McArdle Disease: New Insights into Its Underlying Molecular Mechanisms

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
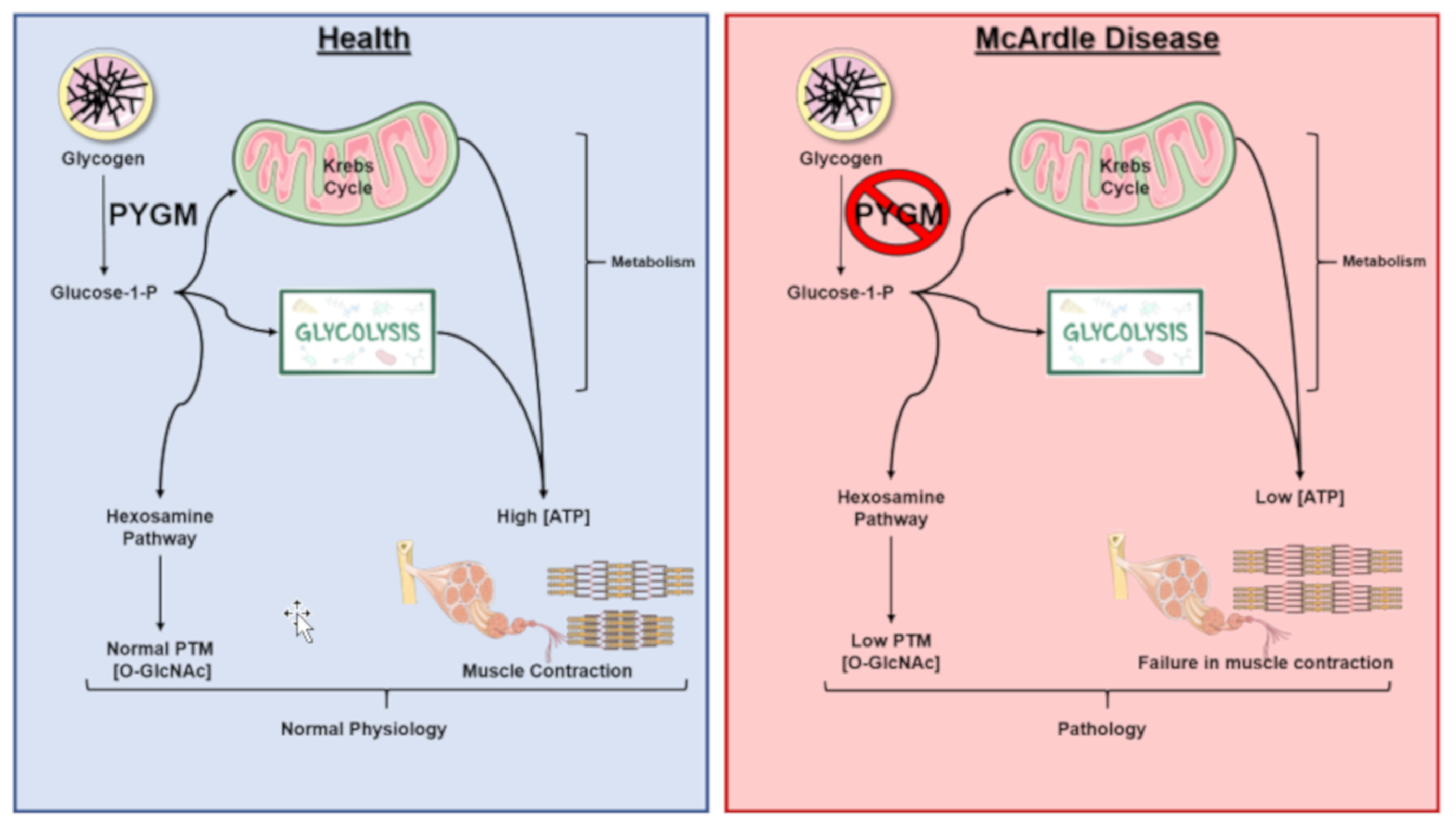
1. General Characteristics of McArdle Disease
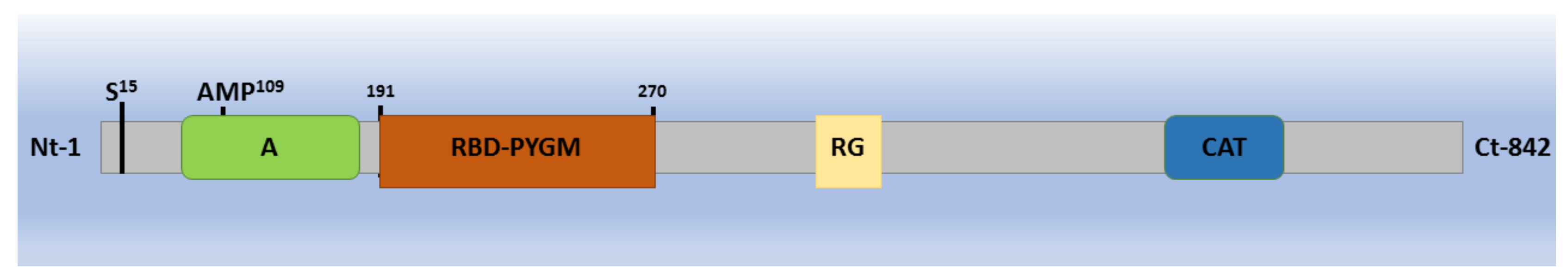
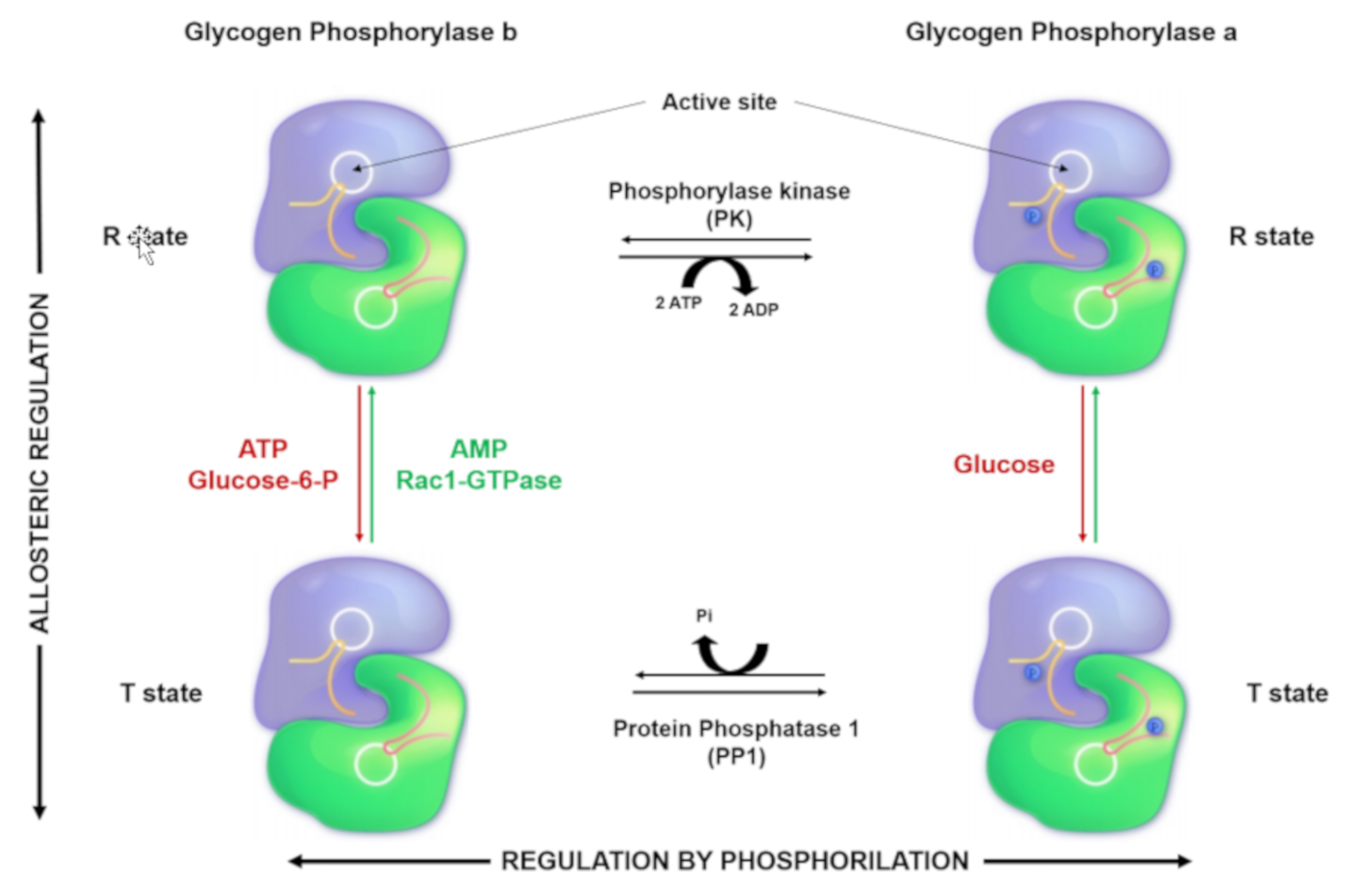
2. Glycogen Phosphorylase: Structure, Function, and Regulation
3. Genetics of McArdle Disease: Mutations and Their Implications
4. PYGM Expression in Other Tissues
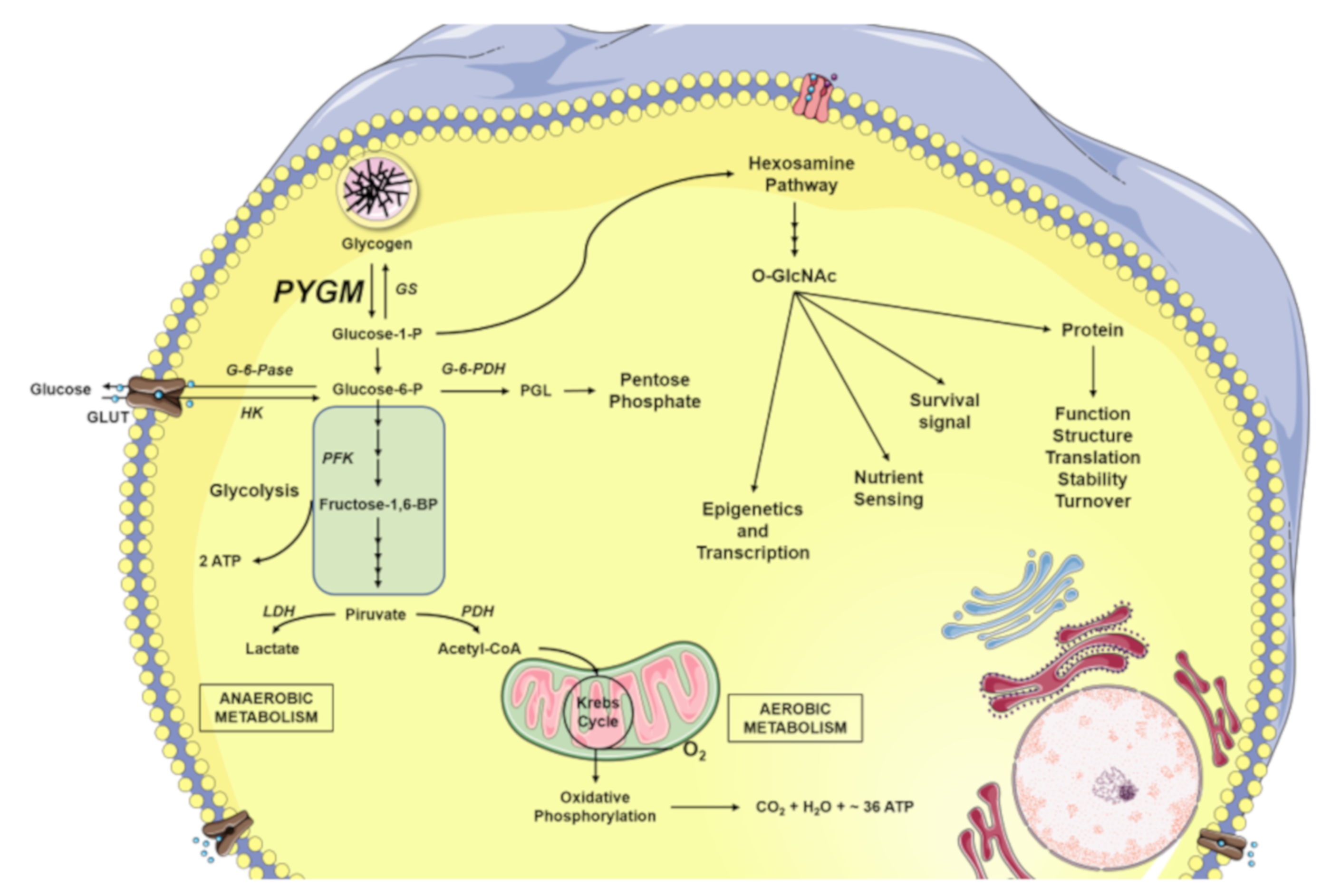
5. Glycogen Degradation: A Source for O-GlcNAcylation
6. Animal Models for the Study of McArdle Disease
7. Clinical Trials in McArdle Disease
8. Future Perspectives
Funding
Conflicts of Interest
References
- Lebo, R.V.; Anderson, L.A.; DiMauro, S.; Lynch, E.; Hwang, P.; Fletterick, R. Rare McArdle disease locus polymorphic site on 11q13 contains CpG sequence. Hum. Genet. 1990, 86, 17–24. [Google Scholar] [CrossRef] [PubMed]
- McArdle, B. Myopathy due to a defect in muscle glycogen breakdown. Clin. Sci. 1951, 10, 13–35. [Google Scholar] [PubMed]
- Mellick, R.S.; Mahler, R.F.; Hughes, B.P. McArdle’s syndrome: Phosphorylase-deficient myopathy. Lancet 1962, 1, 1045–1048. [Google Scholar] [CrossRef]
- De Castro, M.; Johnston, J.; Biesecker, L. Determining the prevalence of McArdle disease from gene frequency by analysis of next-generation sequencing data. Genet. Med. 2015, 17, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Santalla, A.; Nogales-Gadea, G.; Encinar, A.B.; Vieitez, I.; González-Quintana, A.; Serrano-Lorenzo, P. Genotypic and phenotypic features of all Spanish patients with McArdle disease: A 2016 update. BMC Genom. 2017, 18 (Suppl. S8), 819. [Google Scholar] [CrossRef] [PubMed]
- Quinlivan, R.; Buckley, J.; James, M.; Twist, A.; Ball, S.; Duno, M. McArdle disease: A clinical review. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Braakhekke, J.P.; De Bruin, M.I.; Stegeman, D.F.; Wevers, R.A.; Binkhorst, R.A.; Joosten, E.M.G. The second wind phenomenon in McArdle’s disease. Brain 1986, 109, 1087–1101. [Google Scholar] [CrossRef]
- Haller, R.G.; Vissing, J. Spontaneous “second wind” and glucose-induced second “second wind” in McArdle disease: Oxidative mechanisms. Arch. Neurol. 2002, 59, 1395–1402. [Google Scholar] [CrossRef]
- De Luna, N.; Brull, A.; Lucia, A.; Santalla, A.; Garatachea, N.; Martí, R. PYGM expression analysis in white blood cells: A complementary tool for diagnosing McArdle disease? Neuromuscul. Disord. 2014, 24, 1079–1086. [Google Scholar] [CrossRef]
- Santalla, A.; Munguía-Izquierdo, D.; Brea-Alejo, L.; Pagola-Aldazábal, I.; Díez-Bermejo, J.; Fleck, S.J.; Lucia, A. Feasibility of resistance training in adult McArdle patients: Clinical outcomes and muscle strength and mass benefits. Front. Aging Neurosci. 2014, 6, 334. [Google Scholar] [CrossRef]
