Contribution of a Novel B3GLCT Variant to Peters Plus Syndrome Discovered by a Combination of Next-Generation Sequencing and Automated Text Mining

 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Case History and Clinical Findings
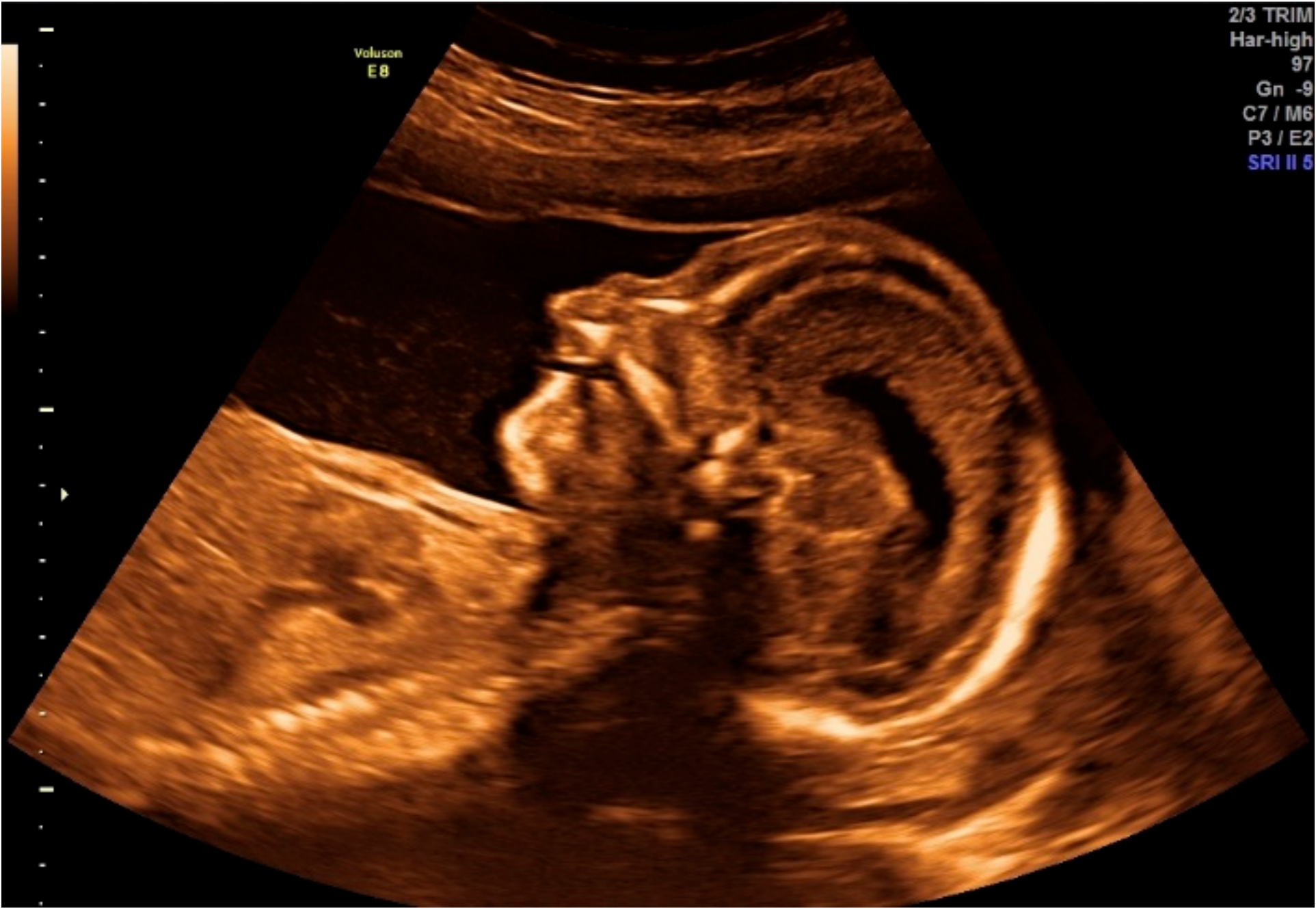
2.1.1. Obstetric Information
2.1.2. Initial Postnatal Examination
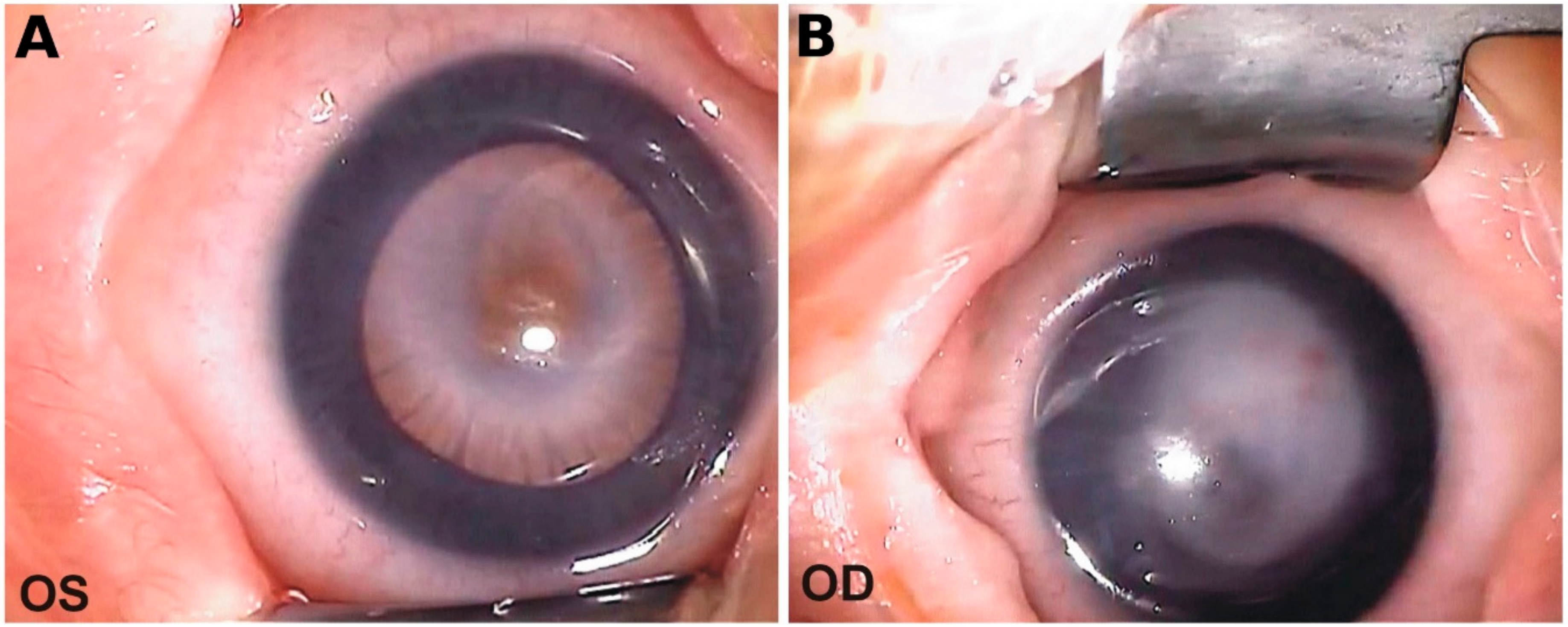
2.1.3. Ophthalmic Evaluation
2.2. High-Throughput Sequencing Results
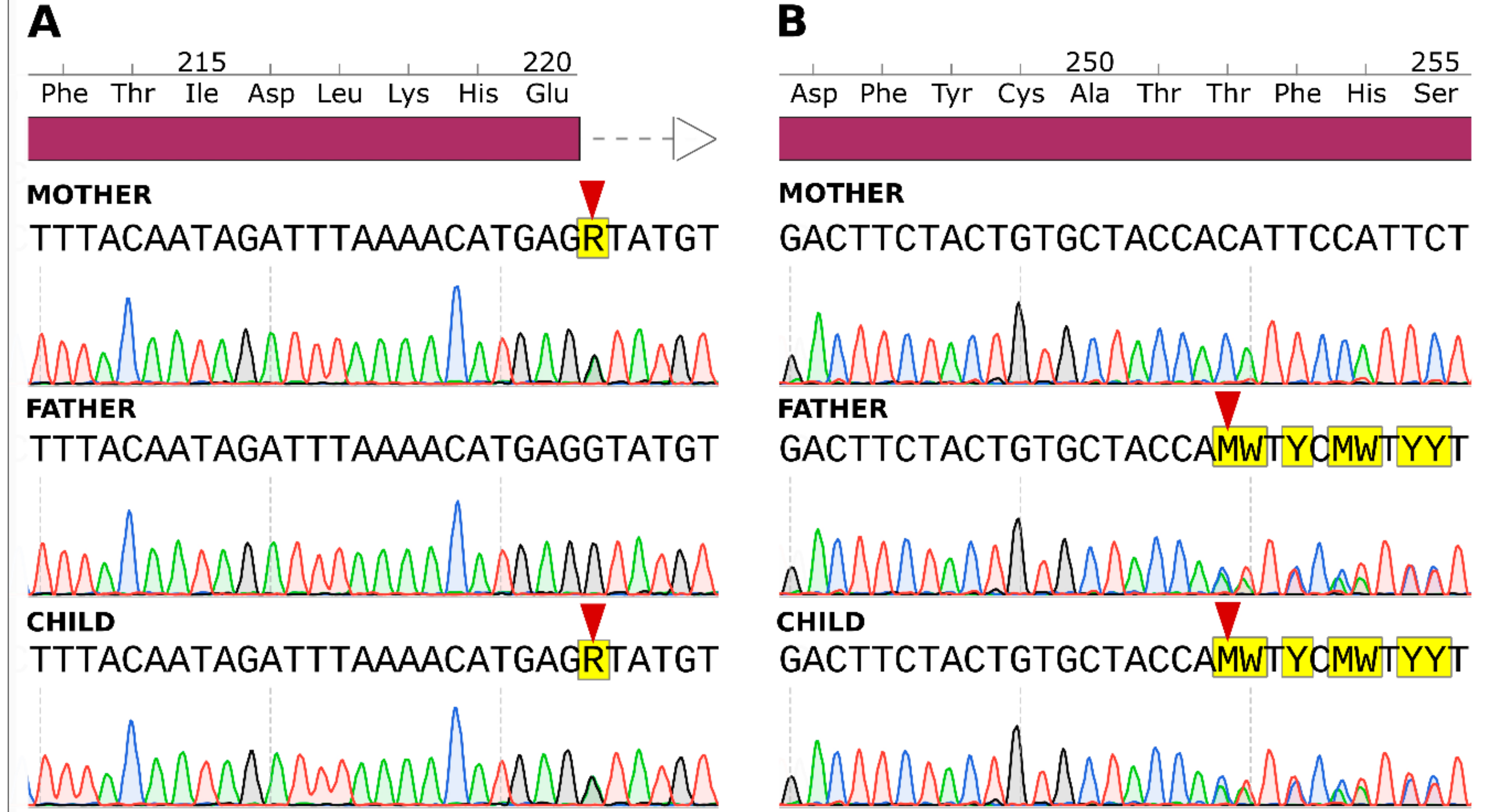
2.3. Sanger Sequencing Results
3. Discussion
4. Methods
4.1. Clinical Assessment
4.2. Ophthalmic Evaluation
4.3. DNA Extraction, Library Preparation, and NGS
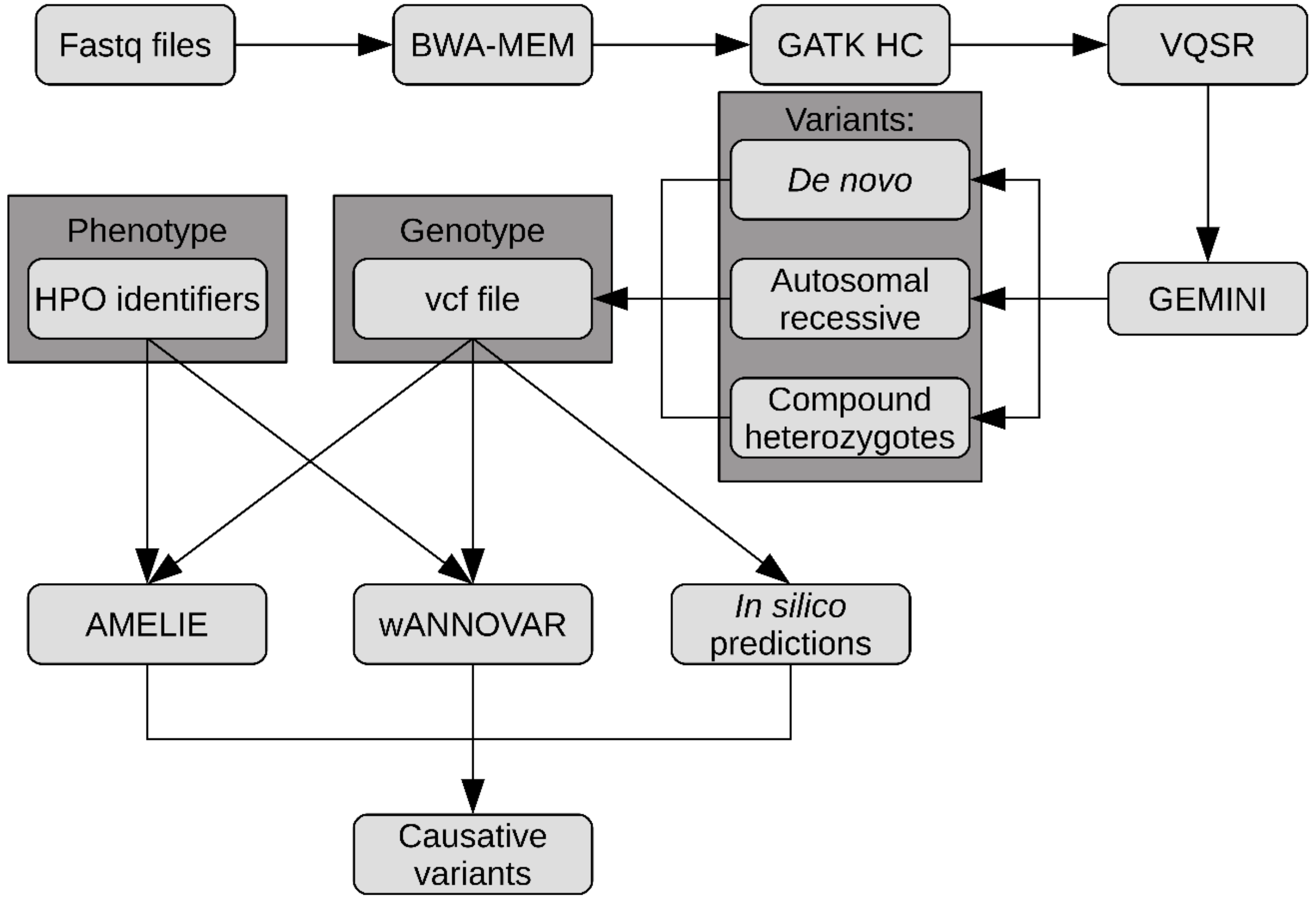
4.4. Data Analysis
4.5. Sanger Sequencing
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.A.; Walter, M.A. Genomics and anterior segment dysgenesis: A review. Clin. Exp. Ophthalmol. 2014, 42, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Jun, A.S.; Broman, K.W.; Do, D.V.; Akpek, E.K.; Stark, W.J.; Gottsch, J.D. Endothelial dystrophy, iris hypoplasia, congenital cataract, and stromal thinning (edict) syndrome maps to chromosome 15q22.1–q25.3. Am. J. Ophthalmol. 2002, 134, 172–176.s. [Google Scholar] [CrossRef]
- Choi, A.; Lao, R.; Ling-Fung Tang, P.; Wan, E.; Mayer, W.; Bardakjian, T.; Shaw, G.M.; Kwok, P.; Schneider, A.; Slavotinek, A. Novel mutations in PXDN cause microphthalmia and anterior segment dysgenesis. Eur. J. Hum. Genet. 2015, 23, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Anand, D.; Agrawal, S.A.; Slavotinek, A.; Lachke, S.A. Mutation update of transcription factor genes FOXE3, HSF4, MAF, and PITX3 causing cataracts and other developmental ocular defects. Hum. Mutat. 2018, 39, 471–494. [Google Scholar] [CrossRef]
