, L.S.; Tsunawaki, S. Expression of NADPH oxidases and enhanced H(2)O(2)-generating activity in human coronary artery endothelial cells upon induction with tumor necrosis factor-alpha. Int. Immunopharmacol. 2008, 8, 1377–1385. [Google Scholar] [CrossRef]
- Beutler, B.; Milsark, I.W.; Cerami, A.C. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science (N. Y.) 1985, 229, 869–871. [Google Scholar] [CrossRef]
- Pfeffer, K.; Matsuyama, T.; Kündig, T.M.; Wakeham, A.; Kishihara, K.; Shahinian, A.; Wiegmann, K.; Ohashi, P.S.; Krönke, M.; Mak, T.W. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 1993, 73, 457–467. [Google Scholar] [CrossRef]
- Eichacker, P.Q.; Parent, C.; Kalil, A.; Esposito, C.; Cui, X.; Banks, S.M.; Gerstenberger, E.P.; Fitz, Y.; Danner, R.L.; Natanson, C. Risk and the efficacy of antiinflammatory agents: Retrospective and confirmatory studies of sepsis. Am. J. Respir. Crit. Care Med. 2002, 166, 1197–1205. [Google Scholar] [CrossRef]
- Dingle, K.; Azizieh, F. Multivariate Comparison of Cytokine Profiles for Normal- and Low-Bone-Density Subjects. Diagnostics (Basel) 2019, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; He, C. TNF-alpha and IL-6: The link between immune and bone system. Curr Drug Targets 2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; He, C.; Yu, X. Pro-Inflammatory Cytokines: New Potential Therapeutic Targets for Obesity-Related Bone Disorders. Curr. Drug Targets 2017, 18, 1664–1675. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lim, D.H.; Oh, J.S.; Kim, Y.G.; Lee, C.K.; Yoo, B.; Hong, S. Effect of TNF inhibitors on bone mineral density in rheumatoid arthritis patients receiving bisphosphonate: A retrospective cohort study. Rheumatol. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Inui, K.; Sugioka, Y.; Mamoto, K.; Okano, T.; Anno, S.; Koike, T. Use of bisphosphonate might be important to improve bone mineral density in patients with rheumatoid arthritis even under tight control: The TOMORROW study. Rheumatol. Int. 2017, 37, 999–1005. [Google Scholar] [CrossRef]
- De Vries, T.J.; El Bakkali, I.; Kamradt, T.; Schett, G.; Jansen, I.D.C.; D’Amelio, P. What Are the Peripheral Blood Determinants for Increased Osteoclast Formation in the Various Inflammatory Diseases Associated With Bone Loss? Front. Immunol. 2019, 10, 505. [Google Scholar] [CrossRef]
- Fiorentino, D.F.; Bond, M.W.; Mosmann, T.R. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J. Exp. Med. 1989, 170, 2081–2095. [Google Scholar] [CrossRef]
- Zdanov, A.; Schalk-Hihi, C.; Gustchina, A.; Tsang, M.; Weatherbee, J.; Wlodawer, A. Crystal structure of interleukin-10 reveals the functional dimer with an unexpected topological similarity to interferon γ. Structure 1995, 3, 591–601. [Google Scholar] [CrossRef]
- Banchereau, J.; Pascual, V.; O’Garra, A. From IL-2 to IL-37: The expanding spectrum of anti-inflammatory cytokines. Nat. Immunol. 2012, 13, 925–931. [Google Scholar] [CrossRef]
- Savan, R.; Ravichandran, S.; Collins, J.R.; Sakai, M.; Young, H.A. Structural conservation of interferon gamma among vertebrates. Cytokine Growth Factor Rev. 2009, 20, 115–124. [Google Scholar] [CrossRef]