- Santalla, A.; Nogales-Gadea, G.; Ørtenblad, N.; Brull, A.; de Luna, N.; Pinós, T.; Lucia, A. McArdle disease: A unique study model in sports medicine. Sports Med. 2014, 44, 1531–1544. [Google Scholar] [CrossRef]
- Fukui, T.; Shimomura, S.; Nakano, K. Potato and rabbit muscle phosphorylases: Comparative studies on the structure, function and regulation of regulatory and nonregulatory enzymes. Mol. Cell. Biochem. 1982, 42, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.N. Glycogen phosphorylase: Control by phosphorylation and allosteric effectors. FASEB J. 1992, 6, 2274–2282. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Bohme, H.J.; Hofmann, E. Developmental changes of glycogen phosphorylase b isozymes in rat tissues. Biomed. Biochim. Acta 1983, 42, 1229–1235. [Google Scholar]
- Sato, K.; Satoh, K.; Sato, T.; Imai, F.; Morris, H.P. Isozyme patterns of glycogen phosphorylase in rat tissues and transplantable hepatomas. Cancer Res. 1976, 36, 487–495. [Google Scholar] [PubMed]
- Barford, D.; Johnson, L.N. The allosteric transition of glycogen phosphorylase. Nature 1989, 340, 609–616. [Google Scholar] [CrossRef]
- Barford, D.; Johnson, L.N. The molecular mechanism for the tetrameric association of glycogen phosphorylase promoted by protein phosphorylation. Protein Sci. 1992, 1, 472–493. [Google Scholar] [CrossRef]
- Llavero, F.; Arrazola Sastre, A.; Luque Montoro, M.; Martín, M.A.; Arenas, J.; Lucia, A.; Zugaza, J.L. Small GTPases of the Ras superfamily and glycogen phosphorylase regulation in T cells. Small GTPases 2019. [Google Scholar] [CrossRef]
- Rozengurt, E. Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell Physiol. 2007, 213, 589–602. [Google Scholar] [CrossRef]
- Andreeva, I.E.; Rice, N.A.; Carlson, G.M. The regulatory alpha subunit of phosphorylase kinase may directly participate in the binding of glycogen phosphorylase. Biochemistry 2002, 67, 1197–1202. [Google Scholar]
- Buchbinder, J.L.; Luong, C.B.; Browner, M.F.; Fletterick, R.J. Partial activation of muscle phosphorylase by replacement of serine 14 with acidic residues at the site of regulatory phosphorylation. Biochemistry 1997, 36, 8039–8044. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.H. Cellular regulation by protein phosphorylation. Biochem. Biophys. Res. Commun. 2013, 430, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Waraich, R.S.; Weigert, C.; Kalbacher, H.; Hennige, A.M.; Lutz, S.Z.; Häring, H.U. Phosphorylation of Ser357 of rat insulin receptor substrate-1 mediates adverse effects of protein kinase C-delta on insulin action in skeletal muscle cells. J. Biol. Chem. 2008, 283, 11226–11233. [Google Scholar] [CrossRef] [PubMed]
- Defer, N.; Best-Belpomme, M.; Hanoune, J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am. J. Physiol. Renal Physiol. 2000, 279, F400–F416. [Google Scholar] [CrossRef]