- Gould, D.B.; Smith, R.S.; John, S.W.M. Anterior segment development relevant to glaucoma. Int. J. Dev. Biol. 2004, 48, 1015–1029. [Google Scholar] [CrossRef]
- Micheal, S.; Siddiqui, S.N.; Zafar, S.N.; Villanueva-Mendoza, C.; Cortés-González, V.; Khan, M.I.; den Hollander, A.I. A Novel Homozygous Mutation in FOXC1 Causes Axenfeld Rieger Syndrome with Congenital Glaucoma. PLoS ONE 2016, 11, e0160016. [Google Scholar] [CrossRef]
- Weh, E.; Reis, L.M.; Happ, H.C.; Levin, A.V.; Wheeler, P.G.; David, K.L.; Carney, E.; Angle, B.; Hauser, N.; Semina, E.V. Whole exome sequence analysis of Peters anomaly. Hum. Genet. 2014, 133, 1497–1511. [Google Scholar] [CrossRef]
- Meuwissen, M.E.C.; Halley, D.J.J.; Smit, L.S.; Lequin, M.H.; Cobben, J.M.; de Coo, R.; van Harssel, J.; Sallevelt, S.; Woldringh, G.; van der Knaap, M.S.; et al. The expanding phenotype of COL4A1 and COL4A2 mutations: Clinical data on 13 newly identified families and a review of the literature. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 843–853. [Google Scholar] [CrossRef]
- Favor, J.; Gloeckner, C.J.; Janik, D.; Klempt, M.; Neuhäuser-Klaus, A.; Pretsch, W.; Schmahl, W.; Quintanilla-Fend, L. Type IV Procollagen Missense Mutations Associated with Defects of the Eye, Vascular Stability, the Brain, Kidney Function and Embryonic or Postnatal Viability in the Mouse, Mus musculus: An Extension of the Col4a1 Allelic Series and the Identification of the First Two Col4a2 Mutant Alleles. Genetics 2007, 175, 725–736. [Google Scholar]
