Genome-Wide Characterization, Expression Profile Analysis of WRKY Family Genes in Santalum album and Functional Identification of Their Role in Abiotic Stress


Abstract
1. Introduction
2. Results
2.1. Identification and Subcellular Localization of SaWRKYs
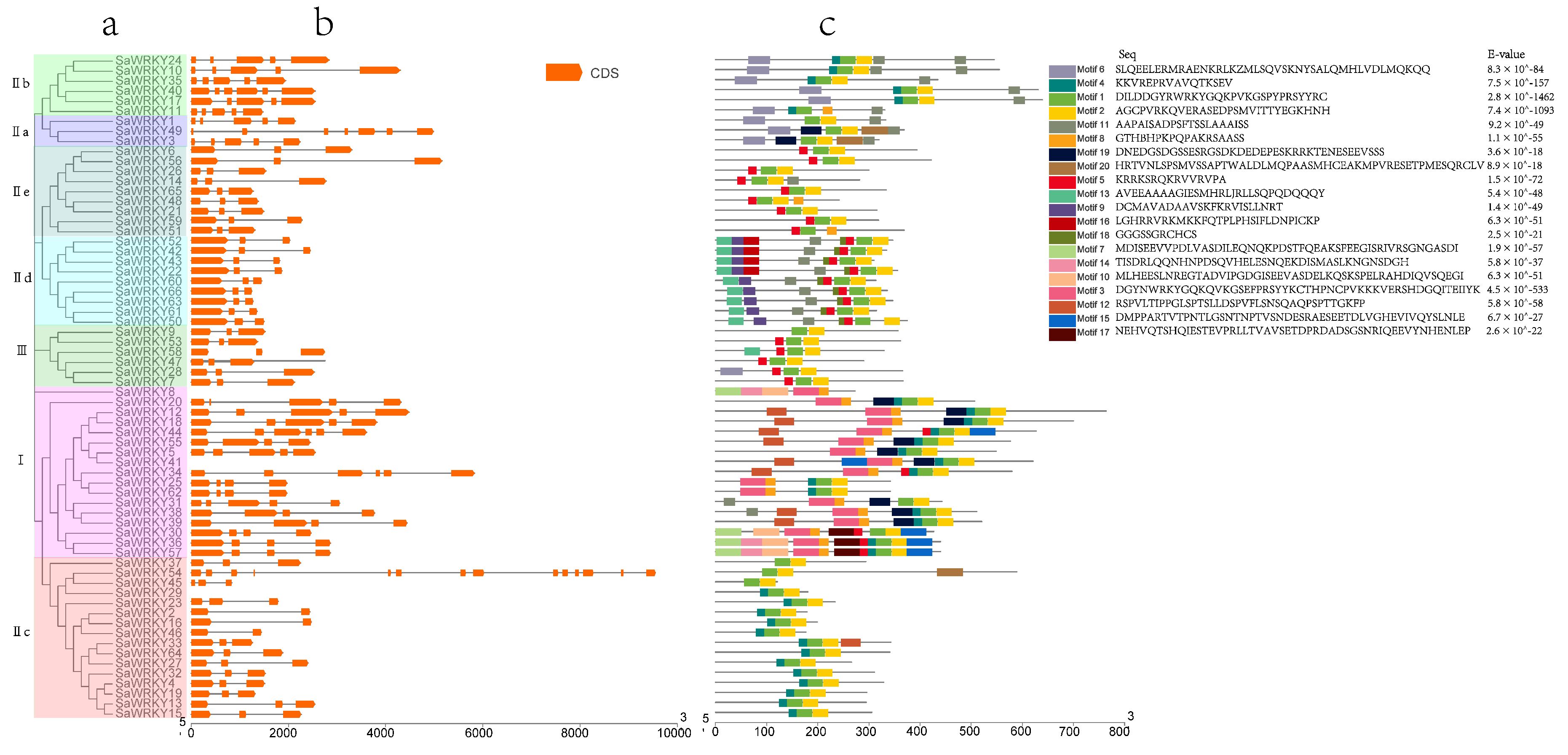
2.2. Phylogenetic Analysis of SaWRKY Proteins
2.3. Exon-Intron Organization of SaWRKY Genes
2.4. Motif Composition of SaWRKY Proteins
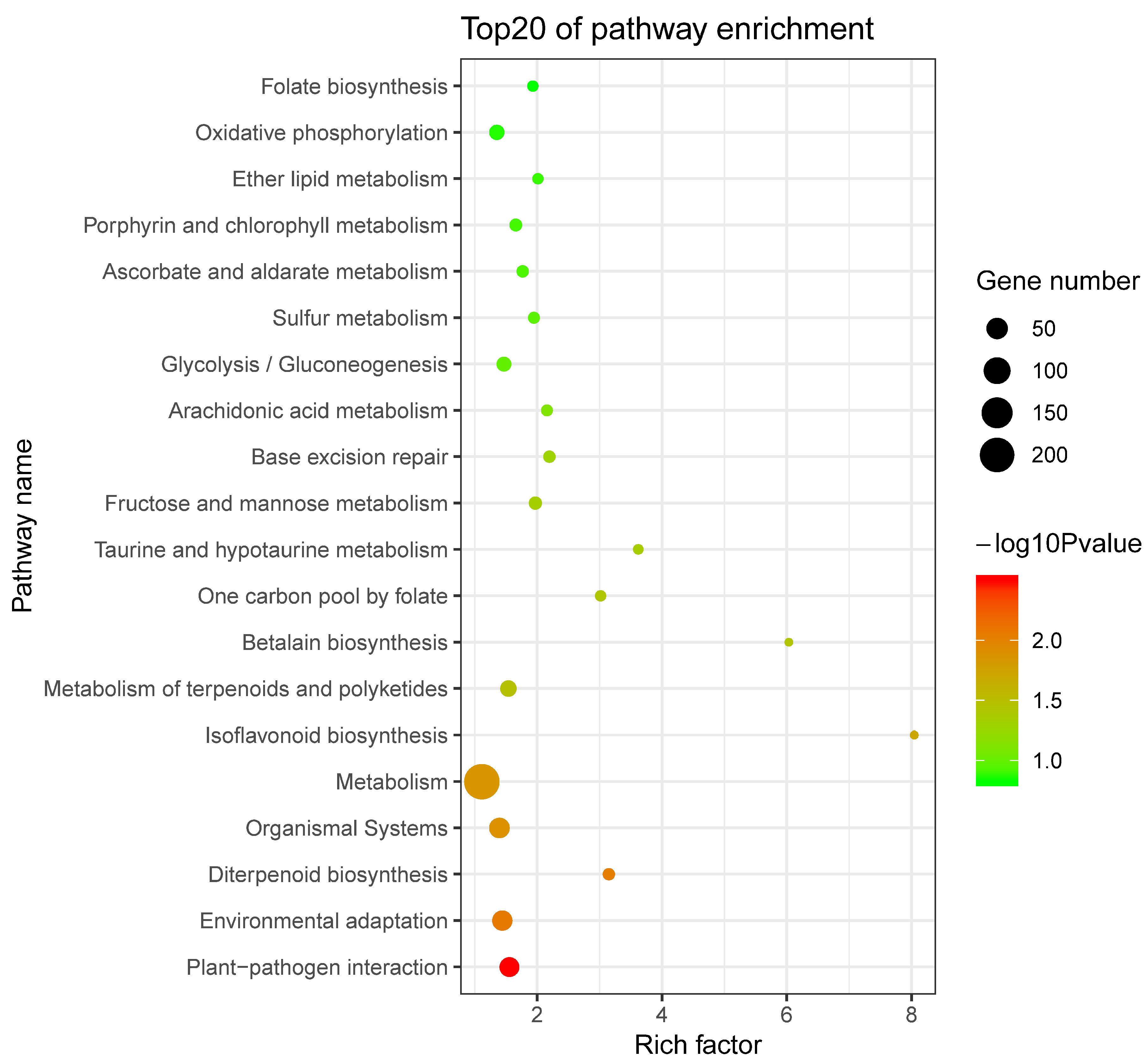
2.5. Prediction and Functional Enrichment Analysis of Potential SaWRKY Target Genes
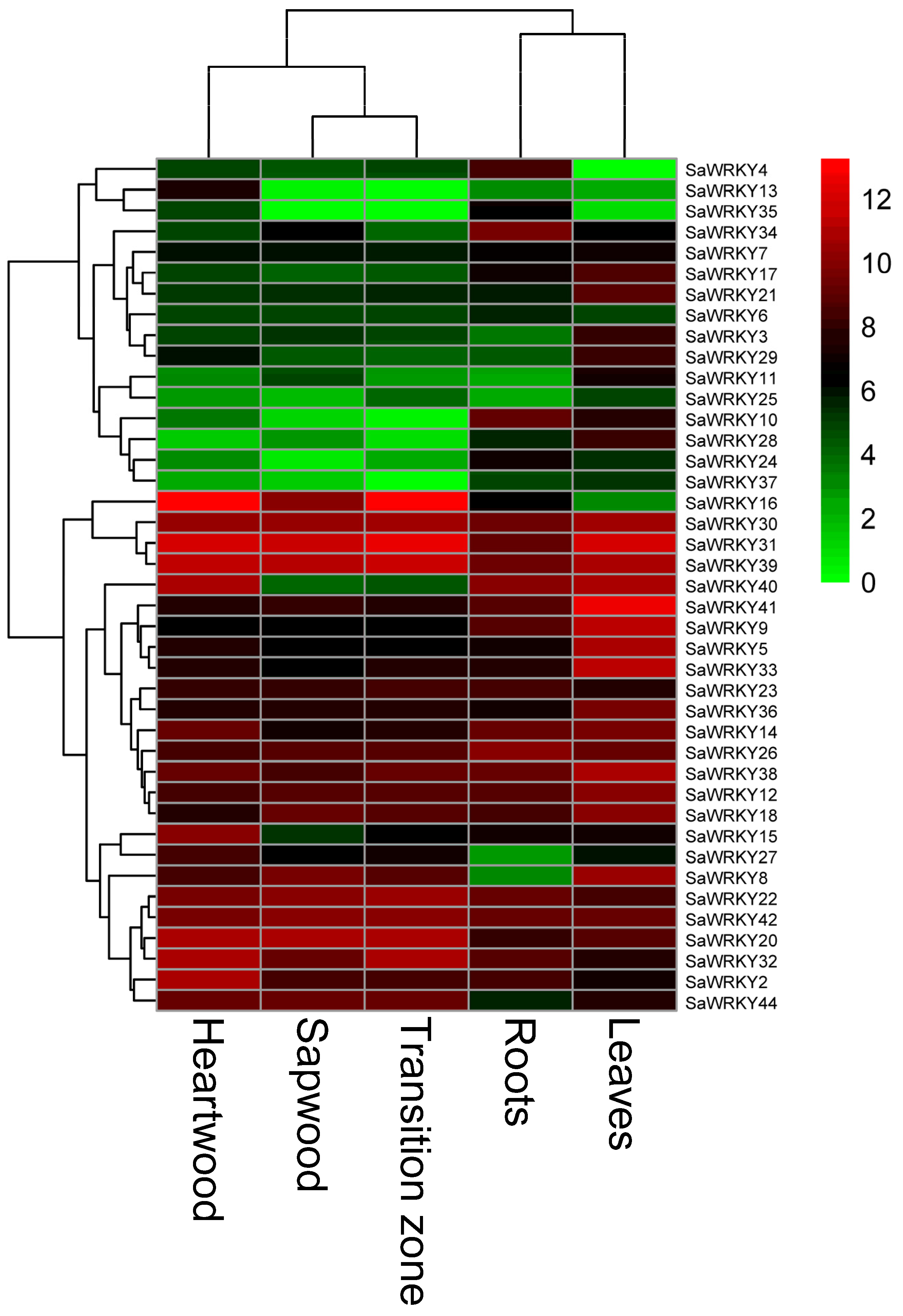
2.6. Expression Patterns of SaWRKY Genes in Different Tissues
2.7. Expression Profiles of SaWRKY Genes in Response to SA and MeJA
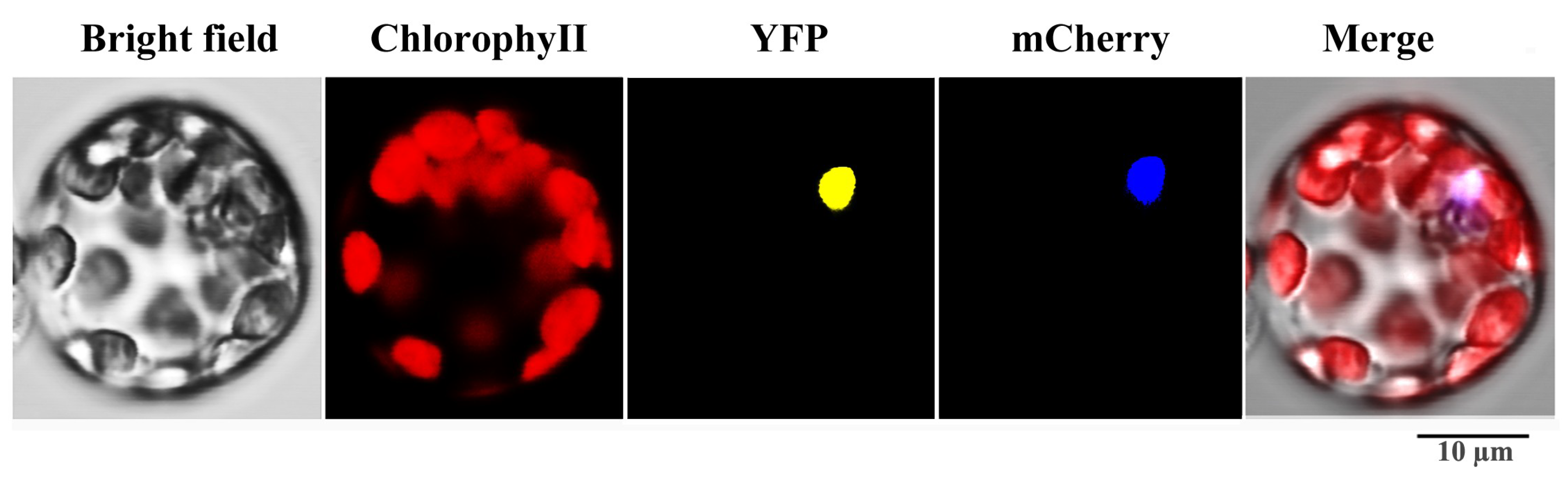
2.8. Characterization, Tissue Expression Patterns and Subcellular Localization of SaWRKY1
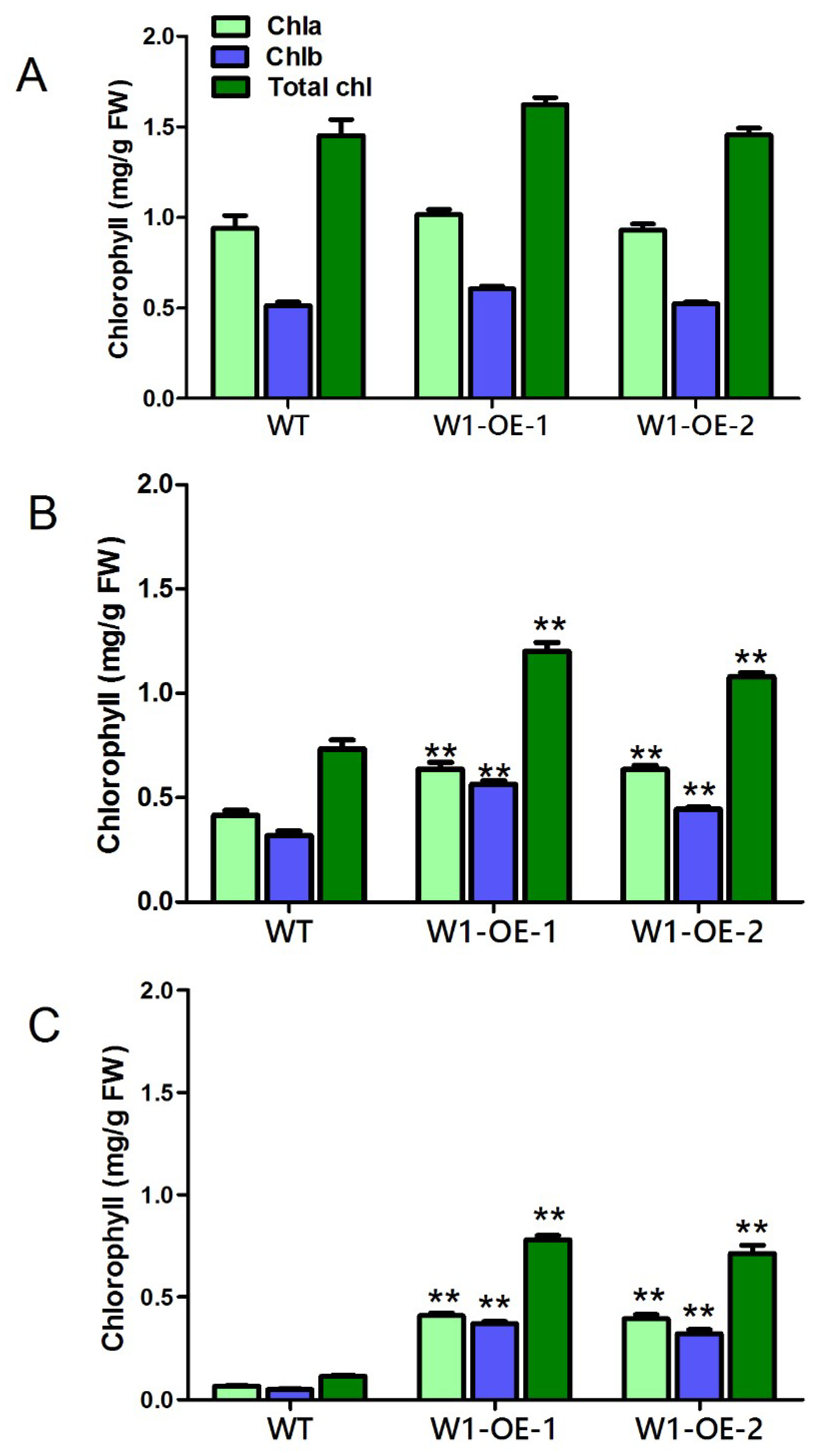
2.9. Overexpression of SaWRKY1 Enhances Salinity Tolerance in Transgenic Arabidopsis Plants
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Hormone Treatments
4.2. Identification of WRKY Genes in S. album and Prediction of Subcellular Localization
4.3. Construction of Phylogenetic Tree and Conserved Motif Analysis of SaWRKY Proteins
4.4. Identification and Functional Enrichment Analysis of Potential SaWRKY Target Genes