- Siewe, L.; Bollati-Fogolin, M.; Wickenhauser, C.; Krieg, T.; Müller, W.; Roers, A. Interleukin-10 derived from macrophages and/or neutrophils regulates the inflammatory response to LPS but not the response to CpG DNA. Eur. J. Immunol. 2006, 36, 3248–3255. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Gagliani, N.; Esplugues, E.; O’Connor, W.; Huber, F.J.; Chaudhry, A.; Kamanaka, M.; Kobayashi, Y.; Booth, C.J.; Rudensky, A.Y.; et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3- and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 2011, 34, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R.; Grütz, G.; Warszawska, K.; Kirsch, S.; Witte, E.; Wolk, K.; Geginat, J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010, 21, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Krause, C.D.; Izotova, L.S.; Pollack, B.P.; Wu, W.; Pestka, S. Identification and functional characterization of a second chain of the interleukin-10 receptor complex. EMBO J. 1997, 16, 5894–5903. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.I.; Logsdon, N.J.; Sheikh, F.; Donnelly, R.P.; Walter, M.R. Conformational changes mediate interleukin-10 receptor 2 (IL-10R2) binding to IL-10 and assembly of the signaling complex. J. Biol. Chem. 2006, 281, 35088–35096. [Google Scholar] [CrossRef] [PubMed]
- Crepaldi, L.; Gasperini, S.; Lapinet, J.A.; Calzetti, F.; Pinardi, C.; Liu, Y.; Zurawski, S.; de Waal Malefyt, R.; Moore, K.W.; Cassatella, M.A. Up-regulation of IL-10R1 expression is required to render human neutrophils fully responsive to IL-10. J. Immunol. (Baltimore Md. 1950) 2001, 167, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Pils, M.C.; Pisano, F.; Fasnacht, N.; Heinrich, J.-M.; Groebe, L.; Schippers, A.; Rozell, B.; Jack, R.S.; Müller, W. Monocytes/macrophages and/or neutrophils are the target of IL-10 in the LPS endotoxemia model. Eur. J. Immunol. 2010, 40, 443–448. [Google Scholar] [CrossRef]
- Sheikh, F.; Baurin, V.V.; Lewis-Antes, A.; Shah, N.K.; Smirnov, S.V.; Anantha, S.; Dickensheets, H.; Dumoutier, L.; Renauld, J.C.; Zdanov, A.; et al. Cutting edge: IL-26 signals through a novel receptor complex composed of IL-20 receptor 1 and IL-10 receptor 2. J. Immunol. 2004, 172, 2006–2010. [Google Scholar] [CrossRef]
- Jones, M.R.; Quinton, L.J.; Simms, B.T.; Lupa, M.M.; Kogan, M.S.; Mizgerd, J.P. Roles of interleukin-6 in activation of STAT proteins and recruitment of neutrophils during Escherichia coli pneumonia. J. Infect. Dis. 2006, 193, 360–369. [Google Scholar] [CrossRef]
- Finbloom, D.S.; Winestock, K.D. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J. Immunol. 1995, 155, 1079–1090. [Google Scholar]
- Kretzschmar, A.K.; Dinger, M.C.; Henze, C.; Brocke-Heidrich, K.; Horn, F. Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem. J. 2004, 377, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Shouval, D.S.; Ouahed, J.; Biswas, A.; Goettel, J.A.; Horwitz, B.H.; Klein, C.; Muise, A.M.; Snapper, S.B. Interleukin 10 Receptor Signaling. In Advances in Immunology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 122, pp. 177–210. [Google Scholar]