- Beazely, M.A.; Alan, J.K.; Watts, V.J. Protein kinase C and epidermal growth factor stimulation of Raf1 potentiates adenylyl cyclase type 6 activation in intact cells. Mol. Pharm. 2005, 67, 250–259. [Google Scholar] [CrossRef]
- Beazely, M.A.; Watts, V.J. Regulatory properties of adenylate cyclases type 5 and 6: A progress report. Eur. J. Pharm. 2006, 535, 1–12. [Google Scholar] [CrossRef]
- Llavero, F.; Montoro, M.L.; Sastre, A.A.; Fernández-Moreno, D.; Lacerda, H.M. Epidermal growth factor receptor controls glycogen phosphorylase in T cells through small GTPases of the RAS family. J. Biol. Chem. 2019, 294, 4345–4358. [Google Scholar] [CrossRef]
- Browner, M.F.; Fauman, E.B.; Fletterick, R.J. Tracking conformational states in allosteric transitions of phosphorylase. Biochemistry 1992, 31, 11297–11304. [Google Scholar] [CrossRef]
- Lukacs, C.M.; Oikonomakos, N.G.; Crowther, R.L.; Hong, L.N.; Kammlott, R.U.; Levin, W. The crystal structure of human muscle glycogen phosphorylase a with bound glucose and AMP: An intermediate conformation with T-state and R-state features. Proteins 2006, 63, 1123–1126. [Google Scholar] [CrossRef]
- Newgard, C.B.; Hwang, P.K.; Fletterick, R.J. The family of glycogen phosphorylases: Structure and function. Crit. Rev. Biochem. Mol. Biol. 1989, 24, 69–99. [Google Scholar] [CrossRef]
- Fletterick, R.J.; Burke, J.A.; Hwang, P.K.; Nakano, K.; Newgard, C.B. Structural relationships in glycogen phosphorylases. Ann. N. Y. Acad. Sci. 1986, 478, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Madsen, N.B.; Avramovic-Zikic, O.; Honikel, K.O. Structure-function relationships in glycogen phosphorylase with respect to its control characteristics. Ann. N. Y. Acad. Sci. 1973, 210, 222–237. [Google Scholar] [CrossRef]
- Arrizabalaga, O.; Lacerda, H.M.; Zubiaga, A.M.; Zugaza, J.L. Rac1 protein regulates glycogen phosphorylase activation and controls interleukin (IL)-2-dependent T cell proliferation. J. Biol. Chem. 2012, 287, 11878–11890. [Google Scholar] [CrossRef]
- Llavero, F.; Artaso, A.; Lacerda, H.M.; Parada, L.A.; Zugaza, J.L. Lck/PLCgamma control migration and proliferation of interleukin (IL)-2-stimulated T cells via the Rac1 GTPase/glycogen phosphorylase pathway. Cell. Signal. 2016, 28, 1713–1724. [Google Scholar] [CrossRef]
- Llavero, F.; Urzelai, B.; Osinalde, N.; Gálvez, P.; Lacerda, H.M.; Parada, L.A.; Zugaza, J.L. Guanine nucleotide exchange factor alphaPIX leads to activation of the Rac 1 GTPase/glycogen phosphorylase pathway in interleukin (IL)-2-stimulated T cells. J. Biol. Chem. 2015, 290, 9171–9182. [Google Scholar] [CrossRef]
- Chasiotis, D. The regulation of glycogen phosphorylase and glycogen breakdown in human skeletal muscle. Acta Physiol. Scand Suppl. 1983, 518, 1–68. [Google Scholar]
- Hue, L.; Bontemps, F.; Hers, H. The effects of glucose and of potassium ions on the interconversion of the two forms of glycogen phosphorylase and of glycogen synthetase in isolated rat liver preparations. Biochem. J. 1975, 152, 105–114. [Google Scholar] [CrossRef]
