Transforming Growth Factor-β Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment


Abstract
1. Introduction
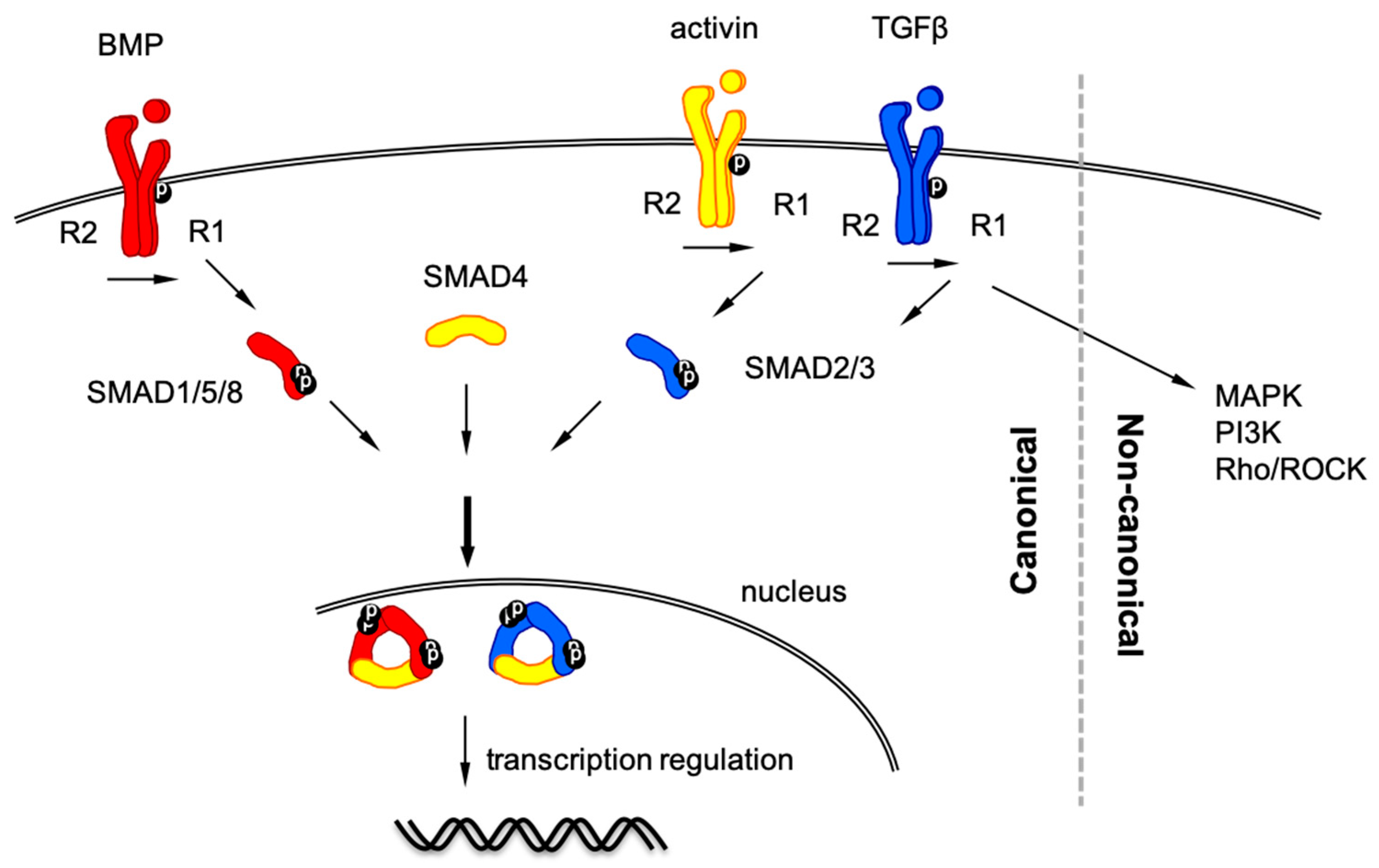
2. TGF-β Signaling Pathway
2.1. TGF-β Signaling in Cell Biology
2.2. TGF-β Signaling in Clinical Situation of CRC Patients
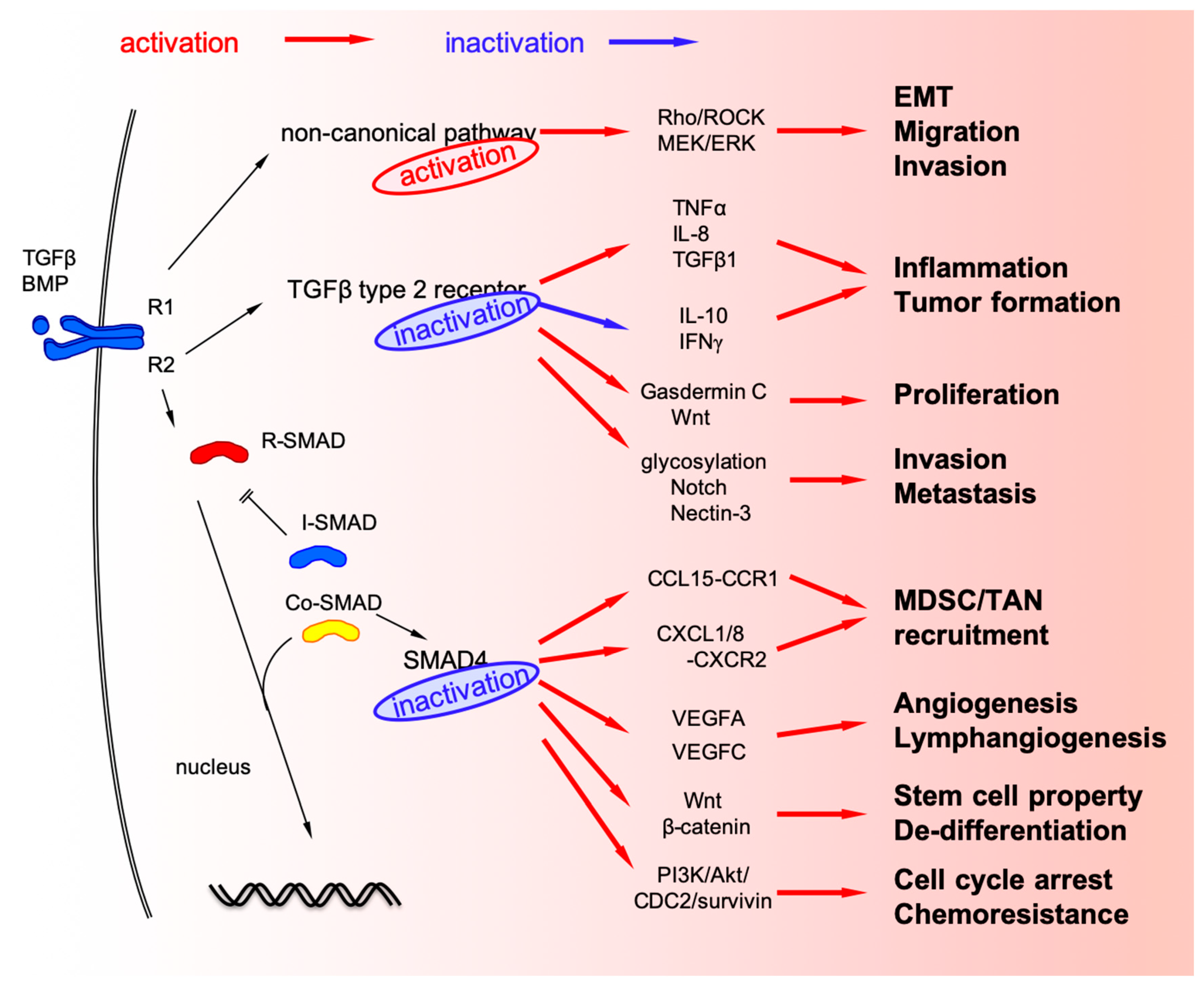
3. TGF-β Signaling in Cancer Cells
3.1. TGFBR2 Mutation in Cancer Cells
3.2. Mutations and Deletions of SMAD4 (Co-SMAD) in CRC Cells
3.3. Non-Canonical TGF-β Signaling Pathways in CRC
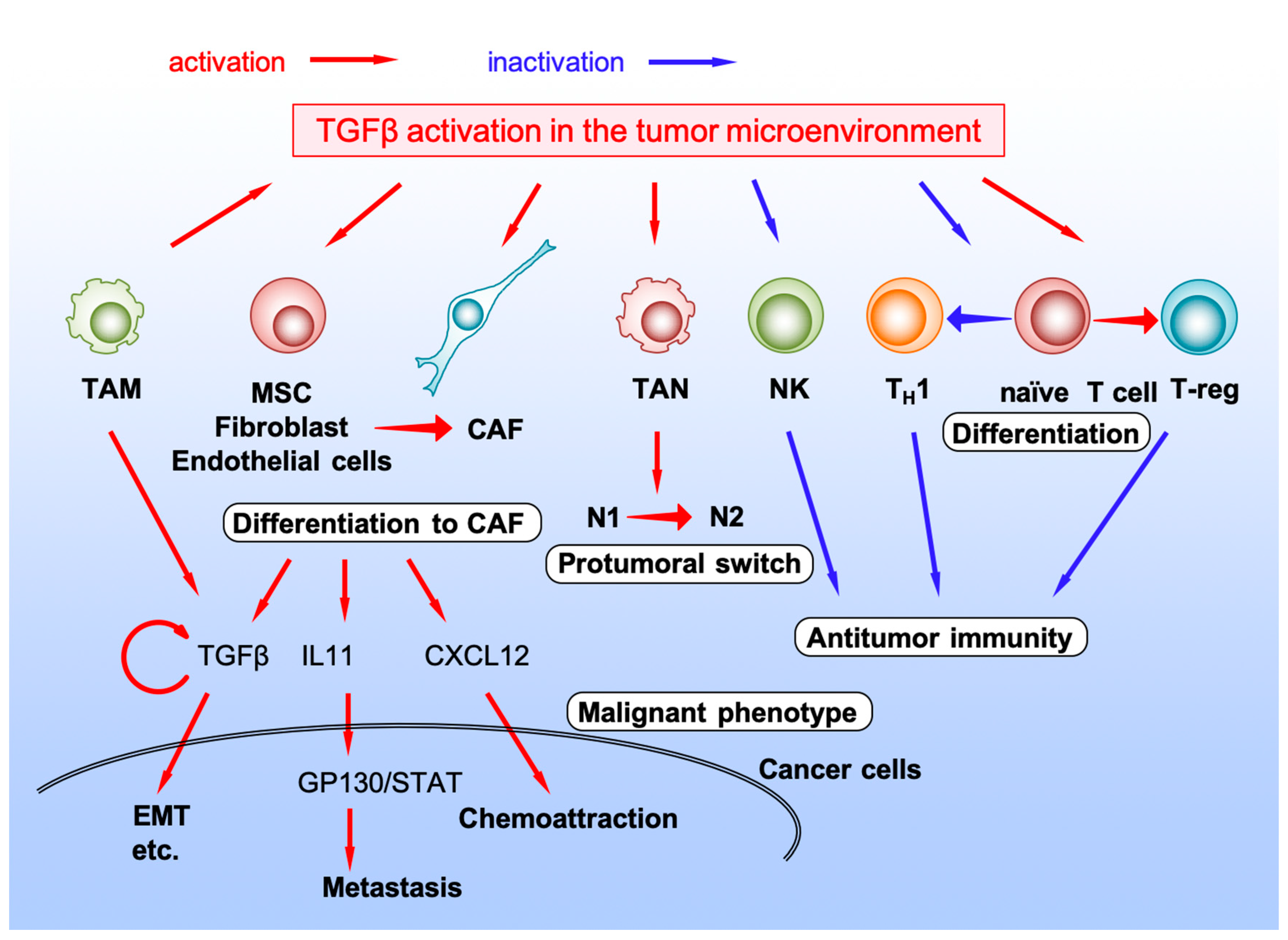
4. TGF-β Signaling in Stromal Cells in the Tumor Microenvironment
4.1. Cancer-Associated Fibroblast
4.2. Natural Killer Cell
4.3. TANs
4.4. TAMs
4.5. T Lymphocyte
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| APC | Adenomatous Polyposis Coli |
| APC | antigen-presenting cell |
| BMP | bone morphogenetic protein |
| BMPR2 | BMP receptor type 2 |
| CAF | cancer-associated fibroblast |
| CCL | C-C motif chemokine ligand |
| CDC | cell-division cycle |
| CMS | consensus molecular subtype |
| Co-SMAD | common-mediator SMAD |
| CRC | colorectal cancer |
| CTLA4 | cytotoxic T lymphocyte antigen 4 |
| CXCL | C-X-C motif chemokine ligand |
| dMMR | mismatch repair deficiency |
| EMT | epithelial to mesenchymal transition |
| FOXP3 | forkhead box P3 |
| GDF15 | growth differentiation factor-15 |
| GEMM | genetically-engineered mouse model |
| IL | interleukin |
| INFγ | interferon-γ |
| I-SMAD | inhibitory SMAD |
| JAK | Janus kinase |
| LOH | loss of heterozygosity |
| MAPK | mitogen-activated protein kinase |
| MDSC | myeloid-derived suppressor cells |
| MHC | major histocompatibility complex |
| MLH | MutL homolog |
| MMP | matrix metalloproteinase |
| MSC | mesenchymal stem cell |
| MSH | MutS homolog |
| MSI | microsatellite instability |
| MSI-H | high level of MSI |
| PCR | polymerase chain reaction |
| PD1 | programmed cell death 1 |
| PI3K | phosphoinositide 3-kinase |
| PIK3CA | PI3K catalytic subunit-α |
| PMS | Postmeiotic segregation increased |
| R-SMAD | receptor-regulated SMAD |
| ROCK | Rho-associated protein kinase |
| SMAD | Caenorhabditis elegans SMA and Drosophila MAD |
| STAT | signal transducer and activator of transcription |
| TAM | tumor-associated macrophages |
| TAN | tumor-associated neutrophils |
| TCR | T-cell receptor |
| TGFβ | Transforming growth factor-beta |
| TGFBR2 | TGFβ receptor type 2 |
| TH1 | type 1 T-helper cell |
| TNFα | tumor necrosis factor-α |
| T-reg | regulatory T-cell |
| VEGF | vascular endothelial growth factor |
References
- Jung, B.; Staudacher, J.J.; Beauchamp, D. Transforming Growth Factor beta Superfamily Signaling in Development of Colorectal Cancer. Gastroenterology 2017, 152, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of Function Between Tumor Prevention and Carcinogenesis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Ihara, S.; Hirata, Y.; Koike, K. TGF-β in inflammatory bowel disease: A key regulator of immune cells, epithelium, and the intestinal microbiota. J. Gastroenterol. 2017, 52, 777–787. [Google Scholar] [CrossRef]
- Batlle, R.; Andrés, E.; Gonzalez, L.; Llonch, E.; Igea, A.; Gutierrez-Prat, N.; Berenguer-Llergo, A.; Nebreda, A.R. Regulation of tumor angiogenesis and mesenchymal–endothelial transition by p38α through TGF-β and JNK signaling. Nat. Commun. 2019, 10, 3071. [Google Scholar] [CrossRef]