4.5. Gene Expression Analysis by Quantitative real-Time PCR (RT-qPCR)
4.6. Cluster Analysis of Expression Data
4.7. Cloning and Subcellular Localization of SaWRKY1
4.8. Arabidopsis thaliana Transformation and Salinity Stress
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.-Z.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis transcription factors: Genome-Wide comparative analysis among eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef]
- Ulker, B.; Somssich, I.E. WRKY transcription factors: From DNA binding towards biological function. Curr. Opin. Plant Biol. 2004, 7, 491–498. [Google Scholar] [CrossRef]
- Pan, Y.J.; Cho, C.C.; Kao, Y.Y.; Sun, C.H. A novel WRKY-Like protein involved in transcriptional activation of cyst wall protein genes in Giardia lamblia. J. Biol. Chem. 2009, 284, 17975–17988. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, S.; Nakamura, K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5' upstream regions of genes coding for sporamin and β-amylase from sweet potato. Mol. Gen. Genet. 1994, 244, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Eulgem, T.; Rushton, P.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Ross, C.A.; Liu, Y.; Shen, Q.J. The WRKY gene family in rice (Oryza sativa). J. Integr. Plant Biol. 2007, 49, 827–842. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Duan, Y.; Yin, J.; Ye, S.; Zhu, J.; Zhang, F.; Lu, W.; Fan, D.; Luo, K. Genome-Wide identification and characterization of the Populus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J. Exp. Bot. 2014, 65, 6629–6644. [Google Scholar] [CrossRef]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef]
- Rinerson, C.I.; Scully, E.D.; Palmer, N.A.; Donze-Reiner, T.; Rabara, R.C.; Tripathi, P.; Shen, Q.J.; Sattler, S.E.; Rohila, J.S.; Sarath, G.; et al. The WRKY transcription factor family and senescence in switchgrass. BMC Genom. 2015, 16, 912. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Vinod, K.; Zheng, Z.; Fan, B.; Chen, Z. Roles of Arabidopsis WRKY3 and WRKY4 transcription factors in plant responses to pathogens. BMC Plant Biol. 2008, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Lai, Z.; Fan, B.; Chen, Z. Arabidopsis WRKY38 and WRKY62 transcription factors interact with histone deacetylase 19 in basal defense. Plant Cell 2008, 20, 2357–2371. [Google Scholar] [CrossRef]
- Liu, X.Q.; Bai, X.Q.; Qian, Q.; Wang, X.J.; Chen, M.S.; Chu, C.C. OsWRKY03, a rice transcriptional activator that functions in defense signaling pathway upstream of OsNPR1. Cell Res. 2005, 15, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, R.; Jiang, S.Y.; Kumar, N.; Venkatesh, P.N.; Ramachandran, S. A comprehensive transcriptional profiling of the WRKY gene family in rice under various abiotic and phytohormone treatments. Plant Cell Physiol. 