- Reis, L.M.; Semina, E.V. Genetics of anterior segment dysgenesis disorders. Curr. Opin. Ophthalmol. 2011, 22, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Birgmeier, J.; Haeussler, M.; Deisseroth, C.A.; Jagadeesh, K.A.; Ratner, A.J.; Guturu, H.; Wenger, A.M.; Stenson, P.D.; Cooper, D.N.; Re, C.; et al. AMELIE accelerates Mendelian patient diagnosis directly from the primary literature. bioRxiv 2017, 171322. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Wang, K. wANNOVAR: Annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, M. The clinical code-breakers. Nature 2018, 562, 291. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef]
- Farwell, K.D.; Shahmirzadi, L.; El-Khechen, D.; Powis, Z.; Chao, E.C.; Davis, B.T.; Baxter, R.M.; Zeng, W.; Mroske, C.; Parra, M.C.; et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model–based analysis: Results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 2015, 17, 578–586. [Google Scholar] [CrossRef]
- Smedley, D.; Jacobsen, J.O.B.; Jäger, M.; Köhler, S.; Holtgrewe, M.; Schubach, M.; Siragusa, E.; Zemojtel, T.; Buske, O.J.; Washington, N.L.; et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc. 2015, 10, 2004–2015. [Google Scholar] [CrossRef]
- Jaeken, J.; Lefeber, D.J.; Matthijs, G. Clinical utility gene card for: Peters plus syndrome. Eur. J. Hum. Genet. EJHG 2016, 24, 1232. [Google Scholar] [CrossRef] [PubMed]
- Gilfix, B.M. Congenital disorders of glycosylation and the challenge of rare diseases. Hum. Mutat. 2019. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.L.; Arno, G.; Poulter, J.A.; Khan, K.N.; Morarji, J.; Hull, S.; Pontikos, N.; Rueda Martin, A.; Smith, K.R.; Ali, M.; et al. Association of Steroid 5α-Reductase Type 3 Congenital Disorder of Glycosylation with Early-Onset Retinal Dystrophy. JAMA Ophthalmol. 