- Muñoz, N.M.; Upton, M.; Rojas, A.; Washington, M.K.; Lin, L.; Chytil, A.; Sozmen, E.G.; Madison, B.B.; Pozzi, A.; Moon, R.T.; et al. Transforming Growth Factor β Receptor Type II Inactivation Induces the Malignant Transformation of Intestinal Neoplasms Initiated by Apc Mutation. Cancer Res. 2006, 66, 9837. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Fujishita, T.; Kakizaki, F.; Hirai, H.; Matsumoto, T.; Iwamoto, M.; Inamoto, S.; Hatano, E.; Hasegawa, S.; et al. Loss of SMAD4 from colorectal cancer cells promotes CCL15 expression to recruit CCR1+ myeloid cells and facilitate liver metastasis. Gastroenterology 2013, 145, 1064–1075. [Google Scholar] [CrossRef]
- Inamoto, S.; Itatani, Y.; Yamamoto, T.; Minamiguchi, S.; Hirai, H.; Iwamoto, M.; Hasegawa, S.; Taketo, M.M.; Sakai, Y.; Kawada, K. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid-Derived Suppressor Cells through the CCL15-CCR1 Chemokine Axis. Clin. Cancer Res. 2016, 22, 492–501. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kawada, K.; Itatani, Y.; Inamoto, S.; Okamura, R.; Iwamoto, M.; Miyamoto, E.; Chen-Yoshikawa, T.F.; Hirai, H.; Hasegawa, S.; et al. Loss of SMAD4 Promotes Lung Metastasis of Colorectal Cancer by Accumulation of CCR1+ Tumor-Associated Neutrophils through CCL15-CCR1 Axis. Clin. Cancer Res. 2017, 23, 833–844. [Google Scholar] [CrossRef]
- Ogawa, R.; Yamamoto, T.; Hirai, H.; Hanada, K.; Kiyasu, Y.; Nishikawa, G.; Mizuno, R.; Inamoto, S.; Itatani, Y.; Sakai, Y.; et al. Loss of SMAD4 Promotes Colorectal Cancer Progression by Recruiting Tumor-Associated Neutrophils via the CXCL1/8-CXCR2 Axis. Clin. Cancer Res. 2019, 25, 2887–2899. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, N.F.C.C.; van Dinther, M.; van den Akker, B.E.W.M.; van Wezel, T.; ten Dijke, P.; Morreau, H. Transforming Growth Factor β Signaling in Colorectal Cancer Cells With Microsatellite Instability Despite Biallelic Mutations in TGFBR2. Gastroenterology 2015, 148, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M. Polymerase Slippage Restoration of Frameshifted TGFBR2 in Colorectal Cancer: A Novel Paradigm. Gastroenterology 2015, 148, 1276–1279. [Google Scholar] [CrossRef]
- Roth, A.D.; Delorenzi, M.; Tejpar, S.; Yan, P.; Klingbiel, D.; Fiocca, R.; d’Ario, G.; Cisar, L.; Labianca, R.; Cunningham, D.; et al. Integrated analysis of molecular and clinical prognostic factors in stage II/III colon cancer. J. Natl. Cancer Inst. 2012, 104, 1635–1646. [Google Scholar] [CrossRef]
- Weiss, A.; Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef]
- Martin-Malpartida, P.; Batet, M.; Kaczmarska, Z.; Freier, R.; Gomes, T.; Aragón, E.; Zou, Y.; Wang, Q.; Xi, Q.; Ruiz, L.; et al. Structural basis for genome wide recognition of 5-bp GC motifs by SMAD transcription factors. Nat. Commun. 2017, 8, 2070. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Chang, C. Agonists and Antagonists of TGF-beta Family Ligands. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Simoneaux, R. The Four Colorectal Cancer Consensus Molecular Subtypes. Oncol. Times 2018, 40, 10–11. [Google Scholar] [CrossRef]
- Shima, K.; Morikawa, T.; Yamauchi, M.; Kuchiba, A.; Imamura, Y.; Liao, X.; Meyerhardt, J.A.; Fuchs, C.S.; Ogino, S. TGFBR2 and BAX mononucleotide tract mutations, microsatellite instability, and prognosis in 1072 colorectal cancers. PLoS ONE 2011, 6, e25062. [Google Scholar] [CrossRef] [PubMed]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Voorneveld, P.W.; Jacobs, R.J.; Kodach, L.L.; Hardwick, J.C. A Meta-Analysis of SMAD4 Immunohistochemistry as a Prognostic Marker in Colorectal Cancer. Transl. Oncol. 2015, 8, 18–24. [Google Scholar] [CrossRef]
