In Situ Analysis of DNA-Protein Complex Formation upon Radiation-Induced DNA Damage



Abstract
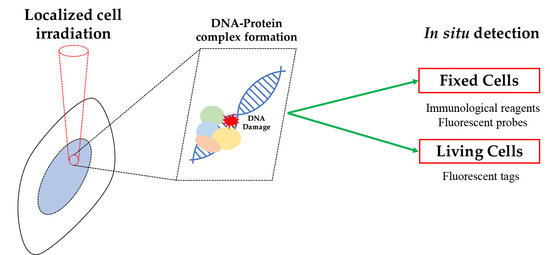
{kind=link}
{kind=link}
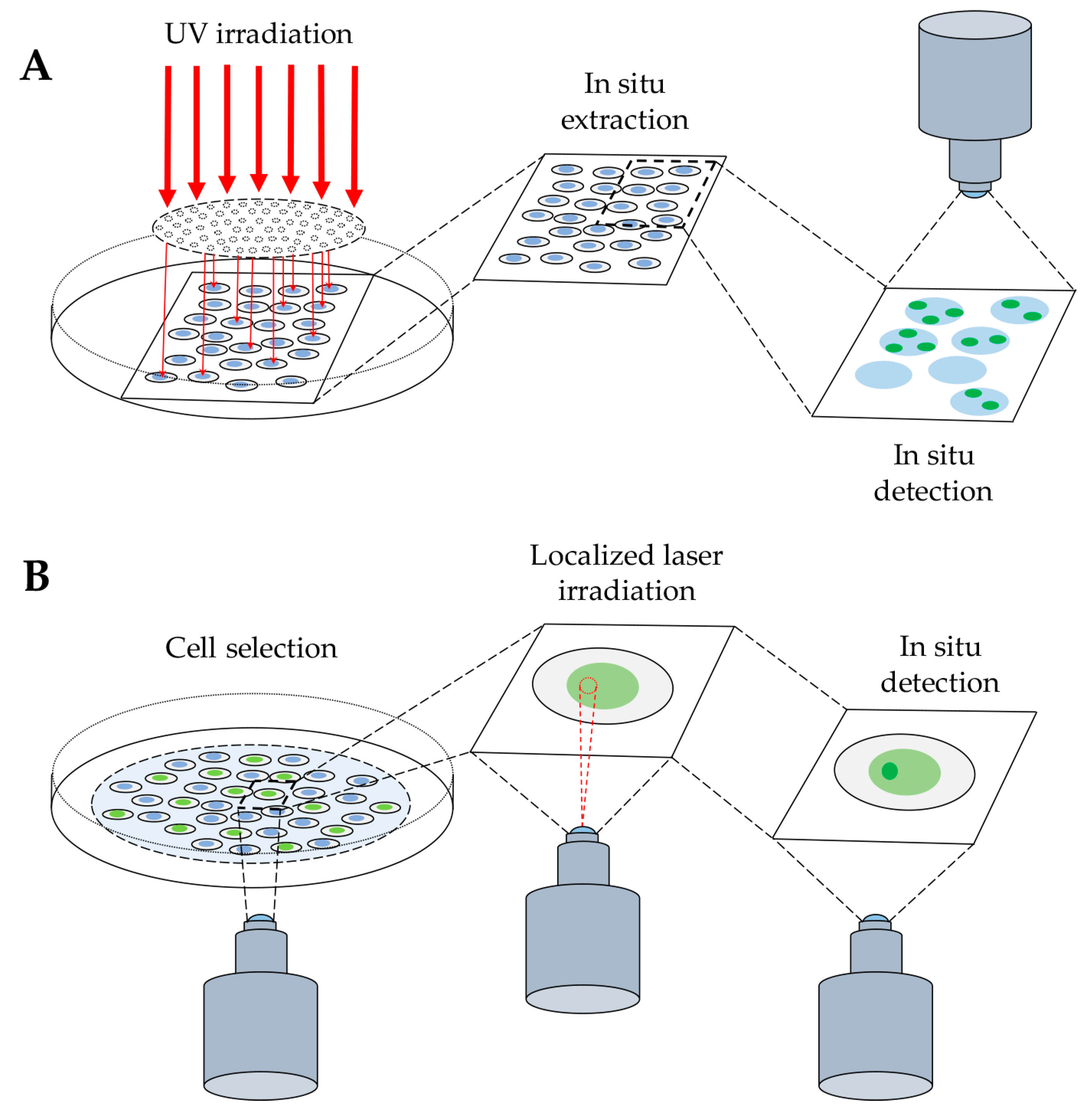
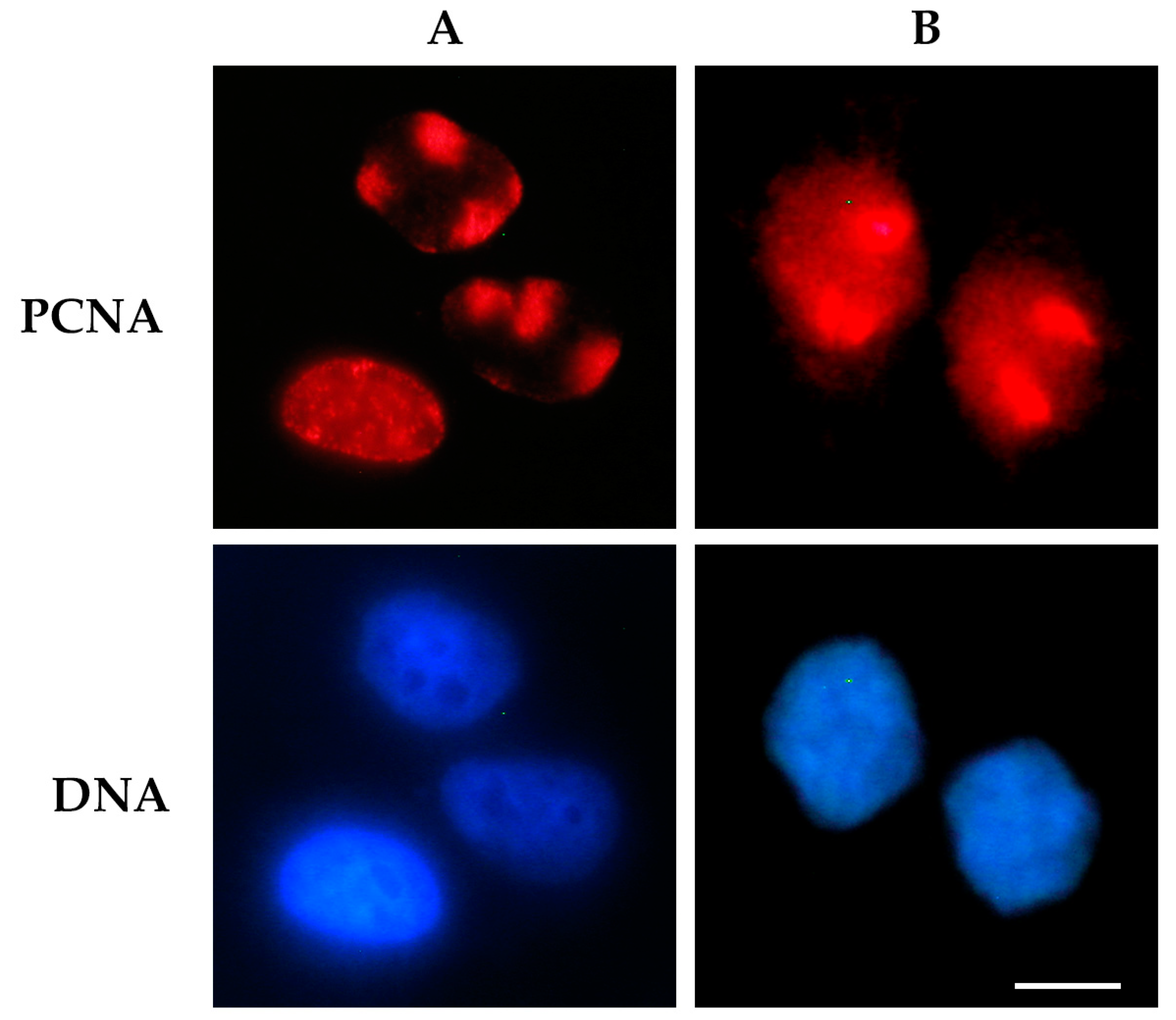{kind=link}
1. Introduction
2. In Situ Detection of DNA-Protein Complex Formation
2.1. Detection of DNA-Protein Complexes in Fixed Cells
2.1.1. Immunofluorescence-Based Techniques
2.1.2. Probe-Based Techniques
2.2. Detection of DNA–Protein Complexes in Living Cells
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BER | Base Excision Repair |
| CPD | Cyclobutane Pyrimidine Dimer |
| DDR | DNA Damage Response |
| DSB | Double Strand Break |
| GFP | Green Fluorescent Protein |
| IR | Ionizing Radiation |
| NER | Nucleotide Excision Repair |
| NHEJ | Non Homologous End Joining |
| PCNA | Proliferating Cell Nuclear Antigen |
| PLA | Proximity Ligation Assay |
| iPOND | Isolation of Protein on Nascent DNA |
| SLA | Subcellular Localization Assay |
| SIRF | In Situ Protein Interactions at Nascent and Stalled Replication Forks |
| SSB | Single Strand Break |
References
- Agarwal, S.; Tafel, A.A.; Kanaar, R. DNA double-strand breaks repair and chromosome translocation. DNA Repair 2006, 5, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H.J. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T. Formation of UV-induced DNA damage contributing to skin cancer development. Photochem. Photobiol. Sci. 2018, 17, 1816–1841. [Google Scholar] [CrossRef]
- Goodhead, D.T. Initial events in the cellular effects of ionizing radiations: Clustered damage in DNA. Int. J. Rad. Biol. 1994, 65, 7–17. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K.J.A.; Medeiros, M.H.G.; Di Mascio, P.; Wagner, J.R. Formation and repair of oxidatively generated damage in cellular DNA. Free Radic. Biol. Med. 2017, 107, 13–34. [Google Scholar] [CrossRef]
- Toulany, M. Targeting DNA double-strand breaks repair pathways to improve radiotherapy. Genes 2019, 10, 25. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making safe to play with knives. Mol. Cell 2010, 22, 179–204. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Kochan, J.A.; Desclos, E.C.B.; Bosch, R.; Meister, L.; Vriend, L.E.M.; van Attikum, H.; Krawczyk, P.M. Meta-analysis of DNA double-strand break response kinetics. Nucleic Acids Res. 2017, 45, 12625–12637. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. Protein dynamics: Implications for nuclear architecture and gene expression. Science 2001, 291, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Toschi, L. Changes in cyclin/proliferating cell nuclear antigen distribution during DNA repair synthesis. J. Cell Biol. 1988, 107, 1623–1628. [Google Scholar] [CrossRef]
- Prosperi, E.; Stivala, L.A.; Sala, E.; Scovassi, A.I.; Bianchi, L. Proliferating Cell Nuclear Antigen Complex Formation Induced by ultraviolet irradiation in human quiescent fibroblasts as detected by immunostaining and flow cytometry. Exp. Cell Res. 1993, 205, 320–325. [Google Scholar] [CrossRef]