2017, 135, 339–347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heinonen, T.Y.K.; Mäki, M. Peters’-plus syndrome is a congenital disorder of glycosylation caused by a defect in the β1,3-glucosyltransferase that modifies thrombospondin type 1 repeats. Ann. Med. 2009, 41, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Sato, M.; Kiyohara, K.; Sogabe, M.; Shikanai, T.; Kikuchi, N.; Togayachi, A.; Ishida, H.; Ito, H.; Kameyama, A.; et al. Molecular cloning and characterization of a novel human β1,3-glucosyltransferase, which is localized at the endoplasmic reticulum and glucosylates O-linked fucosylglycan on thrombospondin type 1 repeat domain. Glycobiology 2006, 16, 1194–1206. [Google Scholar] [CrossRef]
- Vasudevan, D.; Takeuchi, H.; Johar, S.S.; Majerus, E.; Haltiwanger, R.S. Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism. Curr. Biol. CB 2015, 25, 286–295. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar]
- Lesnik Oberstein, S.A.J.; Kriek, M.; White, S.J.; Kalf, M.E.; Szuhai, K.; den Dunnen, J.T.; Breuning, M.H.; Hennekam, R.C.M. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am. J. Hum. Genet. 2006, 79, 562–566. [Google Scholar] [CrossRef]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- Lindeboom, R.G.H.; Supek, F.; Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef]