- Jung, M.; Sabat, R.; Krätzschmar, J.; Seidel, H.; Wolk, K.; Schönbein, C.; Schütt, S.; Friedrich, M.; Döcke, W.-D.; Asadullah, K.; et al. Expression profiling of IL-10-regulated genes in human monocytes and peripheral blood mononuclear cells from psoriatic patients during IL-10 therapy. Eur. J. Immunol. 2004, 34, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Martire-Greco, D.; Rodriguez-Rodrigues, N.; Landoni, V.I.; Rearte, B.; Isturiz, M.A.; Fernández, G.C. Interleukin-10 controls human peripheral PMN activation triggered by lipopolysaccharide. Cytokine 2013, 62, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Huet, O.; Laemmel, E.; Fu, Y.; Dupic, L.; Aprico, A.; Andrews, K.L.; Moore, X.L.; Harrois, A.; Meikle, P.L.; Vicaut, E.; et al. IL-10 anti oxidant effect decreases leukocytes/endothelial interaction induced by TNF-α. Shock 2013, 39, 83–88. [Google Scholar] [CrossRef]
- Thibodeau, J.; Bourgeois-Daigneault, M.-C.; Huppé, G.; Tremblay, J.; Aumont, A.; Houde, M.; Bartee, E.; Brunet, A.; Gauvreau, M.-E.; de Gassart, A.; et al. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur. J. Immunol. 2008, 38, 1225–1230. [Google Scholar] [CrossRef]
- Vicioso, M.A.; Garaud, J.J.; Réglier-Poupet, H.; Lebeaut, A.; Gougerot-Pocidalo, M.A.; Chollet-Martin, S. Moderate inhibitory effect of interleukin-10 on human neutrophil and monocyte chemotaxis in vitro. Eur. Cytokine Netw. 1998, 9, 247–253. [Google Scholar]
- Sikka, G.; Miller, K.L.; Steppan, J.; Pandey, D.; Jung, S.M.; Fraser, C.D.; Ellis, C.; Ross, D.; Vandegaer, K.; Bedja, D.; et al. Interleukin 10 knockout frail mice develop cardiac and vascular dysfunction with increased age. Exp. Gerontol. 2013, 48, 128–135. [Google Scholar] [CrossRef]
- Koch, N.; Jung, M.; Sabat, R.; Krätzschmar, J.; Döcke, W.D.; Asadullah, K.; Volk, H.-D.; Grütz, G. IL-10 protects monocytes and macrophages from complement-mediated lysis. J. Leukoc. Biol. 2009, 86, 155–166. [Google Scholar] [CrossRef]
- Kühn, R.; Löhler, J.; Rennick, D.; Rajewsky, K.; Müller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Ito, S.; Ansari, P.; Sakatsume, M.; Dickensheets, H.; Vazquez, N.; Donnelly, R.P.; Larner, A.C.; Finbloom, D.S. Interleukin-10 inhibits expression of both interferon alpha- and interferon gamma- induced genes by suppressing tyrosine phosphorylation of STAT1. Blood 1999, 93, 1456–1463. [Google Scholar] [CrossRef]
- Heper, Y.; Akalin, E.H.; Mistik, R.; Akgöz, S.; Töre, O.; Göral, G.; Oral, B.; Budak, F.; Helvaci, S. Evaluation of serum C-reactive protein, procalcitonin, tumor necrosis factor alpha, and interleukin-10 levels as diagnostic and prognostic parameters in patients with community-acquired sepsis, severe sepsis, and septic shock. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2006, 25, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Marchant, A.; Alegre, M.L.; Hakim, A.; Piérard, G.; Marécaux, G.; Friedman, G.; De Groote, D.; Kahn, R.J.; Vincent, J.L.; Goldman, M. Clinical and biological significance of interleukin-10 plasma levels in patients with septic shock. J. Clin. Immunol. 1995, 15, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Neidhardt, R.; Keel, M.; Steckholzer, U.; Safret, A.; Ungethuem, U.; Trentz, O.; Ertel, W. Relationship of Interleukin-10 Plasma Levels to Severity of Injury and Clinical Outcome in Injured Patients. J. Trauma Inj. Infect. Crit. Care 1997, 42, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Sherry, R.M.; Cue, J.I.; Goddard, J.K.; Parramore, J.B.; DiPiro, J.T. Interleukin-10 is associated with the development of sepsis in trauma patients. J. Trauma 1996, 40, 613–616. [Google Scholar] [CrossRef]