- Mirzoeva, O.K.; Petrini, J.H.J. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol. Cell. Biol. 2001, 21, 281–288. [Google Scholar] [CrossRef]
- Tsien, R.Y. The green fluorescent protein. Ann. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef]
- Essers, J.; Houtsmuller, A.B.; van Veelen, L.; Paulusma, C.; Nigg, A.L.; Pastink, A.; Vermeulen, W.; Hoeijmakers, J.H.; Kanaar, R. Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J. 2002, 21, 2030–2037. [Google Scholar] [CrossRef]
- Lukas, C.; Falck, J.; Bartkova, J.; Bartek, J.; Lukas, J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat. Cell Biol. 2003, 5, 255–260. [Google Scholar] [CrossRef]
- Moné, M.J.; Bernas, T.; Dinant, C.; Goedvree, F.A.; Manders, E.M.M.; Volker, M.; Houtsmuller, A.B.; Hoeijmakers, J.H.J.; Vermeulen, W.; Van Driel, R. In vivo dynamics of chromatin-associated complex formation in mammalian nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2004, 101, 15933–15937. [Google Scholar] [CrossRef]
- Tobias, F.; Löb, D.; Lengert, N.; Durante, M.; Drossel, B.; Taucher-Scholz, G.; Jakob, B. Spatiotemporal dynamics of early DNA damage response proteins on complex DNA lesions. PLoS ONE 2013, 8, e57953. [Google Scholar] [CrossRef]
- Karanam, K.; Loewer, A.; Lahav, G. Dynamics of the DNA damage response: Insights from live-cell imaging. Brief. Funct. Genomics 2013, 12, 109–117. [Google Scholar] [CrossRef]
- Prosperi, E. The fellowship of the rings: Distinct pools of proliferating cell nuclear antigen trimer at work. FASEB J. 2006, 20, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Miura, M. Detection of Chromatin-bound PCNA in mammalian cells and its use to study DNA excision repair. J. Radiat. Res. 1999, 40, 1–12. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Scovassi, A.I.; Prosperi, E. Analysis of proliferating cell nuclear antigen (PCNA) associated with DNA excision repair sites in mammalian cells. Methods Mol. Biol. 2006, 314, 457–475. [Google Scholar] [CrossRef]
- Katsumi, S.; Kobayashi, N.; Imoto, K.; Nakagawa, A.; Yamashina, Y.; Muramatsu, T.; Shirai, T.; Miyagawa, S.; Sugiura, S.; Hanaoka, F.; et al. In situ visualization of ultraviolet-light-induced DNA damage repair in locally irradiated human fibroblasts. J. Invest. Dermatol. 2001, 117, 1156–1161. [Google Scholar] [CrossRef]
- Moné, M.J.; Volker, M.; Nikaido, O.; Mullenders, L.H.F.; van Zeeland, A.A.; Verschure, P.J.; Manders, E.M.M.; van Driel, R. Local UV-induced DNA damage in cell nuclei results in local transcription inhibition. EMBO Rep. 2001, 2, 1013–1017. [Google Scholar] [CrossRef]
- Volker, M.; Moné, M.J.; Karmakar, P.; van Hoffen, A.; Schul, W.; Vermeulen, W.; Hoeijmakers, J.H.J.; Van Driel, R.; Van Zeeland, A.A.; Mullenders, L.H.F. Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 2001, 8, 213–224. [Google Scholar] [CrossRef]