- Khajavi, M.; Inoue, K.; Lupski, J.R. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. EJHG 2006, 14, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Paila, U.; Chapman, B.A.; Kirchner, R.; Quinlan, A.R. GEMINI: Integrative Exploration of Genetic Variation and Genome Annotations. PLOS Comput. Biol. 2013, 9, e1003153. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinformatics 2018, 34, 511–513. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Robinson, P.N.; Wang, K. Phenolyzer: Phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 2015, 12, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutation | Amino Acid Change | Zygosity | Segregation Analysis | gnomAD European MAF | SIFT | PROVEAN | FATHMM-XF | Mutation Taster | dbSNP |
|---|---|---|---|---|---|---|---|---|---|---|
| LRP8 | NM_004631:c.73_74delCA | p.Gln25fs*10 | Compound Heterozygous | De novo | 3.55 × 10−4 | – | – | – | Disease causing | rs1491461533 |
| NM_004631:c.71delT | p.Leu24fs*50 | De novo | 1.79 × 10−4 | – | – | – | Disease causing | rs761955852 | ||
| TTN | NM_133437:c.71141G>A | p.Arg23714His | Compound Heterozygous | Maternal | 2.88 × 10−3 | Tolerated | Neutral | Benign | Polymorphism | rs200650668 |
| NM_133437:c.25190G>T | p.Ser8397Ile | Paternal | 8.52 × 10−4 | – | Deleterious | – | Disease causing | rs200335120 | ||
| DMXL1 | NM_005509:c.7403_7405delATG | p.Asp2468del | Heterozygous | De novo | 9.30 × 10−5 | – | Deleterious | – | Disease causing | rs200335120 |
| LAMA4 | NM_001105206:c.4665+7T>C | – | Compound Heterozygous | Maternal | 1.80 × 10−5 | – | – | Pathogenic | Disease causing | rs751477013 |
| NM_001105206:c.3239G>A | p.Arg1080Gln | Paternal | 1.39 × 10−2 | Damaging | Neutral | Pathogenic | Disease causing | rs41289902 | ||
| DEAF1 | NM_021008.3:c.69_98del30 | p.Ala24_Ala33del | Heterozygous | De novo | 1.06 × 10−3 | – | – | – | Disease causing | rs766934551 |