- Rios-Arce, N.D.; Dagenais, A.; Feenstra, D.; Coughlin, B.; Kang, H.J.; Mohr, S.; McCabe, L.R.; Parameswaran, N. Loss of interleukin-10 exacerbates early Type-1 diabetes-induced bone loss. J. Cell Physiol. 2019. [Google Scholar] [CrossRef]
- Wang, T.; Jiang, C.; Zhao, P.; Chen, L.; Li, Z. Detection and analysis of serum IL-10 and TGF-beta1 in the SLE patients with osteoporosis or osteonecrosis. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2018, 34, 1032–1035. [Google Scholar]
- Baggiolini, M.; Clark-Lewis, I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992, 307, 97–101. [Google Scholar] [CrossRef]
- Bezzerri, V.; Borgatti, M.; Finotti, A.; Tamanini, A.; Gambari, R.; Cabrini, G. Mapping the Transcriptional Machinery of the IL-8 Gene in Human Bronchial Epithelial Cells. J. Immunol. 2011, 187, 6069–6081. [Google Scholar] [CrossRef]
- Hoffmann, E.; Dittrich-Breiholz, O.; Holtmann, H.; Kracht, M. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 2002, 72, 847–855. [Google Scholar]
- Lehner, M.; Morhart, P.; Stilper, A.; Petermann, D.; Weller, P.; Stachel, D.; Holter, W. Efficient chemokine-dependent migration and primary and secondary IL-12 secretion by human dendritic cells stimulated through Toll-like receptors. J. Immunother. 2007, 30, 312–322. [Google Scholar] [CrossRef]
- Hol, J.; Küchler, A.M.; Johansen, F.E.; Dalhus, B.; Haraldsen, G.; Oynebråten, I. Molecular requirements for sorting of the chemokine interleukin-8/CXCL8 to endothelial Weibel-Palade bodies. J. Biol. Chem. 2009, 284, 23532–23539. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, A.; Voitenok, N.N.; Akalovich, S.; Shaik, S.S.; Randolph, D.A.; Sims, B.; Patel, R.P.; Killingsworth, C.R.; Fallon, M.B.; Ohls, R.K. Developmental changes in circulating IL-8/CXCL8 isoforms in neonates. Cytokine 2009, 46, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Dias, I.H.K.; Marshall, L.; Lambert, P.A.; Chapple, I.L.C.; Matthews, J.B.; Griffiths, H.R. Gingipains from Porphyromonas gingivalis increase the chemotactic and respiratory burst-priming properties of the 77-amino-acid interleukin-8 variant. Infect. Immun. 2008, 76, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Ramjeesingh, R.; Leung, R.; Siu, C.H. Interleukin-8 secreted by endothelial cells induces chemotaxis of melanoma cells through the chemokine receptor CXCR1. FASEB J. 2003, 17, 1292–1294. [Google Scholar] [CrossRef]
- Allen, T.C.; Kurdowska, A. Interleukin 8 and Acute Lung Injury. Arch. Pathol. Lab. Med. 2014, 138, 266–269. [Google Scholar] [CrossRef]
- Kurdowska, A.; Noble, J.M.; Steinberg, K.P.; Ruzinski, J.T.; Hudson, L.D.; Martin, T.R. Anti-interleukin 8 autoantibody: Interleukin 8 complexes in the acute respiratory distress syndrome. Relationship between the complexes and clinical disease activity. Am. J. Respir. Crit. Care Med. 2001, 163, 463–468. [Google Scholar] [CrossRef]
- Kobayashi, Y. The role of chemokines in neutrophil biology. Front. Biosci. 2008, 13, 2400–2407. [Google Scholar] [CrossRef]