- Yan, P.; Klingbiel, D.; Saridaki, Z.; Ceppa, P.; Curto, M.; McKee, T.A.; Roth, A.; Tejpar, S.; Delorenzi, M.; Bosman, F.T.; et al. Reduced Expression of SMAD4 Is Associated with Poor Survival in Colon Cancer. Clin. Cancer Res. 2016, 22, 3037–3047. [Google Scholar] [CrossRef]
- Mizuno, T.; Cloyd, J.M.; Vicente, D.; Omichi, K.; Chun, Y.S.; Kopetz, S.E.; Maru, D.; Conrad, C.; Tzeng, C.D.; Wei, S.H.; et al. SMAD4 gene mutation predicts poor prognosis in patients undergoing resection for colorectal liver metastases. Eur. J. Surg. Oncol. 2018, 44, 684–692. [Google Scholar] [CrossRef]
- Okita, A.; Takahashi, S.; Ouchi, K.; Inoue, M.; Watanabe, M.; Endo, M.; Honda, H.; Yamada, Y.; Ishioka, C. Consensus molecular subtypes classification of colorectal cancer as a predictive factor for chemotherapeutic efficacy against metastatic colorectal cancer. Oncotarget 2018, 9, 18698–18711. [Google Scholar] [CrossRef]
- Mooi, J.K.; Wirapati, P.; Asher, R.; Lee, C.K.; Savas, P.; Price, T.J.; Townsend, A.; Hardingham, J.; Buchanan, D.; Williams, D.; et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: Molecular analysis of the AGITG MAX clinical trial. Ann. Oncol. 2018, 29, 2240–2246. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Bruun, J.; Eide, P.W.; Eilertsen, I.A.; Ramirez, L.; Murumägi, A.; Arjama, M.; Danielsen, S.A.; Kryeziu, K.; Elez, E.; et al. Colorectal Cancer Consensus Molecular Subtypes Translated to Preclinical Models Uncover Potentially Targetable Cancer Cell Dependencies. Clin. Cancer Res. 2018, 24, 794. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.S.; Kikuchi, H.; Zeng, M.; Herraiz, M.T.; Sperduti, I.; Berger, D.; Park, D.Y.; Iafrate, A.J.; Zukerberg, L.R.; Chung, D.C. Epithelial to mesenchymal transition is impaired in colon cancer cells with microsatellite instability. Gastroenterology 2010, 138, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Drost, J.; van Hooff, S.R.; Linnekamp, J.F.; Wang, X.; Jansen, M.; De Sousa E Melo, F.; Prasetyanti, P.R.; Ijspeert, J.E.; Franitza, M.; et al. TGFβ signaling directs serrated adenomas to the mesenchymal colorectal cancer subtype. EMBO Mol. Med. 2016, 8, 745–760. [Google Scholar] [CrossRef]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336. [Google Scholar] [CrossRef]
- Rubenstein, J.H.; Enns, R.; Heidelbaugh, J.; Barkun, A. American Gastroenterological Association Institute Guideline on the Diagnosis and Management of Lynch Syndrome. Gastroenterology 2015, 149, 777–782. [Google Scholar] [CrossRef]
- Vasen, H.F.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut 2013, 62, 812–823. [Google Scholar] [CrossRef]
- Tamura, K.; Kaneda, M.; Futagawa, M.; Takeshita, M.; Kim, S.; Nakama, M.; Kawashita, N.; Tatsumi-Miyajima, J. Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome. Int. J. Clin. Oncol. 2019, 24, 999–1011. [Google Scholar] [CrossRef]
- Haraldsdottir, S.; Hampel, H.; Wu, C.; Weng, D.Y.; Shields, P.G.; Frankel, W.L.; Pan, X.; de la Chapelle, A.; Goldberg, R.M.; Bekaii-Saab, T. Patients with colorectal cancer associated with Lynch syndrome and MLH1 promoter hypermethylation have similar prognoses. Genet. Med. 2016, 18, 863–868. [Google Scholar] [CrossRef]