| GYS2 | NM_021957:c.421G>A | p.Gly141Ser | Homozygous | Maternal, | 2.12 × 10−3 | Tolerated | Deleterious | Pathogenic | Disease causing | rs149533049 |
| Paternal | ||||||||||
| B3GALTL | NM_194318:c.660+1G>A | – | Compound Heterozygous | Paternal | 1.09 × 10−3 | – | – | Pathogenic (high conf.) | Disease causing | rs80338851 |
| NM_194318:c.755delC | p.Thr252fs*13 | Maternal | – | – | – | – | Disease causing | – | ||
| RYR3 | NM_001036:c.9355G>A | p.Glu3119Lys | Compound Heterozygous | Maternal | 3.06 × 10−3 | Tolerated | Deleterious | Pathogenic | Disease causing | rs200830195 |
| NM_001036:c.14110G>A | p.Glu4704Lys | Paternal | 1.08 × 10−2 | Tolerated | Neutral | Pathogenic | Disease causing | rs182257230 | ||
| KIR2DL3 | NM_015868:c.44T>G | p.Leu15Trp | Heterozygous | De novo | 1.07 × 10−3 | Damaging | Deleterious | Benign | Polymorphism | rs149183658 |
| NM_015868:c.70+3G>A | – | Heterozygous | De novo | 1.34 × 10−5 | – | – | Pathogenic | – | rs776793416 | |
| CELSR1 | NM_014246:c.8282C>T | p.Ser2761Leu | Compound Heterozygous | Maternal | 3.24 × 10−3 | Damaging | Deleterious | Pathogenic | Disease causing | rs144039991 |
| NM_014246:c.7313G>A | p.Arg2438Gln | Paternal | 6.34 × 10−4 | Tolerated | Neutral | Benign | Polymorphism | rs199688538 |
| Gene | Best AMELIE Score | AMELIE Rank | Phenolyzer Score | Phenolyzer Rank |
|---|---|---|---|---|
| B3GLCT | 99.3 | 1 | 0.780 | 1 |
| CELSR1 | 94.2 | 2 | 0.001 | 22 |
| LRP8 | 87.8 | 3 | 0.029 | 4 |
| LCAT | 74.0 | 4 | 0.025 | 8 |
| SHANK3 | 48.3 | 5 | 0.026 | 7 |
| LAMA5 | – | – | 0.041 | 2 |
| SP100 | 0 | 11 | 0.037 | 3 |