- Wakasugi, M.; Kawashima, A.; Morioka, H.; Linn, S.; Sancar, A.; Mori, T.; Nikaido, O.; Matsunaga, T. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J. Biol. Chem. 2002, 277, 1637–1640. [Google Scholar] [CrossRef]
- Oh, K.-S.; Imoto, K.; Boyle, J.; Khan, S.G.; Kraemer, K.H. Influence of XPB helicase on recruitment and redistribution of nucleotide excision repair proteins at sites of UV-induced DNA damage. DNA Repair 2007, 6, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Thorel, F.; Constantinou, A.; Dunand-Sauthier, I.; Nouspikel, T.; Lalle, P.; Raams, A.; Jaspers, N.G.J.; Vermeulen, W.; Shivji, M.K.K.; Wood, R.D.; et al. Definition of a short region of XPG necessary for TFIIH interaction and stable recruitment to sites of UV damage. Mol. Cell. Biol. 2004, 24, 10670–10680. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mocquet, V.; Lainé, J.P.; Riedl, T.; Yajin, Z.; Lee, M.Y.; Egly, J.M. Sequential recruitment of the repair factors during NER: The role of XPG in initiating the resynthesis step. EMBO J. 2008, 27, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Chea, J.; Zhang, S.; Zhao, H.; Zhang, Z.; Lee, E.Y.C.; Darzynkiewicz, Z.; Lee, M.Y.W.T. Spatiotemporal recruitment of human DNA polymerase delta to sites of UV damage. Cell Cycle 2012, 11, 2885–2895. [Google Scholar] [CrossRef]
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 2010, 37, 714–727. [Google Scholar] [CrossRef]
- Green, C.M.; Almouzni, G. Local action of the chromatin assembly factor CAF-1 at sites of nucleotide excision repair in vivo. EMBO J. 2003, 22, 5163–5174. [Google Scholar] [CrossRef]
- Al Rashid, S.T.; Dellaire, G.; Cuddihy, A.; Jalali, F.; Vaid, M.; Coackley, C.; Folkard, M.; Xu, Y.; Chen, B.P.C.; Chen, D.J.; et al. Evidence for the direct binding of phosphorylated p53 to sites of DNA breaks in vivo. Cancer Res. 2005, 65, 10810–10821. [Google Scholar] [CrossRef]
- Fitch, M.E.; Cross, I.V.; Ford, J.M. p53 responsive nucleotide excision repair gene products p48 and XPC, but not p53, localize to sites of UV-irradiation-induced DNA damage, in vivo. Carcinogenesis 2003, 24, 843–850. [Google Scholar] [CrossRef]
- Rubbi, C.P.; Milner, J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 2003, 22, 975–986. [Google Scholar] [CrossRef]
- Polo, S.E.; Roche, D.; Almouzni, G. New histone incorporation marks sites of UV repair in human cells. Cell 2006, 127, 481–493. [Google Scholar] [CrossRef]
- Zhu, Q.; Wei, S.; Sharma, N.; Wani, G.; He, J.; Wani, A.A. Human CRL4DDB2 ubiquitin ligase preferentially regulates post-repair chromatin restoration of H3K56Ac through recruitment of histone chaperon CAF-1. Oncotarget 2017, 8, 104525–104542. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-E.; Zhu, Q.; Wani, G.; Chen, J.; Wani, A.A. UV radiation-induced XPC translocation within chromatin is mediated by damaged-DNA binding protein, DDB2. Carcinogenesis 2004, 25, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Dutto, I.; Cazzalini, O.; Stivala, L.A.; Prosperi, E. An improved method for the detection of nucleotide excision repair factors at local UV DNA damage sites. DNA Repair 2017, 51, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Michaelsen, K.; Krishnaswamy, V.; Pogue, B.W.; Brooks, K.; Defreitas, K.; Shaw, I.; Poplack, S.P.; Paulsen, K.D. Characterization of materials for optimal near-infrared and x-ray imaging of the breast. Biomed. Opt. Express 2012, 3, 2078. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gústafsdóttir, S.M.; Östman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar] [CrossRef]
- Pan, Y.; Sackmann, E.K.; Wypisniak, K.; Hornsby, M.; Datwani, S.S.; Herr, A.E. Determination of equilibrium dissociation constants for recombinant antibodies by high-throughput affinity electrophoresis. Sci. Rep. 2016, 6, 39774. [Google Scholar] [CrossRef]
- Klaesson, A.; Grannas, K.; Ebai, T.; Heldin, J.; Koos, B.; Leino, M.; Raykova, D.; Oelrich, J.; Arngården, L.; Söderberg, O.; et al. Improved efficiency of in situ protein analysis by proximity ligation using UnFold probes. Sci. Rep. 2018, 8, 5400. [Google Scholar] [CrossRef]
- Burns, T.J.; Frei, A.P.; Gherardini, P.F.; Bava, F.A.; Batchelder, J.E.; Yoshiyasu, Y.; Yu, J.M.; Groziak, A.R.; Kimmey, S.C.; Gonzalez, V.D.; et al. High-throughput precision measurement of subcellular localization in single cells: High-throughput precision measurement of SLA. Cytometry 2017, 91, 180–189. [Google Scholar] [CrossRef]
- Serebryannyy, L.A.; Misteli, T. HiPLA: High-throughput imaging proximity ligation assay. Methods 2019, 157, 80–87. [Google Scholar] [CrossRef]
- Debaize, L.; Jakobczyk, H.; Rio, A.-G.; Gandemer, V.; Troadec, M.-B. Optimization of proximity ligation assay (PLA) for detection of protein interactions and fusion proteins in non-adherent cells: Application to pre-B lymphocytes. Mol. Cytogenet. 2017, 10, 27. [Google Scholar] [CrossRef]
- Bahjat, M.; Bloedjes, T.A.; van der Veen, A.; de Wilde, G.; Maas, C.; Guikema, J.E.J. Detection and visualization of DNA Damage-induced protein complexes in suspension cell cultures using the proximity ligation assay. JoVE 2017, 124, e55703. [Google Scholar] [CrossRef]
- Rassoolzadeh, H.; Coucoravas, C.; Farnebo, M. The proximity ligation assay reveals that at DNA double-strand breaks WRAP53β associates with γH2AX and controls interactions between RNF8 and MDC1. Nucleus 2015, 6, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Marin-Vicente, C.; Domingo-Prim, J.; Eberle, A.B.; Visa, N. RRP6/EXOSC10 is required for the repair of DNA double-strand breaks by homologous recombination. J. Cell Sci. 2015, 128, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, R.; Xu, K.; Ding, S.; Li, J.; Baek, G.; Ramanand, S.G.; Ding, S.; Liu, Z.; Gao, Y.; et al. Androgen receptor variants mediate DNA repair after prostate cancer irradiation. Cancer Res. 2017, 77, 4745–4754. [Google Scholar] [CrossRef]
