Transcriptomic and Co-Expression Network Profiling of Shoot Apical Meristem Reveal Contrasting Response to Nitrogen Rate between Indica and Japonica Rice Subspecies

Abstract
1. Introduction
2. Results
2.1. N Enrichment Promotes Tillers in a Different Pattern between NPB and YD6
2.2. Tiller Number Is Related with N and Carbohydrate Content
2.3. RNA-Seq Data Quality and Assembly
2.4. Validation of Selected DEG Confirms RNA-Seq Data Reliability
2.5. Differentially Expressed Genes (DEGs) in Response to N Rate
2.6. Gene Ontology (GO) Analysis and KEGG Clustering
2.7. Alternative Splicing Transcripts and Novel Genes
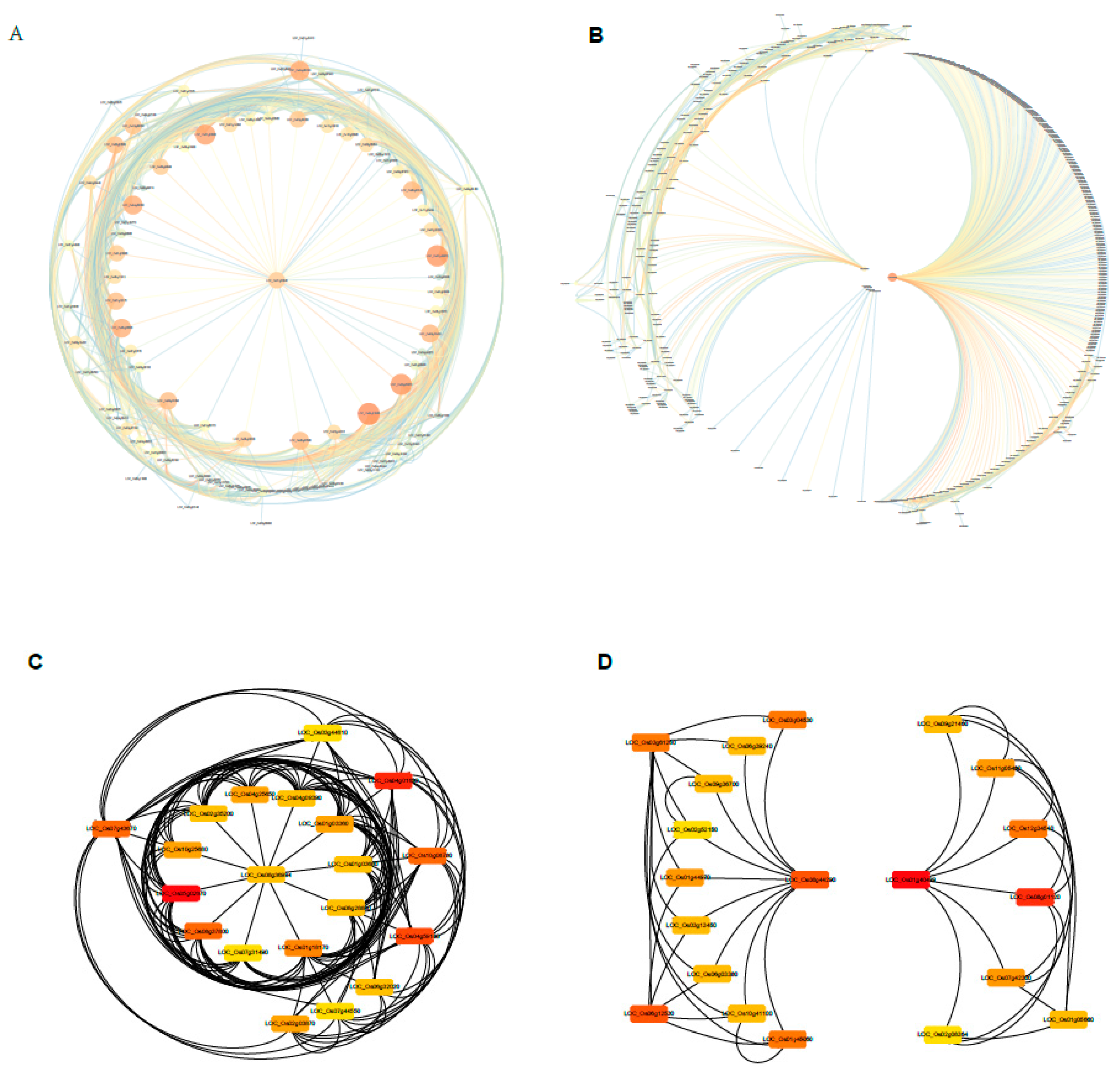
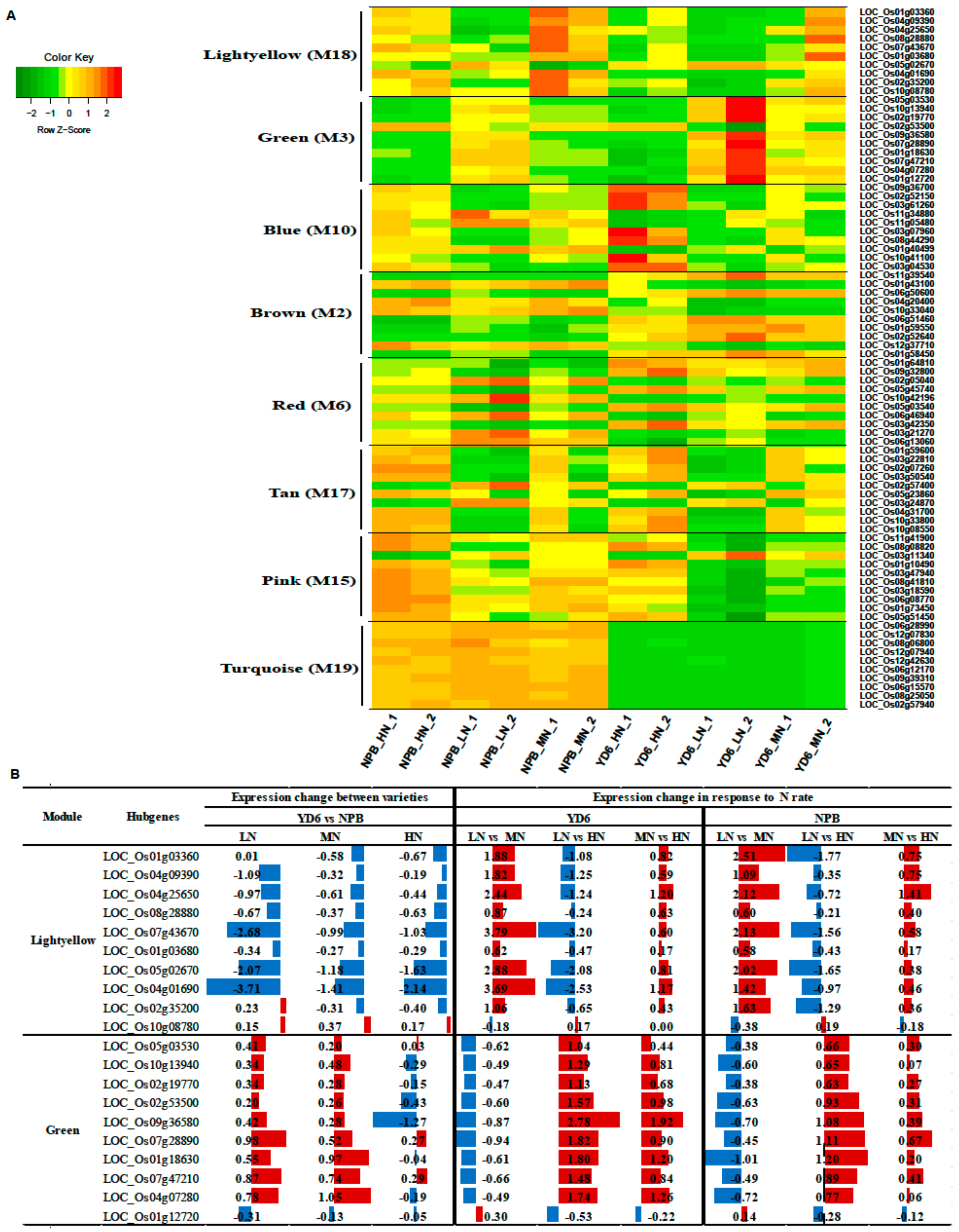
2.8. Identifition of Weighted Gene Co-Expression Network
2.9. Construction of the Gene Co-Expression Networks and Identification of Candidate Hub Genes
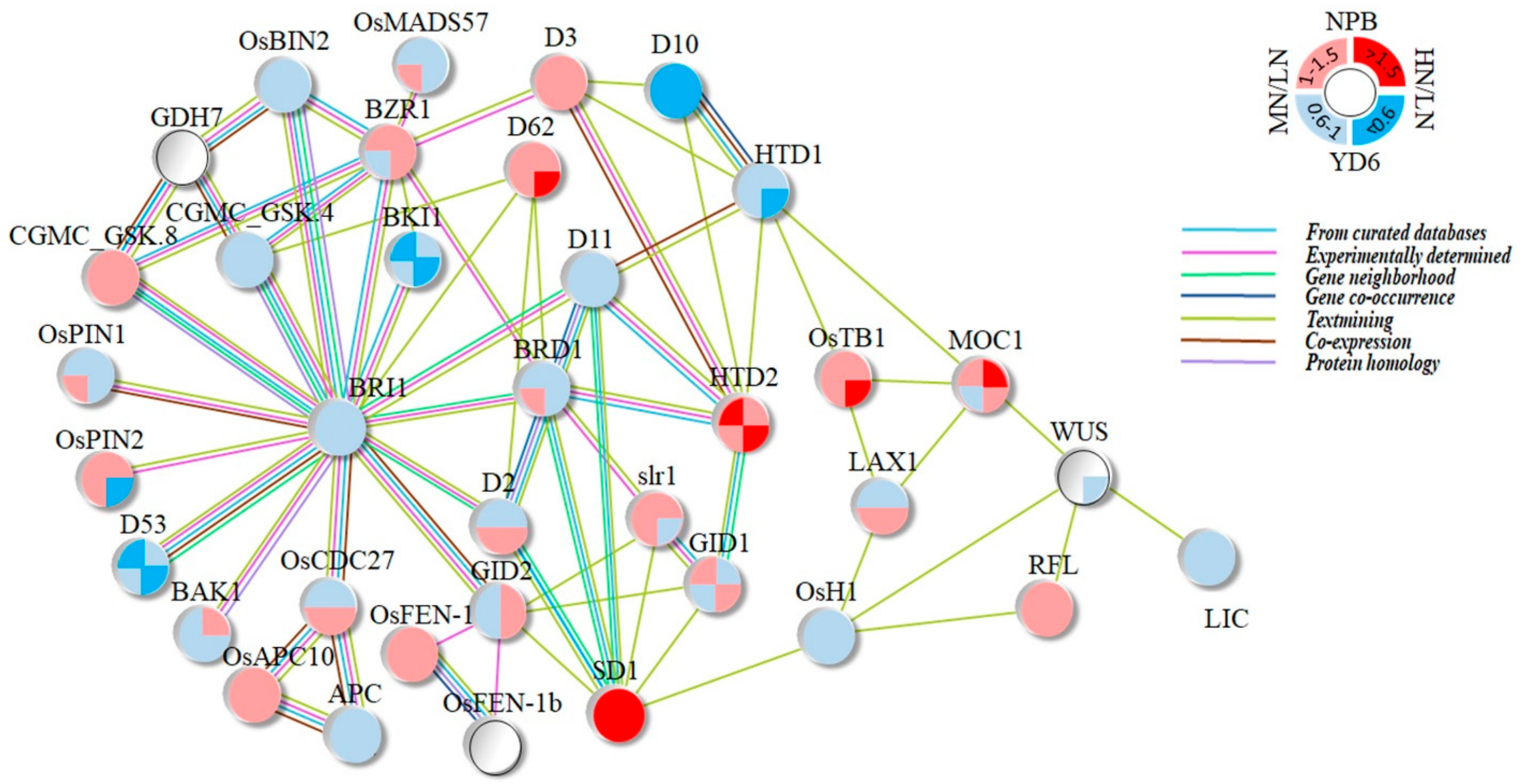
2.10. Expression Profiles of Tiller Related Genes and Their Network
2.11. Expression Profiles of No Apical Meristem Family Genes, Carbohydrate, and N Metabolism and Transport-Related Genes
3. Discussion
3.1. Reducing N Input to Low or Moderate Rate Is Still Good to Promote Enough Tillers
3.2. Most of the Tiller Genes Are Not Drastically Respond to N Rate
4. Materials and Methods
4.1. Plant Materials
4.2. Growth Conditions, N Rate Treatment and Measurement
4.3. Samples Preparation for RNA Extraction, cDNA Library Construction and Sequencing
4.4. RNA-Seq Data Processing and Gene Expression Calculation
4.5. Novel Gene, Alternative Splicing and Enrichment Analysis
4.6. Quantitative Real Time RT-PCR Validation
4.7. Co-Expression Network Analysis for Construction of Modules
4.8. Statistical Analysis of Genes Expression Data in a Specific Pathway
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AS | Alternative splicing |
| DEGs | Differentially expressed genes |
| FPKM | Fragments per kilo-base per million reads |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| N | Nitrogen |
| NAM | No apical meristem |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| RNA-Seq | RNA-sequencing |
| SAM | Shoot apical meristem |
| WSC | Water soluble carbohydrate |