- García-Consuegra, I.; Rubio, J.C.; Nogales-Gadea, G.; Bautista, J.; Jimenez, S.; Cabello, A.; Lucía, A.; Andreu, A.L.; Arenas, J.; Martin, M.A. Novel mutations in patients with McArdle disease by analysis of skeletal muscle mRNA. J. Med. Genet. 2009, 46, 198–202. [Google Scholar] [CrossRef]
- Nogales-Gadea, G.; Brull, A.; Santalla, A.; Andreu, A.L.; Arenas, J.; Martín, M.; Lucia, A.; de Luna, N.; Pinós, T.A. McArdle Disease: Update of Reported Mutations and Polymorphisms in the PYGM Gene. Hum. Mutat. 2015, 36, 669–678. [Google Scholar] [CrossRef]
- Tsujino, S.; Shanske, S.; DiMauro, S. Molecular genetic heterogeneity of myophosphorylase deficiency (McArdle’s disease). N. Engl. J. Med. 1993, 329, 241–245. [Google Scholar] [CrossRef]
- DiMauro, S.; Hartlage, P.L. Fatal infantile form of muscle phosphorylase deficiency. Neurology 1978, 28, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.A.; Rubio, J.C.; Buchbinder, J.; Fernández-Hojas, R.; Del Hoyo, P.; Teijeira, S.; Gámez, J.; Navarro, C.; Fernández, J.M.; Cabello, A.; et al. Molecular heterogeneity of myophosphorylase deficiency (McArdle’s disease): A genotype-phenotype correlation study. Ann. Neurol. 2001, 50, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Weber, J.L.; Vladutiu, G.D.; Tarnopolsky, M.A. Six novel mutations in the myophosphorylase gene in patients with McArdle disease and a family with pseudo-dominant inheritance pattern. Mol. Genet. Metab. 2011, 104, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.M.; Walker, K.R.; Creed, K.E.; Dunton, E.; Davies, L.; Quinlivan, R.; Karpati, G. Phosphorylase re-expression, increase in the force of contraction and decreased fatigue following notexin-induced muscle damage and regeneration in the ovine model of McArdle disease. Neuromuscul. Disord. 2014, 24, 167–177. [Google Scholar] [CrossRef]
- Martinuzzi, A.; Vergani, L.; Carrozzo, R.; Fanin, M.; Bartoloni, L.; Angelini, C.; Askanas, V.; Engel, W.K. Expression of muscle-type phosphorylase in innervated and aneural cultured muscle of patients with myophosphorylase deficiency. J. Clin. Investig. 1993, 92, 1774–1780. [Google Scholar] [CrossRef]
- Pfeiffer-Guglielmi, B.; Bröer, S.; Bröer, A.; Hamprecht, B. Isozyme pattern of glycogen phosphorylase in the rat nervous system and rat astroglia-rich primary cultures: Electrophoretic and polymerase chain reaction studies. Neurochem. Res. 2000, 25, 1485–1491. [Google Scholar] [CrossRef]
- Pinacho, R.; Vila, E.; Prades, R.; Tarragó, T.; Castro, E.; Ferrer, I.; Ramos, B. The glial phosphorylase of glycogen isoform is reduced in the dorsolateral prefrontal cortex in chronic schizophrenia. Schizophr. Res. 2016, 177, 37–43. [Google Scholar] [CrossRef]
- Schmid, H.; Dolderer, B.; Thiess, U.; Verleysdonk, S.; Hamprecht, B. Renal expression of the brain and muscle isoforms of glycogen phosphorylase in different cell types. Neurochem. Res. 2008, 33, 2575–2582. [Google Scholar] [CrossRef]