- Song, K.-H.; Jung, S.-Y.; Park, J.-I.; Ahn, J.; Park, J.K.; Um, H.-D.; Park, I.-C.; Hwang, S.-G.; Ha, H.; Song, J.-Y. Inhibition of karyopherin-α2 augments radiation-induced cell death by perturbing BRCA1-mediated DNA repair. Int. J. Mol. Sci. 2019, 20, 2843. [Google Scholar] [CrossRef]
- Galbiati, A.; Beauséjour, C.; d’Adda di Fagagna, F. A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell 2017, 16, 422–427. [Google Scholar] [CrossRef]
- Roy, S.; Luzwick, J.W.; Schlacher, K. SIRF: Quantitative in situ analysis of protein interactions at DNA replication forks. J. Cell Biol. 2018, 217, 1521–1536. [Google Scholar] [CrossRef]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef]
- Weibrecht, I.; Gavrilovic, M.; Lindbom, L.; Landegren, U.; Wählby, C.; Söderberg, O. Visualising individual sequence-specific protein–DNA interactions in situ. New Biotechnol. 2012, 29, 589–598. [Google Scholar] [CrossRef]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef]
- Cremer, C.; Cremer, T.; Fukuda, M.; Nakanishi, K. Detection of laser-UV microirradiation-induced DNA photolesions by immunofluorescent staining. Hum. Genet. 1980, 54, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, S.; Walter, J.; Shinohara, A.; Kamada, N.; Cremer, T. Rad51 Accumulation at sites of DNA damage and in postreplicative chromatin. J. Cell Biol. 2000, 150, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Dinant, C.; de Jager, M.; Essers, J.; van Cappellen, W.A.; Kanaar, R.; Houtsmuller, A.B.; Vermeulen, W. Activation of multiple DNA repair pathways by sub-nuclear damage induction methods. J. Cell Sci. 2007, 120, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Houtsmuller, A.B.; Vermeulen, W. Macromolecular dynamics in living cell nuclei revealed by fluorescence redistribution after photobleaching. Histochem. Cell Biol. 2001, 115, 13–21. [Google Scholar] [CrossRef]
- Vermeulen, W. Dynamics of mammalian NER proteins. DNA Repair 2011, 10, 760–771. [Google Scholar] [CrossRef]
- Lan, L.; Nakajima, S.; Oohata, Y.; Takao, M.; Okano, S.; Masutani, M.; Wilson, S.H.; Yasui, A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 13738–13743. [Google Scholar] [CrossRef]
- Luijsterburg, M.S.; von Bornstaedt, G.; Gourdin, A.M.; Politi, A.Z.; Moné, M.J.; Warmerdam, D.O.; Goedhart, J.; Vermeulen, W.; van Driel, R.; Höfer, T. Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J. Cell Biol. 2010, 189, 445–463. [Google Scholar] [CrossRef]
- Cazzalini, O.; Sommatis, S.; Tillhon, M.; Dutto, I.; Bachi, A.; Rapp, A.; Nardo, T.; Scovassi, A.I.; Necchi, D.; Cardoso, M.C.; et al. CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res. 2014, 42, 8433–8448. [Google Scholar] [CrossRef]
- Lengert, L.; Lengert, N.; Drossel, B.; Cardoso, M.C.; Muster, B.; Nowak, D.; Rapp, A. Discrimination of kinetic models by a combination of microirradiation and fluorescence photobleaching. Biophys. J. 2015, 109, 1551–1564. [Google Scholar] [CrossRef][Green Version]