References
- Ding, Y.; Huang, P.; Lin, Q. Relationship between emergence of tiller and nitrogen concentration of leaf blade or leaf sheath on specific node of rice. J. Nanjing Agric. Univ. 1995, 18, 14–18. [Google Scholar]
- Jin, Z.; Zhang, Y.; Pan, D.; Tong, L.; Li, D.; Li, M.; Wang, H.; Han, Y.; Zhang, Z.; Agriculture, S.O. Correlation analysis of plant carbon-nitrogen content and tiller-related gene expression with rice tiller formation. J. Northeast Agric. Univ. 2017, 48, 10–18. [Google Scholar]
- Mathieu, S.; Bernhard, M.; Bernard, N.; Gilles, P.; André, M. Long-term fate of nitrate fertilizer in agricultural soils. Proc. Natl. Acad. Sci. USA 2013, 110, 18185–18189. [Google Scholar]
- Yang, X.; Nian, J.; Xie, Q.; Feng, J.; Zhang, F.; Jing, H.; Zhang, J.; Dong, G.; Liang, Y.; Peng, J. Rice ferredoxin-dependent glutamate synthase regulates nitrogen-carbon metabolomes and is genetically differentiated between japonica and indica subspecies. Mol. Plant 2016, 9, 1520–1534. [Google Scholar] [CrossRef]
- Chu, G.; Chen, S.; Xu, C.; Wang, D.; Zhang, X. Agronomic and physiological performance of indica/japonica hybrid rice cultivar under low nitrogen conditions. Field Crop. Res. 2019, 243, 107625. [Google Scholar] [CrossRef]
- Pinson, S.R.M.; Wang, Y.; Tabien, R.E. Mapping and validation of quantitative trait loci associated with tiller production in rice. Crop Sci. 2015, 55, 1537. [Google Scholar] [CrossRef]
- Minakuchi, K.; Kameoka, H.; Yasuno, N.; Umehara, M.; Luo, L.; Kobayashi, K.; Hanada, A.; Ueno, K.; Asami, T.; Yamaguchi, S. FINE CULM1 (FC1) works downstream of strigolactones to inhibit the outgrowth of axillary buds in rice. Plant Cell Physiol. 2010, 51, 1127–1135. [Google Scholar] [CrossRef]
- Choi, M.S.; Woo, M.O.; Koh, E.B.; Lee, J.; Ham, T.H.; Seo, H.S.; Koh, H.J. Teosinte Branched 1 modulates tillering in rice plants. Plant Cell Rep. 2012, 31, 57–65. [Google Scholar] [CrossRef]
- Li, X.; Qian, Q.; Fu, Z.; Wang, Y.; Xiong, G.; Zeng, D.; Wang, X.; Liu, X.; Teng, S.; Hiroshi, F.; et al. Control of tillering in rice. Nature 2003, 422, 618–621. [Google Scholar] [CrossRef]