2008, 49, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, Y.; Li, S.; Zhang, L.; Zou, C.; Yu, D. The role of WRKY transcription factors in plant abiotic stresses. Biochim. Biophys. Acta 2012, 1819, 120–128. [Google Scholar] [CrossRef]
- Ding, Z.J.; Yan, J.Y.; Li, G.X.; Wu, Z.C.; Zhang, S.Q.; Zheng, S.J. WRKY41 controls Arabidopsis seed dormancy via direct regulation of ABI3 transcript levels not downstream of ABA. Plant J. 2014, 79, 810–823. [Google Scholar] [CrossRef]
- Guan, Y.; Meng, X.; Khanna, R.; LaMontagne, E.; Liu, Y.; Zhang, S. Phosphorylation of a WRKY transcription factor by MAPKs is required for pollen development and function in Arabidopsis. PLoS Genet. 2014, 10, e1004384. [Google Scholar] [CrossRef]
- Devaiah, B.N.; Karthikeyan, A.S.; Raghothama, K.G. WRKY75 transcription factor is a modulator of phosphate acquisition and root development in Arabidopsis. Plant Physiol. 2007, 143, 1789–1801. [Google Scholar] [CrossRef]
- Jiang, Y.; Liang, G.; Yang, S.; Yu, D. Arabidopsis WRKY57 functions as a node of convergence for jasmonic acid- and auxin-mediated signaling in jasmonic acid-induced leaf senescence. Plant Cell 2014, 26, 230–245. [Google Scholar] [CrossRef]