- Xu, Y.; Pasche, B. TGF-beta signaling alterations and susceptibility to colorectal cancer. Hum. Mol. Genet. 2007, 16 Spec No 1, R14–R20. [Google Scholar] [CrossRef]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y.; et al. Combined Mutation of Apc, Kras, and Tgfbr2 Effectively Drives Metastasis of Intestinal Cancer. Cancer Res. 2018, 78, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Kataoka, S.; Nakayama, M.; Ali, M.A.E.; Oshima, H.; Yamamoto, D.; Park, J.-W.; Takegami, Y.; An, T.; Jenkins, N.A.; et al. CRISPR-Cas9–mediated gene knockout in intestinal tumor organoids provides functional validation for colorectal cancer driver genes. Proc. Natl. Acad. Sci. USA 2019, 116, 15635. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, M.; Pinto, C.; Peixoto, A.; Veiga, I.; Lopes, P.; Henrique, R.; Baldaia, H.; Carneiro, F.; Seruca, R.; Tomlinson, I.; et al. Target gene mutational pattern in Lynch syndrome colorectal carcinomas according to tumour location and germline mutation. Br. J. Cancer 2015, 113, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Giannakis, M. Immunoscore for (colorectal) cancer precision medicine. Lancet 2018, 391, 2084–2086. [Google Scholar] [CrossRef]
- Principe, D.R.; DeCant, B.; Staudacher, J.; Vitello, D.; Mangan, R.J.; Wayne, E.A.; Mascariñas, E.; Diaz, A.M.; Bauer, J.; McKinney, R.D.; et al. Loss of TGFβ signaling promotes colon cancer progression and tumor-associated inflammation. Oncotarget 2017, 8, 3826. [Google Scholar] [CrossRef]
- Oshima, H.; Nakayama, M.; Han, T.S.; Naoi, K.; Ju, X.; Maeda, Y.; Robine, S.; Tsuchiya, K.; Sato, T.; Sato, H.; et al. Suppressing TGFbeta signaling in regenerating epithelia in an inflammatory microenvironment is sufficient to cause invasive intestinal cancer. Cancer Res. 2015, 75, 766–776. [Google Scholar] [CrossRef]
- Morris, S.M.; Davison, J.; Carter, K.T.; O’Leary, R.M.; Trobridge, P.; Knoblaugh, S.E.; Myeroff, L.L.; Markowitz, S.D.; Brett, B.T.; Scheetz, T.E.; et al. Transposon mutagenesis identifies candidate genes that cooperate with loss of transforming growth factor-beta signaling in mouse intestinal neoplasms. Int. J. Cancer 2017, 140, 853–863. [Google Scholar] [CrossRef]
- Miguchi, M.; Hinoi, T.; Shimomura, M.; Adachi, T.; Saito, Y.; Niitsu, H.; Kochi, M.; Sada, H.; Sotomaru, Y.; Ikenoue, T.; et al. Gasdermin C Is Upregulated by Inactivation of Transforming Growth Factor β Receptor Type II in the Presence of Mutated Apc, Promoting Colorectal Cancer Proliferation. PLoS ONE 2016, 11, e0166422. [Google Scholar] [CrossRef]
- Ferreira, I.G.; Pucci, M.; Venturi, G.; Malagolini, N.; Chiricolo, M.; Dall’Olio, F. Glycosylation as a Main Regulator of Growth and Death Factor Receptors Signaling. Int. J. Mol. Sci. 2018, 19, 580. [Google Scholar] [CrossRef]
- Lee, J.; Katzenmaier, E.M.; Kopitz, J.; Gebert, J. Reconstitution of TGFBR2 in HCT116 colorectal cancer cells causes increased LFNG expression and enhanced N-acetyl-d-glucosamine incorporation into Notch1. Cell. Signal. 2016, 28, 1105–1113. [Google Scholar] [CrossRef]
- Lee, J.; Fricke, F.; Warnken, U.; Schnölzer, M.; Kopitz, J.; Gebert, J. Reconstitution of TGFBR2-Mediated Signaling Causes Upregulation of GDF-15 in HCT116 Colorectal Cancer Cells. PLoS ONE 2015, 10, e0131506. [Google Scholar] [CrossRef] [PubMed]
- Fricke, F.; Michalak, M.; Warnken, U.; Hausser, I.; Schnolzer, M.; Kopitz, J.; Gebert, J. SILAC-Based Quantification of TGFBR2-Regulated Protein Expression in Extracellular Vesicles of Microsatellite Unstable Colorectal Cancers. Int. J. Mol. Sci. 2019, 20, 4162. [Google Scholar] [CrossRef] [PubMed]