- Lin, Q.; Wang, D.; Dong, H.; Gu, S.; Cheng, Z.; Gong, J.; Qin, R.; Jiang, L.; Li, G.; Wang, J.L. Rice APC/CTE controls tillering by mediating the degradation of MONOCULM 1. Nat. Commun. 2012, 3, 752. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Yu, Y.; Duan, J.; Liao, Z.; Xiong, G.; Meng, X.; Liu, G.; Qian, Q.; Li, J. Degradation of MONOCULM 1 by APC/CTAD1 regulates rice tillering. Nat. Commun. 2012, 3, 750. [Google Scholar] [CrossRef] [PubMed]
- Keishi, K.; Masahiko, M.; Shin, U.; Yuzuki, S.; Ikuyo, F.; Hironobu, O.; Ko, S.; Junko, K. LAX and SPA: Major regulators of shoot branching in rice. Proc. Natl. Acad. Sci. USA 2003, 100, 11765–11770. [Google Scholar]
- Liang, W.; Shang, F.; Lin, Q.; Lou, C.; Zhang, J. Tillering and panicle branching genes in rice. Gene 2014, 537, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Smith, S.M.; Li, J. Genetic regulation of shoot architecture. Annu. Rev. Plant Biol. 2018, 69, 437–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Pan, Y.; Luo, M. Detection and correction of assembly errors of rice Nipponbare reference sequence. Plant Biol. 2014, 16, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Streets, A.M.; Huang, Y. How deep is enough in single-cell RNA-seq? Nat. Biotechnol. 2014, 32, 1005–1006. [Google Scholar] [CrossRef]
- Cole, T.; Williams, B.A.; Geo, P.; Ali, M.; Gordon, K.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Lior, P. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar]
- Sammeth, M.; Foissac, S.; Guigo, R. A general definition and nomenclature for alternative splicing events. PLoS Comput. Biol. 2008, 4, e1000147. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Cheng, X.; Peng, J.; Ma, J.; Tang, Y.; Chen, R.; Mysore, K.S.; Wen, J. NO APICAL MERISTEM (MtNAM) regulates floral organ identity and lateral organ separation in Medicago truncatula. New Phytol. 2012, 195, 71–84. [Google Scholar] [CrossRef]
- Krahmer, J.; Ganpudi, A.; Abbas, A.; Romanowski, A.; Halliday, K.J. Phytochrome, carbon sensing, metabolism, and plant growth plasticity. Plant Physiol. 2017, 176, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yao, Q.; Gao, X.; Jiang, C.; Harberd, N.P.; Fu, X. Shoot-to-root mobile transcription factor HY5 coordinates plant carbon and nitrogen acquisition. Curr. Biol. 2016, 26, 640–646. [Google Scholar] [CrossRef]