- Jones, C.G.; Keeling, C.I.; Ghisalberti, E.L.; Barbour, E.L.; Plummer, J.A.; Bohlmann, J. Isolation of cDNAs and functional characterisation of two multi-product terpene synthase enzymes from sandalwood, Santalum album L. Arch. Biochem. Biophys. 2008, 477, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Ito, H.; Hayashi, K.; Hasegawa, T.; Machiguchi, T.; Yoshida, T. Aromatic constituents from the heartwood of Santalum album L. Chem. Pharm. Bull. 2005, 53, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Baldovini, N.; Delasalle, C.; Joulain, D. Phytochemistry of the heartwood from fragrant Santalum species: A review. Flavour Fragr. J. 2011, 26, 7–26. [Google Scholar] [CrossRef]
- Rashkow, E.D. Perfumed the axe that laid it low: The endangerment of sandalwood in southern India. Indian Econ. Soc. Hist. Rev. 2014, 51, 41–70. [Google Scholar] [CrossRef]
- Teixeira da Silva, J.A.; Kher, M.M.; Soner, D.; Page, T.; Zhang, X.; Nataraj, M.; Ma, G. Sandalwood: Basic biology, tissue culture, and genetic transformation. Planta 2016, 243, 847–887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Teixeira da Silva, J.A.; Niu, M.; Li, M.; He, C.; Zhao, J.; Zeng, S.; Duan, J.; Ma, G. Physiological and transcriptomic analyses reveal a response mechanism to cold stress in Santalum album L. leaves. Sci. Rep. 2017, 7, 42165. [Google Scholar] [CrossRef]