- Jakobsen, E.; Bak, L.K.; Walls, A.B.; Reuschlein, A.K.; Schousboe, A.; Waagepetersen, H.S. Glycogen Shunt Activity and Glycolytic Supercompensation in Astrocytes May Be Distinctly Mediated via the Muscle Form of Glycogen Phosphorylase. Neurochem. Res. 2017, 42, 2490–2494. [Google Scholar] [CrossRef]
- Pfeiffer-Guglielmi, B.; Fleckenstein, B.; Jung, G.; Hamprecht, B. Immunocytochemical localization of glycogen phosphorylase isozymes in rat nervous tissues by using isozyme-specific antibodies. J. Neurochem. 2003, 85, 73–81. [Google Scholar] [CrossRef]
- Alsberge, J.B.; Chen, J.J.; Zaidi, A.A.; Fu, A.D. Retinal Dystrophy in a Patient with Mcardle Disease. Retin. Cases Brief Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Leonardy, N.J.; Harbin, R.L.; Sternberg, P., Jr. Pattern dystrophy of the retinal pigment epithelium in a patient with McArdle’s disease. Am. J. Ophthalmol. 1988, 106, 741–742. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, I.; Santalla, A.; Diez-Bermejo, J.; Munguía-Izquierdo, D.; Alegre, L.M.; Nogales-Gadea, G. A New Condition in McArdle Disease: Poor Bone Health-Benefits of an Active Lifestyle. Med. Sci. Sports Exerc. 2018, 50, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Carlson, G.M.; Dienel, G.A.; Colbran, R.J. Introduction to the Thematic Minireview Series: Brain glycogen metabolism. J. Biol. Chem. 2018, 293, 7087–7088. [Google Scholar] [CrossRef]
- Kanungo, S.; Wells, K.; Tribett, T.; El-Gharbawy, A. Glycogen metabolism and glycogen storage disorders. Ann. Transl. Med. 2018, 6, 474. [Google Scholar] [CrossRef]
- Prats, C.; Graham, T.E.; Shearer, J. The dynamic life of the glycogen granule. J. Biol. Chem. 2018, 293, 7089–7098. [Google Scholar] [CrossRef]
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessières, B.; Gao, V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018, 66, 1244–1262. [Google Scholar] [CrossRef]
- Wender, R.; Brown, A.M.; Fern, R.; Swanson, R.A.; Farrell, K.; Ransom, B.R. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J. Neurosci. 2000, 20, 6804–6810. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Morrison, J.H.; Shoemaker, W.J.; Sapin, V.; Bloom, F.E. Vasoactive intestinal polypeptide induces glycogenolysis in mouse cortical slices: A possible regulatory mechanism for the local control of energy metabolism. Proc. Natl. Acad. Sci. USA 1981, 78, 6535–6539. [Google Scholar] [CrossRef]
- Sorg, O.; Magistretti, P.J. Characterization of the glycogenolysis elicited by vasoactive intestinal peptide, noradrenaline and adenosine in primary cultures of mouse cerebral cortical astrocytes. Brain Res. 1991, 563, 227–233. [Google Scholar] [CrossRef]
- Sorg, O.; Magistretti, P.J. Vasoactive intestinal peptide and noradrenaline exert long-term control on glycogen levels in astrocytes: Blockade by protein synthesis inhibition. J. Neurosci. 1992, 12, 4923–4931. [Google Scholar] [CrossRef] [PubMed]