- Brooks, J.R.; Griffin, V.K.; Kattan, M.W. A modified method for total carbohydrate analysis of glucose syrups, maltodextrins, and other starch hydrolysis products. Cereal. Chem. 1986, 63, 465–466. [Google Scholar]
- Zhou, W.; Yang, Z.; Wang, T.; Fu, Y.; Chen, Y.; Hu, B.; Yamagishi, J.; Ren, W. Environmental compensation effect and synergistic mechanism of optimized nitrogen management increasing nitrogen use efficiency in indica hybrid rice. Front. Plant Sci. 2019, 10, 245. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zha, M.; Li, Y.; Ding, Y.; Chen, L.; Ding, C.; Wang, S. The interaction between nitrogen availability and auxin, cytokinin, and strigolactone in the control of shoot branching in rice (Oryza sativa L.). Plant Cell Rep. 2015, 34, 1647–1662. [Google Scholar] [CrossRef]
- Lin, C.M.; Koh, S.; Stacey, G.; Yu, S.; Lin, T.; Tsay, T. Cloning and functional characterization of a constitutively expressed nitrate transporter gene, OsNRT1, from rice. Plant Physiol. 2000, 122, 379–388. [Google Scholar] [CrossRef]
- Tang, Z.; Fan, X.; Li, Q.; Feng, H.; Miller, A.; Shen, Q.; Xu, G. Knockdown of a rice stelar nitrate transporter alters long-distance translocation but not root influx. Plant Physiol. 2012, 160, 2052–2063. [Google Scholar] [CrossRef]
- Fan, X.; Tang, Z.; Tan, Y.; Zhang, Y.; Luo, B.; Yang, M.; Lian, X.; Shen, Q.; Miller, A.J.; Xu, G. Overexpression of a pH-sensitive nitrate transporter in rice increases crop yields. Proc. Natl. Acad. Sci. USA 2016, 113, 7118–7123. [Google Scholar] [CrossRef]
- Ishikawa, S.; Ishimaru, Y.; Igura, M.; Kuramata, M.; Abe, T.; Senoura, T.; Hase, Y.; Arao, T.; Nishizawa, N.; Nakanishi, H. Ion-beam irradiation, gene identification, and marker-assisted breeding in the development of low-cadmium rice. Proc. Natl. Acad. Sci. USA 2018, 115, E4950–E4951. [Google Scholar] [CrossRef]