- Teixeira da Silva, J.A.; Kher, M.M.; Soner, D.; Nataraj, M.; Dobránszki, J.; Millar, M.A. Santalum molecular biology: Molecular markers for genetic diversity, phylogenetics and taxonomy, and genetic transformation. Agrofor. Syst. 2017, 9, 1301–1315. [Google Scholar] [CrossRef]
- Zhang, X.; Berkowitz, O.; Teixeira da Silva, J.A.; Zhang, M.; Ma, G.; Whelan, J.; Duan, J. RNA-Seq analysis identifies key genes associated with haustorial development in the root hemiparasite Santalum album. Front. Plant Sci. 2015, 6, 661. [Google Scholar] [CrossRef]
- Celedon, J.M.; Chiang, A.; Yuen, M.M.; Diaz-Chavez, M.L.; Madilao, L.L.; Finnegan, P.M.; Barbour, E.L.; Bohlmann, J. Heartwood-Specific transcriptome and metabolite signatures of tropical sandalwood (Santalum album) reveal the final step of (Z)-santalol fragrance biosynthesis. Plant J. 2016, 86, 289–299. [Google Scholar] [CrossRef]
- Mahesh, H.B.; Subba, P.; Advani, J.; Shirke, M.D.; Loganathan, R.M.; Chandana, S.L.; Shilpa, S.; Chatterjee, O.; Pinto, S.M.; Prasad, T.S.K.; et al. Multi-omics driven assembly and annotation of the sandalwood (Santalum album) genome. Plant Physiol. 2018, 176, 2772–2788. [Google Scholar] [CrossRef]