- Perucca, P.; Cazzalini, O.; Mortusewicz, O.; Necchi, D.; Savio, M.; Nardo, T.; Stivala, L.A.; Leonhardt, H.; Cardoso, M.C.; Prosperi, E. Spatiotemporal dynamics of p21 CDKN1A protein recruitment to DNA-damage sites and interaction with proliferating cell nuclear antigen. J. Cell Sci. 2006, 119, 1517–1527. [Google Scholar] [CrossRef]
- Mortusewicz, O.; Leonhardt, H. XRCC1 and PCNA are loading platforms with distinct kinetic properties and different capacities to respond to multiple DNA lesions. BMC Mol. Biol. 2007, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Leonhardt, H.; Cardoso, M.C. Spatiotemporal dynamics of regulatory protein recruitment at DNA damage sites. J. Cell. Biochem. 2008, 104, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Campalans, A.; Kortulewski, T.; Amouroux, R.; Menoni, H.; Vermeulen, W.; Radicella, J.P. Distinct spatiotemporal patterns and PARP dependence of XRCC1 recruitment to single-strand break and base excision repair. Nucleic Acids Res. 2013, 41, 3115–3129. [Google Scholar] [CrossRef]
- Mortusewicz, O.; Amé, J.-C.; Schreiber, V.; Leonhardt, H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007, 35, 7665–7675. [Google Scholar] [CrossRef]
- Strickfaden, H.; McDonald, D.; Kruhlak, M.J.; Haince, J.-F.; Th’ng, J.P.H.; Rouleau, M.; Ishibashi, T.; Corry, G.N.; Ausio, J.; Underhill, D.A.; et al. Poly(ADP-ribosyl)ation-dependent transient chromatin decondensation and histone displacement following laser microirradiation. J. Biol. Chem. 2016, 291, 1789–1802. [Google Scholar] [CrossRef]
- Garbrecht, J.; Hornegger, H.; Herbert, S.; Kaufmann, T.; Gotzmann, J.; Elsayad, K.; Slade, D. Simultaneous dual-channel imaging to quantify interdependent protein recruitment to laser-induced DNA damage sites. Nucleus 2018, 9, 474–491. [Google Scholar] [CrossRef]
- Haince, J.-F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.-Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef]
- Weterings, E.; Verkaik, N.S.; Keijzers, G.; Florea, B.I.; Wang, S.-Y.; Ortega, L.G.; Uematsu, N.; Chen, D.J.; van Gent, D.C. The Ku80 carboxy terminus stimulates joining and artemis-mediated processing of DNA ends. Mol. Cell. Biol. 2009, 29, 1134–1142. [Google Scholar] [CrossRef]
- Reynolds, P.; Anderson, J.A.; Harper, J.V.; Hill, M.A.; Botchway, S.W.; Parker, A.W.; O’Neill, P. The dynamics of Ku70/80 and DNA-PKcs at DSBs induced by ionizing radiation is dependent on the complexity of damage. Nucleic Acids Res. 2012, 40, 10821–10831. [Google Scholar] [CrossRef]
- Mortusewicz, O.; Rothbauer, U.; Cardoso, M.C.; Leonhardt, H. Differential recruitment of DNA Ligase I and III to DNA repair sites. Nucleic Acids Res. 2006, 34, 3523–3532. [Google Scholar] [CrossRef]
- Gassman, N.R.; Holton, N.W. Targets for repair: Detecting and quantifying DNA damage with fluorescence-based methodologies. Curr. Opin. Biotech. 2019, 55, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, R.S.; Karemore, G.; Gudjonsson, T.; Rask, M.-B.; Neumann, B.; Hériché, J.-K.; Pepperkok, R.; Ellenberg, J.; Gerlich, D.W.; Lukas, J.; et al. Profiling DNA damage response following mitotic perturbations. Nat. Commun. 2016, 7, 13887. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.S.; Kohler, R.H.; Landon, M.; Giedt, R.; Weissleder, R. Single cell resolution in vivo imaging of DNA damage following PARP inhibition. Sci. Rep. 2015, 5, 10129. [Google Scholar] [CrossRef] [PubMed]