- He, F.; Karve, A.; Maslov, S.; Babst, B. Large-scale public transcriptomic data mining reveals a tight connection between the transport of nitrogen and other transport processes in Arabidopsis. Front. Plant Sci. 2016, 7, 1207. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goel, P.; Sharma, N.K.; Bhuria, M.; Sharma, V.; Chauhan, R.; Pathania, S.; Swarnkar, M.K.; Chawla, V.; Acharya, V.; Shankar, R. Transcriptome and co-expression network analyses identify key genes regulating nitrogen use efficiency in Brassica juncea L. Sci. Rep. 2018, 8, 7451. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chao, W.; Fu, Y.; Hu, G.; Si, H.; Li, Z.; Luan, W.; He, Z.; Sun, Z. Identification and characterization of HTD2: a novel gene negatively regulating tiller bud outgrowth in rice. Planta 2009, 230, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Qian, Q.; Liu, X.; Yan, M.; Feng, Q.; Dong, G.; Liu, J.; Han, B. Dwarf 88, a novel putative esterase gene affecting architecture of rice plant. Plant Mol. Biol. 2009, 71, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Arite, T.; Iwata, H.; Ohshima, K.; Maekawa, M.; Nakajima, M.M.; Sakakibara, H.; Kyozuka, J. DWARF10, an RMS1/MAX4/DAD1 ortholog, controls lateral bud outgrowth in rice. Plant J. 2010, 51, 1019–1029. [Google Scholar] [CrossRef]
- Zhang, S.; Li, G.; Fang, J.; Chen, W.; Jiang, H.; Zou, J.; Liu, X.; Zhao, X.; Li, X.; Chu, C. The interactions among DWARF10, auxin and cytokinin underlie lateral bud outgrowth in rice. J. Integr. Plant Biol. 2010, 52, 626–638. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, Y.; Wang, L.; Yang, K.; Xiao, N.; Liu, Y.; Fu, Y.; Sun, Z.; Fang, R.; Chen, X. A novel rice gene, NRR responds to macronutrient deficiency and regulates root growth. Mol. Plant 2012, 5, 63–72. [Google Scholar] [CrossRef]
- Morita, R.; Inoue, K.; Ikeda, K.I.; Hatanaka, T.; Misoo, S.; Fukayama, H. Starch content in leaf sheath controlled by CO2 responsive CCT protein is a potential determinant of photosynthetic capacity in rice. Plant Cell Physiol. 2016, 57, 2334–2341. [Google Scholar] [CrossRef]
- Nishida, S.; Kakei, Y.; Shimada, Y.; Fujiwara, T. Genome-wide analysis of specific alterations in transcript structure and accumulation caused by nutrient deficiencies in Arabidopsis thaliana. Plant J. 2017, 91, 741–753. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Gong, W. Genome-wide identification and comparative analysis of alternative splicing across four legume species. Planta. 2019, 249, 1133–1142. [Google Scholar] [CrossRef]