- Ning, Y.; Manegold, P.C.; Hong, Y.K.; Zhang, W.; Pohl, A.; Lurje, G.; Winder, T.; Yang, D.; LaBonte, M.J.; Wilson, P.M.; et al. Interleukin-8 is associated with proliferation, migration, angiogenesis and chemosensitivity in vitro and in vivo in colon cancer cell line models. Int. J. Cancer 2011, 128, 2038–2049. [Google Scholar] [CrossRef]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef]
- Kraft, R.; Herndon, D.N.; Finnerty, C.C.; Cox, R.A.; Song, J.; Jeschke, M.G. Predictive Value of IL-8 for Sepsis and Severe Infections After Burn Injury: A Clinical Study. Shock (AugustaGa.) 2015, 43, 222–227. [Google Scholar] [CrossRef]
- Macdonald, S.P.J.; Stone, S.F.; Neil, C.L.; van Eeden, P.E.; Fatovich, D.M.; Arendts, G.; Brown, S.G.A. Sustained elevation of resistin, NGAL and IL-8 are associated with severe sepsis/septic shock in the emergency department. PLoS ONE 2014, 9, e110678. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.C.B.; Regner, A.; Miotto, K.D.L.; Moura, S.d.; Ikuta, N.; Vargas, A.E.; Chies, J.A.B.; Simon, D. Increased levels of interleukin-6, -8 and -10 are associated with fatal outcome following severe traumatic brain injury. Brain Inj. 2014, 28, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Gopcevic, A.; Mazul-Sunko, B.; Marout, J.; Sekulic, A.; Antoljak, N.; Siranovic, M.; Ivanec, Z.; Margaritoni, M.; Bekavac-Beslin, M.; Zarkovic, N. Plasma interleukin-8 as a potential predictor of mortality in adult patients with severe traumatic brain injury. Tohoku J. Exp. Med. 2007, 211, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Al-Daghri, N.M.; Yakout, S.; Al-Shehri, E.; Al-Fawaz, H.A.; Aljohani, N.; Al-Saleh, Y. Inflammatory and bone turnover markers in relation to PTH and vitamin D status among saudi postmenopausal women with and without osteoporosis. Int. J. Clin. Exp. Med. 2014, 7, 3528–3535. [Google Scholar] [PubMed]
- Sousa, L.H.; Linhares, E.V.; Alexandre, J.T.; Lisboa, M.R.; Furlaneto, F.; Freitas, R.; Ribeiro, I.; Val, D.; Marques, M.; Chaves, H.V.; et al. Effects of Atorvastatin on Periodontitis of Rats Subjected to Glucocorticoid-Induced Osteoporosis. J. Periodontol. 2016, 87, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Sun, Y.; Xu, W.; Lin, T.; Zeng, H. Expression of RANKL by peripheral neutrophils and its association with bone mineral density in COPD. Respirology 2017, 22, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 2063. [Google Scholar] [CrossRef]
- Rittirsch, D.; Flierl, M.A.; Ward, P.A. Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 2008, 8, 776–787. [Google Scholar] [CrossRef]
- Scheidt-Nave, C.; Bismar, H.; Leidig-Bruckner, G.; Woitge, H.; Seibel, M.J.; Ziegler, R.; Pfeilschifter, J. Serum interleukin 6 is a major predictor of bone loss in women specific to the first decade past menopause. J. Clin. Endocrinol. Metab 2001, 86, 2032–2042. [Google Scholar] [CrossRef]
- Moffett, S.P.; Zmuda, J.M.; Cauley, J.A.; Stone, K.L.; Nevitt, M.C.; Ensrud, K.E.; Hillier, T.A.; Hochberg, M.C.; Joslyn, G.; Morin, P.; et al. Association of the G-174C variant in the interleukin-6 promoter region with bone loss and fracture risk in older women. J. Bone Min. Res. 2004, 19, 1612–1618. [Google Scholar] [CrossRef]