- Fricke, F.; Mussack, V.; Buschmann, D.; Hausser, I.; Pfaffl, M.W.; Kopitz, J.; Gebert, J. TGFBR2-dependent alterations of microRNA profiles in extracellular vesicles and parental colorectal cancer cells. Int. J. Oncol. 2019, 55, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. Tumor suppressor genes. Biol. Cancer 2013, 2, 231–274. [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Huang, D.; Sun, W.; Zhou, Y.; Li, P.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef]
- Takaku, K.; Oshima, M.; Miyoshi, H.; Matsui, M.; Seldin, M.F.; Taketo, M.M. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 1998, 92, 645–656. [Google Scholar] [CrossRef]
- Kitamura, T.; Kometani, K.; Hashida, H.; Matsunaga, A.; Miyoshi, H.; Hosogi, H.; Aoki, M.; Oshima, M.; Hattori, M.; Takabayashi, A.; et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat. Genet. 2007, 39, 467–475. [Google Scholar] [CrossRef]
- Riggins, G.J.; Kinzler, K.W.; Vogelstein, B.; Thiagalingam, S. Frequency of Smad gene mutations in human cancers. Cancer Res. 1997, 57, 2578–2580. [Google Scholar]
- Salovaara, R.; Roth, S.; Loukola, A.; Launonen, V.; Sistonen, P.; Avizienyte, E.; Kristo, P.; Jarvinen, H.; Souchelnytskyi, S.; Sarlomo-Rikala, M.; et al. Frequent loss of SMAD4/DPC4 protein in colorectal cancers. Gut 2002, 51, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Zhao, S.; Tang, H.; Zhang, D.; Sun, H.; Yu, F.; Jiang, W.; Yue, B.; Wang, J.; Zhang, M.; et al. MicroRNA-20a-5p promotes colorectal cancer invasion and metastasis by downregulating Smad4. Oncotarget 2016, 7, 45199–45213. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Lin, X.; Feng, X.H. Posttranslational Regulation of Smads. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Fei, T.; Xia, K.; Li, Z.; Zhou, B.; Zhu, S.; Chen, H.; Zhang, J.; Chen, Z.; Xiao, H.; Han, J.-D.J.; et al. Genome-wide mapping of SMAD target genes reveals the role of BMP signaling in embryonic stem cell fate determination. Genome Res. 2010, 20, 36–44. [Google Scholar] [CrossRef]
- Kennedy, B.A.; Deatherage, D.E.; Gu, F.; Tang, B.; Chan, M.W.; Nephew, K.P.; Huang, T.H.; Jin, V.X. ChIP-seq defined genome-wide map of TGFbeta/SMAD4 targets: Implications with clinical outcome of ovarian cancer. PLoS ONE 2011, 6, e22606. [Google Scholar] [CrossRef]
- Means, A.L.; Freeman, T.J.; Zhu, J.; Woodbury, L.G.; Marincola-Smith, P.; Wu, C.; Meyer, A.R.; Weaver, C.J.; Padmanabhan, C.; An, H.; et al. Epithelial Smad4 Deletion Up-Regulates Inflammation and Promotes Inflammation-Associated Cancer. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 257–276. [Google Scholar] [CrossRef]
- Kitamura, T.; Fujishita, T.; Loetscher, P.; Revesz, L.; Hashida, H.; Kizaka-Kondoh, S.; Aoki, M.; Taketo, M.M. Inactivation of chemokine (C-C motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 13063–13068. [Google Scholar] [CrossRef]
- Hirai, H.; Fujishita, T.; Kurimoto, K.; Miyachi, H.; Kitano, S.; Inamoto, S.; Itatani, Y.; Saitou, M.; Maekawa, T.; Taketo, M.M. CCR1-mediated accumulation of myeloid cells in the liver microenvironment promoting mouse colon cancer metastasis. Clin. Exp. Metastasis 2014, 31, 977–989. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Roelands, J.; Kuppen, P.J.K.; Vermeulen, L.; Maccalli, C.; Decock, J.; Wang, E.; Marincola, F.M.; Bedognetti, D.; Hendrickx, W. Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications. Int. J. Mol. Sci. 2017, 18, 2229. [Google Scholar] [CrossRef]