- Li, W.; Lin, W.; Ray, P.; Lan, P.; Schmidt, W. Genome-wide detection of condition-sensitive alternative splicing in Arabidopsis roots. Plant Physiol. 2013, 162, 1750–1763. [Google Scholar] [CrossRef] [PubMed]
- Calixto, C.; Guo, W.; James, A.; Tzioutziou, N.; Entizne, J.; Panter, P.; Knight, H.; Nimmo, H.; Zhang, R.; Brown, J. Rapid and dynamic alternative splicing impacts the Arabidopsis cold response transcriptome. Plant Cell. 2018, 30, 1424–1444. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Li, W.; Zhang, F.; Guo, W. RNA-seq analysis reveals alternative splicing under salt stress in cotton, Gossypium davidsonii. BMC Genom. 2018, 19, 73. [Google Scholar] [CrossRef] [PubMed]
- Clegg, K.M. The application of the anthrone reagent to the estimation of starch in cereals. J. Sci. Food Agric. 1956, 7, 40–44. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter LSalzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B.J. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Sylvain, F.; Michael, S. ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35, W297–W299. [Google Scholar]
- Michael, S. Complete alternative splicing events are bubbles in splicing graphs. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2009, 16, 1117–1140. [Google Scholar]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, N.; Mo, L.; Lv, M.; Gao, Y.; Wang, J.; Liu, C.; Yin, S.; Zhou, J.; Xiao, N.; et al. Global transcriptome and co-expression network analysis reveal contrasting response of japonica and indica rice cultivar to γ radiation. Int. J. Mol. Sci. 2019, 20, 4358. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, N.; Cadeiras, M.; Bondar, G.; Cheng, R.K.; Shahzad, K.; Onat, D.; Latif, F.; Korin, Y.; Reed, E.; Fakhro, R. Weighted gene coexpression network analysis (WGCNA) modeling of multiorgan dysfunction syndrome after mechanical circulatory support therapy. J. Heart Lung Transpl. 2013, 32, S223. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Description | KME Value |