- Fajar, J.K.; Azharuddin, A. The association between interleukin 6 -174 G/C gene polymorphism and the risk of osteoporosis: A meta-analysis. J. Taibah. Univ. Med. Sci. 2017, 12, 212–220. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Czerny, B.; Kaminski, A.; Kurzawski, M.; Kotrych, D.; Safranow, K.; Dziedziejko, V.; Bohatyrewicz, A.; Pawlik, A. The association of IL-1beta, IL-2, and IL-6 gene polymorphisms with bone mineral density and osteoporosis in postmenopausal women. Eur. J. Obs. Gynecol. Reprod. Biol. 2010, 149, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Parameswaran, V.; Udayan, R.; Burgess, J.; Jones, G. Circulating levels of inflammatory markers predict change in bone mineral density and resorption in older adults: A longitudinal study. J. Clin. Endocrinol Metab 2008, 93, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.X.; Vrindts, Y.; Lopez, M.; De Groote, D.; Zangerle, P.F.; Collette, J.; Franchimont, N.; Geenen, V.; Albert, A.; Reginster, J.Y. Increase in cytokine production (IL-1 beta, IL-6, TNF-alpha but not IFN-gamma, GM-CSF or LIF) by stimulated whole blood cells in postmenopausal osteoporosis. Maturitas 1997, 26, 63–71. [Google Scholar] [CrossRef]
- Jilka, R.L.; Hangoc, G.; Girasole, G.; Passeri, G.; Williams, D.C.; Abrams, J.S.; Boyce, B.; Broxmeyer, H.; Manolagas, S.C. Increased osteoclast development after estrogen loss: Mediation by interleukin-6. Science 1992, 257, 88–91. [Google Scholar] [CrossRef]
- Chao, T.H.; Yu, H.N.; Huang, C.C.; Liu, W.S.; Tsai, Y.W.; Wu, W.T. Association of interleukin-1 beta (-511C/T) polymorphisms with osteoporosis in postmenopausal women. Ann. Saudi. Med. 2010, 30, 437–441. [Google Scholar] [CrossRef]
- Al-Daghri, N.M.; Aziz, I.; Yakout, S.; Aljohani, N.J.; Al-Saleh, Y.; Amer, O.E.; Sheshah, E.; Younis, G.Z.; Al-Badr, F.B. Inflammation as a contributing factor among postmenopausal Saudi women with osteoporosis. Medicine (Baltim.) 2017, 96, e5780. [Google Scholar] [CrossRef]
- Inanir, A.; Ozoran, K.; Tutkak, H.; Mermerci, B. The effects of calcitriol therapy on serum interleukin-1, interleukin-6 and tumour necrosis factor-alpha concentrations in post-menopausal patients with osteoporosis. J. Int. Med. Res. 2004, 32, 570–582. [Google Scholar] [CrossRef]
- Tsuboi, M.; Kawakami, A.; Nakashima, T.; Matsuoka, N.; Urayama, S.; Kawabe, Y.; Fujiyama, K.; Kiriyama, T.; Aoyagi, T.; Maeda, K.; et al. Tumor necrosis factor-alpha and interleukin-1beta increase the Fas-mediated apoptosis of human osteoblasts. J. Lab. Clin. Med. 1999, 134, 222–231. [Google Scholar] [CrossRef]
- Kimble, R.B.; Vannice, J.L.; Bloedow, D.C.; Thompson, R.C.; Hopfer, W.; Kung, V.T.; Brownfield, C.; Pacifici, R. Interleukin-1 receptor antagonist decreases bone loss and bone resorption in ovariectomized rats. J. Clin. Investig. 1994, 93, 1959–1967. [Google Scholar] [CrossRef]