- Steurer, B.; Turkyilmaz, Y.; Van Toorn, M.; Van Leeuwen, W.; Escudero-Ferruz, P.; Marteijn, J.A. Fluorescently-labelled CPD and 6-4PP photolyases: New tools for live-cell DNA damage quantification and laser-assisted repair. Nucleic Acids Res. 2019, 47, 3536–3549. [Google Scholar] [CrossRef]
- Woodrick, J.; Gupta, S.; Khatkar, P.; Dave, K.; Levashova, D.; Choudhury, S.; Elias, H.; Saha, T.; Mueller, S.; Roy, R. A novel method for monitoring functional lesion-specific recruitment of repair proteins in live cells. Mutat. Res. 2015, 775, 48–58. [Google Scholar] [CrossRef][Green Version]
- Herce, H.D.; Deng, W.; Helma, J.; Leonhardt, H.; Cardoso, M.C. Visualization and targeted disruption of protein interactions in living cells. Nat. Commun. 2013, 4, 2660. [Google Scholar] [CrossRef]
- Lan, L.; Nakajima, S.; Wei, L.; Sun, L.; Hsieh, C.-L.; Sobol, R.W.; Bruchez, M.; Van Houten, B.; Yasui, A.; Levine, A.S. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Res. 2014, 42, 2330–2345. [Google Scholar] [CrossRef]
- Smith, R.; Lebeaupin, T.; Juhász, S.; Chapuis, C.; D’Augustin, O.; Dutertre, S.; Burkovics, P.; Biertümpfel, C.; Timinszky, G.; Huet, S. Poly(ADP-ribose)-dependent chromatin unfolding facilitates the association of DNA-binding proteins with DNA at sites of damage. Nucleic Acids Res. 2019, in press. [Google Scholar] [CrossRef]
- Reynolds, P.; Botchway, S.W.; Parker, A.W.; O’Neill, P. Spatiotemporal dynamics of DNA repair proteins following laser microbeam induced DNA damage – When is a DSB not a DSB? Mutat. Res. 2013, 756, 14–20. [Google Scholar] [CrossRef]
- Muster, B.; Rapp, A.; Cardoso, M.C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics 2017, 4, 47–68. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R.; Angelov, D. Biphotonic ionization of DNA: From model studies to cell. Photochem. Photobiol. 2019, 95, 59–72. [Google Scholar] [CrossRef]
- Jakob, B.; Rudolph, J.H.; Gueven, N.; Lavin, M.F.; Taucher-Scholz, G. Live cell imaging of heavy-ion-induced radiation responses by beamline microscopy. Radiat. Res. 2005, 163, 681–690. [Google Scholar] [CrossRef]
- Tsien, R.Y.; Ernst, L.; Waggoner, A. Fluorophores for confocal microscopy: Photophysics and photochemistry. In Handbook of Biological Confocal Microscopy; Pawley, J.B., Ed.; Springer: Boston, MA, USA, 2006; pp. 338–352. ISBN 978-0-387-25921-5. [Google Scholar] [CrossRef]
- Bernas, T.; Zarebski, M.; Cook, R.R.; Dobrucki, J.W. Minimizing photobleaching during confocal microscopy of fluorescent probes bound to chromatin: Role of anoxia and photon flux. J. Microsc. 2004, 215, 281–296. [Google Scholar] [CrossRef]