- Zonneville, J.; Safina, A.; Truskinovsky, A.M.; Arteaga, C.L.; Bakin, A.V. TGF-β signaling promotes tumor vasculature by enhancing the pericyte-endothelium association. BMC Cancer 2018, 18, 670. [Google Scholar] [CrossRef] [PubMed]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sheng, J.; Dai, D.; Liu, T.; Qi, F. Smad4 acts as tumor suppressor by antagonizing lymphangiogenesis in colorectal cancer. Pathol. Res. Pract. 2015, 211, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, X.; Lv, X.; Xiao, J.; Liu, B.; Zhang, Y. Smad4 Inhibits VEGF-A and VEGF-C Expressions via Enhancing Smad3 Phosphorylation in Colon Cancer. Anat. Rec. 2017, 300, 1560–1569. [Google Scholar] [CrossRef]
- Perekatt, A.O.; Shah, P.P.; Cheung, S.; Jariwala, N.; Wu, A.; Gandhi, V.; Kumar, N.; Feng, Q.; Patel, N.; Chen, L.; et al. SMAD4 Suppresses WNT-Driven Dedifferentiation and Oncogenesis in the Differentiated Gut Epithelium. Cancer Res. 2018, 78, 4878–4890. [Google Scholar] [CrossRef]
- Freeman, T.J.; Smith, J.J.; Chen, X.; Washington, M.K.; Roland, J.T.; Means, A.L.; Eschrich, S.A.; Yeatman, T.J.; Deane, N.G.; Beauchamp, R.D. Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of beta-catenin. Gastroenterology 2012, 142, 562–571. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, B.; Chen, X.; Bae, S.; Singh, K.; Washington, M.K.; Datta, P.K. Loss of Smad4 in colorectal cancer induces resistance to 5-fluorouracil through activating Akt pathway. Br. J. Cancer 2014, 110, 946–957. [Google Scholar] [CrossRef]
- Papageorgis, P.; Cheng, K.; Ozturk, S.; Gong, Y.; Lambert, A.W.; Abdolmaleky, H.M.; Zhou, J.R.; Thiagalingam, S. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res. 2011, 71, 998–1008. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-beta Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Zhang, B.; Leng, C.; Wu, C.; Zhang, Z.; Dou, L.; Luo, X.; Zhang, B.; Chen, X. Smad4 sensitizes colorectal cancer to 5-fluorouracil through cell cycle arrest by inhibiting the PI3K/Akt/CDC2/survivin cascade. Oncol. Rep. 2016, 35, 1807–1815. [Google Scholar] [CrossRef][Green Version]
- Voorneveld, P.W.; Kodach, L.L.; Jacobs, R.J.; Liv, N.; Zonnevylle, A.C.; Hoogenboom, J.P.; Biemond, I.; Verspaget, H.W.; Hommes, D.W.; de Rooij, K.; et al. Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology 2014, 147, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Wu, Y.; Zhang, W.; Zhang, Z.; Jin, G.; Zhao, J.; Yu, J.; Lin, Y.; Zhang, W.; Liang, H.; et al. Targeting the ERK pathway reduces liver metastasis of Smad4-inactivated colorectal cancer. Cancer Biol. Ther. 2013, 14, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Halder, S.K.; Kashikar, N.D.; Cho, Y.J.; Datta, A.; Gorden, D.L.; Datta, P.K. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology 2010, 138, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.X.; Cao, Z.B.; He, T.T.; Huang, T.; Xiang, C.L.; Liu, Y. TGFbeta1 is essential for MSCs-CAFs differentiation and promotes HCT116 cells migration and invasion via JAK/STAT3 signaling. Onco Targets Ther. 2019, 12, 5323–5334. [Google Scholar] [CrossRef]
- Koliaraki, V.; Pallangyo, C.K.; Greten, F.R.; Kollias, G. Mesenchymal Cells in Colon Cancer. Gastroenterology 2017, 152, 964–979. [Google Scholar] [CrossRef]