- D’Amelio, P.; Grimaldi, A.; Di Bella, S.; Brianza, S.Z.M.; Cristofaro, M.A.; Tamone, C.; Giribaldi, G.; Ulliers, D.; Pescarmona, G.P.; Isaia, G. Estrogen deficiency increases osteoclastogenesis up-regulating T cells activity: A key mechanism in osteoporosis. Bone 2008, 43, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Kotrych, D.; Dziedziejko, V.; Safranow, K.; Sroczynski, T.; Staniszewska, M.; Juzyszyn, Z.; Pawlik, A. TNF-alpha and IL10 gene polymorphisms in women with postmenopausal osteoporosis. Eur. J. Obs. Gynecol. Reprod. Biol. 2016, 199, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Zha, L.; He, L.; Liang, Y.; Qin, H.; Yu, B.; Chang, L.; Xue, L. TNF-alpha contributes to postmenopausal osteoporosis by synergistically promoting RANKL-induced osteoclast formation. Biomed. Pharm. 2018, 102, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Zhou, Z.; Zhu, L.; Hu, X.; Lu, J.; Shi, C.; Chen, F.; Chen, A. TNF-alpha suppresses osteogenic differentiation of MSCs by accelerating P2Y2 receptor in estrogen-deficiency induced osteoporosis. Bone 2018, 117, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Su, X.; Yang, X.; Hu, C.; Li, B.; Lv, Y.; Shuai, Y.; Jing, H.; Deng, Z.; Jin, Y. TNF-alpha Inhibits FoxO1 by Upregulating miR-705 to Aggravate Oxidative Damage in Bone Marrow-Derived Mesenchymal Stem Cells during Osteoporosis. Stem. Cells 2016, 34, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, D.R.; Nedwin, G.E.; Bringman, T.S.; Smith, D.D.; Mundy, G.R. Stimulation of bone resorption and inhibition of bone formation in vitro by human tumour necrosis factors. Nature 1986, 319, 516–518. [Google Scholar] [CrossRef]
- Tural, S.; Alayli, G.; Kara, N.; Tander, B.; Bilgici, A.; Kuru, O. Association between osteoporosis and polymorphisms of the IL-10 and TGF-beta genes in Turkish postmenopausal women. Hum. Immunol. 2013, 74, 1179–1183. [Google Scholar] [CrossRef]
- Azizieh, F.; Raghupathy, R.; Shehab, D.; Al-Jarallah, K.; Gupta, R. Cytokine profiles in osteoporosis suggest a proresorptive bias. Menopause 2017, 24, 1057–1064. [Google Scholar] [CrossRef]
- Gur, A.; Denli, A.; Nas, K.; Cevik, R.; Karakoc, M.; Sarac, A.J.; Erdogan, F. Possible pathogenetic role of new cytokines in postmenopausal osteoporosis and changes during calcitonin plus calcium therapy. Rheumatol. Int. 2002, 22, 194–198. [Google Scholar] [CrossRef]
- Evans, K.E.; Fox, S.W. Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 2007, 8, 4. [Google Scholar] [CrossRef]
- Dresner-Pollak, R.; Gelb, N.; Rachmilewitz, D.; Karmeli, F.; Weinreb, M. Interleukin 10-deficient mice develop osteopenia, decreased bone formation, and mechanical fragility of long bones. Gastroenterology 2004, 127, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Pathak, J.L.; Bakker, A.D.; Verschueren, P.; Lems, W.F.; Luyten, F.P.; Klein-Nulend, J.; Bravenboer, N. CXCL8 and CCL20 Enhance Osteoclastogenesis via Modulation of Cytokine Production by Human Primary Osteoblasts. PLoS ONE 2015, 10, e0131041. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. https://doi.org/10.3390/ijms20236008
Kany S, Vollrath JT, Relja B. Cytokines in Inflammatory Disease. International Journal of Molecular Sciences. 2019; 20(23):6008. https://doi.org/10.3390/ijms20236008
Chicago/Turabian StyleKany, Shinwan, Jan Tilmann Vollrath, and Borna Relja. 2019. "Cytokines in Inflammatory Disease" International Journal of Molecular Sciences 20, no. 23: 6008. https://doi.org/10.3390/ijms20236008
APA StyleKany, S., Vollrath, J. T., & Relja, B. (2019). Cytokines in Inflammatory Disease. International Journal of Molecular Sciences, 20(23), 6008. https://doi.org/10.3390/ijms20236008