|---|---|---|
| Light yellow module (M18) with positive correlation associated with the tillers | ||
| LOC_Os04g09390 | HEV3 - Hevein family protein precursor, expressed | 0.97 |
| LOC_Os01g03680 | BBTI8 - Bowman-Birk type bran trypsin inhibitor precursor, expressed | 0.95 |
| LOC_Os08g28880 | patatin, putative, expressed | 0.94 |
| LOC_Os10g08780 | expressed protein | 0.94 |
| LOC_Os01g03360 | BBTI5 - Bowman-Birk type bran trypsin inhibitor precursor, expressed | 0.94 |
| LOC_Os02g35200 | VP15, putative, expressed | 0.88 |
| LOC_Os07g43670 | ribonuclease T2 family domain containing protein, expressed | 0.85 |
| LOC_Os04g01690 | pyridoxal-dependent decarboxylase protein, putative, expressed | 0.85 |
| LOC_Os04g25650 | cysteine-rich receptor-like protein kinase, putative, expressed | 0.85 |
| LOC_Os05g02670 | kinesin motor domain containing protein, putative, expressed | −0.80 |
| Green module (M3) with negative correlation associated with the tillers, N rate in leaf and stem | ||
| LOC_Os10g13940 | MATE efflux protein, putative, expressed | 0.98 |
| LOC_Os07g28890 | ethylene-responsive protein related, putative, expressed | 0.98 |
| LOC_Os01g12720 | protein kinase domain containing protein, expressed | 0.98 |
| LOC_Os07g47210 | GDSL-like lipase/acylhydrolase, putative, expressed | 0.97 |
| LOC_Os01g18630 | aspartic proteinase oryzasin-1 precursor, putative, expressed | 0.95 |
| LOC_Os02g19770 | eukaryotic translation initiation factor 1A, putative, expressed | 0.94 |
| LOC_Os09g36580 | thaumatin, putative, expressed | 0.93 |
| LOC_Os05g03530 | tetraspanin family protein, putative, expressed | 0.90 |
| LOC_Os04g07280 | AGAP002737-PA, putative, expressed | 0.90 |
| LOC_Os02g53500 | RFC5 - Putative clamp loader of PCNA, replication factor C subunit 5, expressed | −0.92 |
| Blue module (M10) with positive correlation associated with the N rate in leaf and stem | ||
| LOC_Os08g44290 | RNA recognition motif containing protein, putative, expressed | 0.98 |
| LOC_Os03g61260 | ribosomal L18p/L5e family protein, putative, expressed | 0.98 |
| LOC_Os03g04530 | cytochrome P450, putative, expressed | 0.95 |