| LAMA4 | – | – | 0.028 | 5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Totoń-Żurańska, J.; Kapusta, P.; Rybak-Krzyszkowska, M.; Lorenc, K.; Machlowska, J.; Skalniak, A.; Filipek, E.; Pawlik, D.; Wołkow, P.P. Contribution of a Novel B3GLCT Variant to Peters Plus Syndrome Discovered by a Combination of Next-Generation Sequencing and Automated Text Mining. Int. J. Mol. Sci. 2019, 20, 6006. https://doi.org/10.3390/ijms20236006
Totoń-Żurańska J, Kapusta P, Rybak-Krzyszkowska M, Lorenc K, Machlowska J, Skalniak A, Filipek E, Pawlik D, Wołkow PP. Contribution of a Novel B3GLCT Variant to Peters Plus Syndrome Discovered by a Combination of Next-Generation Sequencing and Automated Text Mining. International Journal of Molecular Sciences. 2019; 20(23):6006. https://doi.org/10.3390/ijms20236006
Chicago/Turabian StyleTotoń-Żurańska, Justyna, Przemysław Kapusta, Magda Rybak-Krzyszkowska, Katarzyna Lorenc, Julita Machlowska, Anna Skalniak, Erita Filipek, Dorota Pawlik, and Paweł P. Wołkow. 2019. "Contribution of a Novel B3GLCT Variant to Peters Plus Syndrome Discovered by a Combination of Next-Generation Sequencing and Automated Text Mining" International Journal of Molecular Sciences 20, no. 23: 6006. https://doi.org/10.3390/ijms20236006
APA StyleTotoń-Żurańska, J., Kapusta, P., Rybak-Krzyszkowska, M., Lorenc, K., Machlowska, J., Skalniak, A., Filipek, E., Pawlik, D., & Wołkow, P. P. (2019). Contribution of a Novel B3GLCT Variant to Peters Plus Syndrome Discovered by a Combination of Next-Generation Sequencing and Automated Text Mining. International Journal of Molecular Sciences, 20(23), 6006. https://doi.org/10.3390/ijms20236006