- Bouzier-Sore, A.K.; Pellerin, L. Unraveling the complex metabolic nature of astrocytes. Front. Cell Neurosci. 2013, 7, 179. [Google Scholar] [CrossRef]
- Pérez-Escuredo, J.; Van Hee, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 2016, 1863, 2481–2497. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, L.; Pellerin, L. Monocarboxylate transporters: New players in body weight regulation. Obes. Rev. 2015, 16 (Suppl. S1), 55–66. [Google Scholar] [CrossRef]
- Love, D.C.; Hanover, J.A. The hexosamine signaling pathway: Deciphering the “O-GlcNAc code”. Sci. STKE 2005, 312, re13. [Google Scholar] [CrossRef]
- Kang, J.G.; Park, S.Y.; Ji, S.; Jang, I.; Park, S.; Kim, H.S. O-GlcNAc protein modification in cancer cells increases in response to glucose deprivation through glycogen degradation. J. Biol. Chem. 2009, 284, 34777–34784. [Google Scholar] [CrossRef]
- Hurtado-Guerrero, R.; Dorfmueller, H.C.; van Aalten, D.M. Molecular mechanisms of O-GlcNAcylation. Curr. Opin. Struct. Biol. 2008, 18, 551–557. [Google Scholar] [CrossRef]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef]
- Zachara, N.E.; Hart, G.W. The emerging significance of O-GlcNAc in cellular regulation. Chem. Rev. 2002, 102, 431–438. [Google Scholar] [CrossRef]
- Zachara, N.E.; Hart, G.W. Cell signaling, the essential role of O-GlcNAc! Biochim. Biophys. Acta 2006, 1761, 599–617. [Google Scholar] [CrossRef]
- Chatham, J.C.; Marchase, R.B. Protein O-GlcNAcylation: A critical regulator of the cellular response to stress. Curr. Signal. Transduct. Ther. 2010, 5, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Udeshi, N.D.; Slawson, C.; Compton, P.D.; Sakabe, K.; Cheung, W.D. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci. Signal. 2010, 3, ra2. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.T.; Luo, H.; Guan, W.J.; Zhang, H.; Chen, C.; Wang, Z.; Li, J.D. O-GlcNAcylation of BMAL1 regulates circadian rhythms in NIH3T3 fibroblasts. Biochem. Biophys. Res. Commun. 2013, 431, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Vocadlo, D.J.; Vosseller, K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappaB activity in pancreatic cancer cells. J. Biol. Chem. 2013, 288, 15121–15130. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, Q.; Hart, G.W. The intersections between O-GlcNAcylation and phosphorylation: Implications for multiple signaling pathways. J. Cell Sci. 2010, 123, 13–22. [Google Scholar] [CrossRef]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef]
- Mi, W.; Gu, Y.; Han, C.; Liu, H.; Fan, Q.; Zhang, X.; Cong, Q.; Yu, W. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim. Biophys. Acta 2011, 1812, 514–519. [Google Scholar] [CrossRef]
- Slawson, C.; Hart, G.W. O-GlcNAc signalling: Implications for cancer cell biology. Nat. Rev. Cancer 2011, 11, 678–684. [Google Scholar] [CrossRef]