- Bernas, T.; Robinson, J.P.; Asem, E.K.; Rajwa, B. Loss of image quality in photobleaching during microscopic imaging of fluorescent probes bound to chromatin. J. Biomed. Opt. 2005, 10, 064015. [Google Scholar] [CrossRef]
- Anonymous. Artifacts of light. Nat. Methods 2013, 10, 1135. [Google Scholar] [CrossRef]
- Daddysman, M.K.; Tycon, M.A.; Fecko, C.J. Photoinduced damage resulting from fluorescence imaging of live cells. In Photoswitching Proteins; Cambridge, S., Ed.; Springer: New York, NY, USA, 2014; Volume 1148, pp. 1–17. ISBN 978-1-4939-0469-3. [Google Scholar] [CrossRef]
- Vogel, A.; Noack, J.; Hüttman, G.; Paltauf, G. Mechanisms of femtosecond laser nanosurgery of cells and tissues. Appl. Phys. B 2005, 81, 1015–1047. [Google Scholar] [CrossRef]
- Laissue, P.P.; Alghamdi, R.A.; Tomancak, P.; Reynaud, E.G.; Shroff, H. Assessing phototoxicity in live fluorescence imaging. Nat. Methods 2017, 14, 657–661. [Google Scholar] [CrossRef]
- Bogdanov, A.M.; Bogdanova, E.A.; Chudakov, D.M.; Gorodnicheva, T.V.; Lukyanov, S.; Lukyanov, K.A. Cell culture medium affects GFP photostability: A solution. Nat. Methods 2009, 6, 859–860. [Google Scholar] [CrossRef]
- Stockley, J.H.; Evans, K.; Matthey, M.; Volbracht, K.; Agathou, S.; Mukanowa, J.; Burrone, J.; Káradóttir, R.T. Surpassing light-induced cell damage in vitro with novel cell culture media. Sci. Rep. 2017, 7, 849. [Google Scholar] [CrossRef]
- Bogdanov, A.M.; Kudryavtseva, E.I.; Lukyanov, K.A. Anti-fading media for live cell GFP imaging. PLoS ONE 2012, 7, e53004. [Google Scholar] [CrossRef]
- Magidson, V.; Khodjakov, A. Circumventing photodamage in live-cell microscopy. Methods Cell Biol. 2013, 114, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, C.; Wee, T.-L.; Duh, Y.-R.; Couto, M.P.; Ardakani, K.H.; Brown, C.M. Excitation light dose engineering to reduce photo-bleaching and photo-toxicity. Sci. Rep. 2016, 6, 30892. [Google Scholar] [CrossRef] [PubMed]
- Icha, J.; Weber, M.; Waters, J.C.; Norden, C. Phototoxicity in live fluorescence microscopy, and how to avoid it. BioEssays 2017, 39, 1700003. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ticli, G.; Prosperi, E. In Situ Analysis of DNA-Protein Complex Formation upon Radiation-Induced DNA Damage. Int. J. Mol. Sci. 2019, 20, 5736. https://doi.org/10.3390/ijms20225736
Ticli G, Prosperi E. In Situ Analysis of DNA-Protein Complex Formation upon Radiation-Induced DNA Damage. International Journal of Molecular Sciences. 2019; 20(22):5736. https://doi.org/10.3390/ijms20225736
Chicago/Turabian StyleTicli, Giulio, and Ennio Prosperi. 2019. "In Situ Analysis of DNA-Protein Complex Formation upon Radiation-Induced DNA Damage" International Journal of Molecular Sciences 20, no. 22: 5736. https://doi.org/10.3390/ijms20225736
APA StyleTicli, G., & Prosperi, E. (2019). In Situ Analysis of DNA-Protein Complex Formation upon Radiation-Induced DNA Damage. International Journal of Molecular Sciences, 20(22), 5736. https://doi.org/10.3390/ijms20225736