- Lamprecht, S.; Sigal-Batikoff, I.; Shany, S.; Abu-Freha, N.; Ling, E.; Delinasios, G.J.; Moyal-Atias, K.; Delinasios, J.G.; Fich, A. Teaming Up for Trouble: Cancer Cells, Transforming Growth Factor-beta1 Signaling and the Epigenetic Corruption of Stromal Naive Fibroblasts. Cancers 2018, 10, 61. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Zhou, J.; Lu, L.; Wang, H.; Zhang, G.; Wan, G.; Cai, S.; Du, J. Nodal Facilitates Differentiation of Fibroblasts to Cancer-Associated Fibroblasts that Support Tumor Growth in Melanoma and Colorectal Cancer. Cells 2019, 8, 538. [Google Scholar] [CrossRef]
- Ciszewski, W.M.; Sobierajska, K.; Wawro, M.E.; Klopocka, W.; Chefczynska, N.; Muzyczuk, A.; Siekacz, K.; Wujkowska, A.; Niewiarowska, J. The ILK-MMP9-MRTF axis is crucial for EndMT differentiation of endothelial cells in a tumor microenvironment. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2283–2296. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.; Paauwe, M.; Verspaget, H.W.; Wiercinska, E.; van der Zon, J.M.; van der Ploeg, K.; Koelink, P.J.; Lindeman, J.H.; Mesker, W.; ten Dijke, P.; et al. Interaction with colon cancer cells hyperactivates TGF-beta signaling in cancer-associated fibroblasts. Oncogene 2014, 33, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.A.; Chen, Y.F.; Bao, Y.; Mahara, S.; Yatim, S.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1alpha and TGF-beta2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Otegbeye, F.; Ojo, E.; Moreton, S.; Mackowski, N.; Lee, D.A.; de Lima, M.; Wald, D.N. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE 2018, 13, e0191358. [Google Scholar] [CrossRef]
- Dell’Aquila, E.; Cremolini, C.; Zeppola, T.; Lonardi, S.; Bergamo, F.; Masi, G.; Stellato, M.; Marmorino, F.; Schirripa, M.; Urbano, F.; et al. Prognostic and predictive role of neutrophil/lymphocytes ratio in metastatic colorectal cancer: A retrospective analysis of the TRIBE study by GONO. Ann. Oncol. 2018, 29, 924–930. [Google Scholar] [CrossRef]
- Haram, A.; Boland, M.R.; Kelly, M.E.; Bolger, J.C.; Waldron, R.M.; Kerin, M.J. The prognostic value of neutrophil-to-lymphocyte ratio in colorectal cancer: A systematic review. J. Surg. Oncol. 2017, 115, 470–479. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Y.; Zheng, F. Prognostic significance of elevated preoperative neutrophil-to-lymphocyte ratio for patients with colorectal cancer undergoing curative surgery: A meta-analysis. Medicine 2019, 98, e14126. [Google Scholar] [CrossRef]
- Inamoto, S.; Kawada, K.; Okamura, R.; Hida, K.; Sakai, Y. Prognostic impact of the combination of neutrophil-to-lymphocyte ratio and Glasgow prognostic score in colorectal cancer: A retrospective cohort study. Int. J. Colorectal Dis. 2019, 34, 1303–1315. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef] [PubMed]
- Giese, M.A.; Hind, L.E.; Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 2019, 133, 2159–2167. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Ang, C.W.; Nedjadi, T.; Sheikh, A.A.; Tweedle, E.M.; Tonack, S.; Honap, S.; Jenkins, R.E.; Park, B.K.; Schwarte-Waldhoff, I.; Khattak, I.; et al. Smad4 loss is associated with fewer S100A8-positive monocytes in colorectal tumors and attenuated response to S100A8 in colorectal and pancreatic cancer cells. Carcinogenesis 2010, 31, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Gulubova, M.; Ananiev, J.; Yovchev, Y.; Julianov, A.; Karashmalakov, A.; Vlaykova, T. The density of macrophages in colorectal cancer is inversely correlated to TGF-beta1 expression and patients’ survival. J. Mol. Histol. 2013, 44, 679–692. [Google Scholar] [CrossRef]