- Butkinaree, C.; Park, K.; Hart, G.W. O-linked beta-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim. Biophys. Acta 2010, 1800, 96–106. [Google Scholar] [CrossRef]
- Angelos, S.; Valberg, S.J.; Smith, B.P.; McQuarrie, P.S.; Shanske, S.; Tsujino, S.; Mauro, S.; Cardinet, G.H., III. Myophosphorylase deficiency associated with rhabdomyolysis and exercise intolerance in 6 related Charolais cattle. Muscle Nerve 1995, 18, 736–740. [Google Scholar] [CrossRef]
- Tsujino, S.; Shanske, S.; Valberg, S.J.; Cardinet, G.H., III; Smith, B.P.; DiMauro, S. Cloning of bovine muscle glycogen phosphorylase cDNA and identification of a mutation in cattle with myophosphorylase deficiency, an animal model for McArdle’s disease. Neuromuscul. Disord. 1996, 6, 19–26. [Google Scholar] [CrossRef]
- Walker, K.R. Characterization of the Ovine Model of McArdle Disease: Development of Therapeutic Strategies. Ph.D. Thesis, Murdoch University, Perth Campus, Murdoch, Western Australia, 2006. [Google Scholar]
- Tan, P.; Allen, J.G.; Wilton, S.D.; Akkari, P.A.; Huxtable, C.R.; Laing, N.G. A splice-site mutation causing ovine McArdle’s disease. Neuromuscul. Disord. 1997, 7, 336–342. [Google Scholar] [CrossRef]
- Howell, J.M.; Dunton, E.; Creed, K.E.; Quinlivan, R.; Sewry, C. Investigating sodium valproate as a treatment for McArdle disease in sheep. Neuromuscul. Disord. 2015, 25, 111–119. [Google Scholar] [CrossRef]
- Nogales-Gadea, G.; Pinos, T.; Lucia, A.; Arenas, J.; Camara, Y.; Brull, A.; de Luna, N.; Martín, M.A.; Garcia-Arumí, E.; Martí, R.; et al. Knock-in mice for the R50X mutation in the PYGM gene present with McArdle disease. Brain 2012, 135, 2048–2057. [Google Scholar] [CrossRef]
- Brull, A.; de Luna, N.; Blanco-Grau, A.; Lucia, A.; Martin, M.A.; Arenas, J.; Martí, R.; Andreu, A.L.; Pinós, T. Phenotype consequences of myophosphorylase dysfunction: Insights from the McArdle mouse model. J. Physiol. 2015, 593, 2693–2706. [Google Scholar] [CrossRef]
- Fiuza-Luces, C.; Nogales-Gadea, G.; García-Consuegra, I.; Pareja-Galeano, H.; Rufián-Vázquez, L.; Pérez, L.M.; Andreu, A.L.; Arenas, J.; Martín, M.A.; Pinós, T.; et al. Muscle Signaling in Exercise Intolerance: Insights from the McArdle Mouse Model. Med. Sci. Sports Exerc. 2016, 48, 1448–1458. [Google Scholar] [CrossRef]
- Birch, K.E.; Quinlivan, R.M.; Morris, G.E. Cell models for McArdle disease and aminoglycoside-induced read-through of a premature termination codon. Neuromuscul. Disord. 2013, 23, 43–51. [Google Scholar] [CrossRef]
- Steele, I.C.; Patterson, V.H.; Nicholls, D.P. A double blind, placebo controlled, crossover trial of D-ribose in McArdle’s disease. J. Neurol. Sci. 1996, 136, 174–177. [Google Scholar] [CrossRef]