| LOC_Os09g36700 | ribonuclease T2 family domain containing protein, expressed | 0.94 |
| LOC_Os02g52150 | heat shock 22 kDa protein, mitochondrial precursor, putative, expressed | 0.91 |
| LOC_Os03g07960 | expressed protein | 0.88 |
| LOC_Os10g41100 | CCT motif family protein, expressed | 0.84 |
| LOC_Os11g34880 | NB-ARC domain containing protein, expressed | −0.70 |
| LOC_Os11g05480 | transcription factor, putative, expressed | −0.76 |
| LOC_Os01g40499 | S-locus lectin protein kinase family protein, putative, expressed | −0.82 |


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Zhou, J.; Huang, N.; Mo, L.; Lv, M.; Gao, Y.; Chen, C.; Yin, S.; Ju, J.; Dong, G.; et al. Transcriptomic and Co-Expression Network Profiling of Shoot Apical Meristem Reveal Contrasting Response to Nitrogen Rate between Indica and Japonica Rice Subspecies. Int. J. Mol. Sci. 2019, 20, 5922. https://doi.org/10.3390/ijms20235922
Zhang X, Zhou J, Huang N, Mo L, Lv M, Gao Y, Chen C, Yin S, Ju J, Dong G, et al. Transcriptomic and Co-Expression Network Profiling of Shoot Apical Meristem Reveal Contrasting Response to Nitrogen Rate between Indica and Japonica Rice Subspecies. International Journal of Molecular Sciences. 2019; 20(23):5922. https://doi.org/10.3390/ijms20235922
Chicago/Turabian StyleZhang, Xiaoxiang, Juan Zhou, Niansheng Huang, Lanjing Mo, Minjia Lv, Yingbo Gao, Chen Chen, Shuangyi Yin, Jing Ju, Guichun Dong, and et al. 2019. "Transcriptomic and Co-Expression Network Profiling of Shoot Apical Meristem Reveal Contrasting Response to Nitrogen Rate between Indica and Japonica Rice Subspecies" International Journal of Molecular Sciences 20, no. 23: 5922. https://doi.org/10.3390/ijms20235922
APA StyleZhang, X., Zhou, J., Huang, N., Mo, L., Lv, M., Gao, Y., Chen, C., Yin, S., Ju, J., Dong, G., Zhou, Y., Yang, Z., Li, A., Wang, Y., Huang, J., & Yao, Y. (2019). Transcriptomic and Co-Expression Network Profiling of Shoot Apical Meristem Reveal Contrasting Response to Nitrogen Rate between Indica and Japonica Rice Subspecies. International Journal of Molecular Sciences, 20(23), 5922. https://doi.org/10.3390/ijms20235922