- Zhang, D.; Qiu, X.; Li, J.; Zheng, S.; Li, L.; Zhao, H. TGF-beta secreted by tumor-associated macrophages promotes proliferation and invasion of colorectal cancer via miR-34a-VEGF axis. Cell Cycle 2018, 17, 2766–2778. [Google Scholar] [CrossRef]
- Cai, J.; Xia, L.; Li, J.; Ni, S.; Song, H.; Wu, X. Tumor-Associated Macrophages Derived TGF-betaInduced Epithelial to Mesenchymal Transition in Colorectal Cancer Cells through Smad2,3-4/Snail Signaling Pathway. Cancer Res. Treat. 2019, 51, 252–266. [Google Scholar] [CrossRef]
- Ling, A.; Lundberg, I.V.; Eklöf, V.; Wikberg, M.L.; Öberg, Å.; Edin, S.; Palmqvist, R. The infiltration, and prognostic importance, of Th1 lymphocytes vary in molecular subgroups of colorectal cancer. J. Pathol. Clin. Res. 2015, 2, 21–31. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Sun, X.; Cui, Y.; Feng, H.; Liu, H.; Liu, X. TGF-beta signaling controls Foxp3 methylation and T reg cell differentiation by modulating Uhrf1 activity. J. Exp. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shitara, K.; Nishikawa, H. Regulatory T cells: A potential target in cancer immunotherapy. Ann. N. Y. Acad. Sci. 2018, 1417, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Kuranaga, Y.; Kumazaki, M.; Shinohara, H.; Taniguchi, K.; Akao, Y. Colorectal cancer cell-derived extracellular vesicles induce phenotypic alteration of T cells into tumor-growth supporting cells with transforming growth factor-beta1-mediated suppression. Oncotarget 2016, 7, 27033–27043. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.-J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef]
- Hu, G.; Li, Z.A.; Wang, S. Tumor-infiltrating FoxP3(+) Tregs predict favorable outcome in colorectal cancer patients: A meta-analysis. Oncotarget 2017, 8, 75361–75371. [Google Scholar] [CrossRef]
- Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.-M.; Howlader, N.; Henley, S.J.; Anderson, R.N.; Firth, A.U.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 2018, 124, 2785–2800. [Google Scholar] [CrossRef]
- Malvezzi, M.; Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2019 with focus on breast cancer. Ann. Oncol. 2019, 30, 781–787. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Models | Factors Changed | Function Observed |
|---|---|---|
| GEMM 1 [59,67] | Ccl9 upregulation | CCR1+ myeloid cell recruitment |
| Xenograft [8,9,10] | CCL15 upregulation | CCR1+ TAN/MDSC recruitment |
| GEMM [75] | Wnt activation | Dedifferentiation Stem cell characteristics |
| Allograft [80] | PI3K/Akt/CDC2/survivin activation | 5-FU resistance |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-β Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. https://doi.org/10.3390/ijms20235822
Itatani Y, Kawada K, Sakai Y. Transforming Growth Factor-β Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. International Journal of Molecular Sciences. 2019; 20(23):5822. https://doi.org/10.3390/ijms20235822
Chicago/Turabian StyleItatani, Yoshiro, Kenji Kawada, and Yoshiharu Sakai. 2019. "Transforming Growth Factor-β Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment" International Journal of Molecular Sciences 20, no. 23: 5822. https://doi.org/10.3390/ijms20235822
APA StyleItatani, Y., Kawada, K., & Sakai, Y. (2019). Transforming Growth Factor-β Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. International Journal of Molecular Sciences, 20(23), 5822. https://doi.org/10.3390/ijms20235822