- De Luna, N.; Brull, A.; Guiu, J.M.; Lucia, A.; Martin, M.A.; Arenas, J. Sodium valproate increases the brain isoform of glycogen phosphorylase: Looking for a compensation mechanism in McArdle disease using a mouse primary skeletal-muscle culture in vitro. Dis. Model. Mech. 2015, 8, 467–472. [Google Scholar] [CrossRef]
- Vorgerd, M.; Grehl, T.; Jäger, M.; Müller, K.; Freitag, G.; Patzold, T. Creatine therapy in myophosphorylase deficiency (McArdle disease): A placebo-controlled crossover trial. Arch. Neurol. 2000, 57, 956–963. [Google Scholar] [CrossRef]
- Vorgerd, M.; Zange, J.; Kley, R.; Grehl, T.; Hüsing, A.; Jäger, M. Effect of high-dose creatine therapy on symptoms of exercise intolerance in McArdle disease: Double-blind, placebo-controlled crossover study. Arch. Neurol. 2002, 59, 97–101. [Google Scholar] [CrossRef]
- Vorgerd, M.; Zange, J. Treatment of glycogenosys type V (McArdle disease) with creatine and ketogenic diet with clinical scores and with 31P-MRS on working leg muscle. Acta Myol. 2007, 26, 61–63. [Google Scholar]
- Quinlivan, R.; Martinuzzi, A.; Schoser, B. Pharmacological and nutritional treatment for McArdle disease (Glycogen Storage Disease type V). Cochrane Database Syst. Rev. 2014, 11, CD003458. [Google Scholar] [CrossRef]
- Quinlivan, R.M.; Beynon, R.J. Pharmacological and nutritional treatment trials in McArdle disease. Acta Myol. 2007, 26, 58–60. [Google Scholar]
- Vissing, J.; Haller, R.G. The effect of oral sucrose on exercise tolerance in patients with McArdle’s disease. N. Engl. J. Med. 2003, 349, 2503–2509. [Google Scholar] [CrossRef]
- Quinlivan, R.; Lucia, A.; Scalco, R.S.; Santana, A.; Parini, J.; Godfrey, R.; Marti, R. Report on the EUROMAC McArdle Exercise Testing Workshop, Madrid, Spain, 11–12 July 2014. Neuromuscul. Disord. 2015, 25, 739–745. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llavero, F.; Arrazola Sastre, A.; Luque Montoro, M.; Gálvez, P.; Lacerda, H.M.; Parada, L.A.; Zugaza, J.L. McArdle Disease: New Insights into Its Underlying Molecular Mechanisms. Int. J. Mol. Sci. 2019, 20, 5919. https://doi.org/10.3390/ijms20235919
Llavero F, Arrazola Sastre A, Luque Montoro M, Gálvez P, Lacerda HM, Parada LA, Zugaza JL. McArdle Disease: New Insights into Its Underlying Molecular Mechanisms. International Journal of Molecular Sciences. 2019; 20(23):5919. https://doi.org/10.3390/ijms20235919
Chicago/Turabian StyleLlavero, Francisco, Alazne Arrazola Sastre, Miriam Luque Montoro, Patricia Gálvez, Hadriano M Lacerda, Luis A. Parada, and José Luis Zugaza. 2019. "McArdle Disease: New Insights into Its Underlying Molecular Mechanisms" International Journal of Molecular Sciences 20, no. 23: 5919. https://doi.org/10.3390/ijms20235919
APA StyleLlavero, F., Arrazola Sastre, A., Luque Montoro, M., Gálvez, P., Lacerda, H. M., Parada, L. A., & Zugaza, J. L. (2019). McArdle Disease: New Insights into Its Underlying Molecular Mechanisms. International Journal of Molecular Sciences, 20(23), 5919. https://doi.org/10.3390/ijms20235919