- Wan, Y.; Mao, M.; Wan, D.; Yang, Q.; Yang, F.; Mandlaa; Li, G.; Wang, R. Identification of the WRKY gene family and functional analysis of two genes in Caragana intermedia. BMC Plant Biol. 2018, 18, 31. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Shi, H.; Xia, Z.; Tie, W.; Ding, Z.; Yan, Y.; Wang, W.; Hu, W.; Li, K. Genome-wide identification and expression analysis of the WRKY gene family in cassava. Front Plant Sci. 2016, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, I.; de Vos, R.C.; Bones, A.M.; Hall, R.D. Plant molecular stress responses face climate change. Trends Plant Sci. 2010, 15, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Santner, A.; Estelle, M. Recent advances and emerging trends in plant hormone signalling. Nature. 2009, 459, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Halim, V.A.; Vess, A.; Scheel, D.; Rosahl, S. The role of salicylic acid and jasmonic acid in pathogen defence. Plant Biol. 2006, 8, 307–313. [Google Scholar] [CrossRef]
- Miura, K.; Tada, Y. Regulation of water, salinity, and cold stress responses by salicylic acid. Front Plant Sci. 2014, 5, 4. [Google Scholar] [CrossRef]
- Guo, C.; Guo, R.; Xu, X.; Gao, M.; Li, X.; Song, J.; Zheng, Y.; Wang, X. Evolution and expression analysis of the grape (Vitis vinifera L.) WRKY gene family. J. Exp. Bot. 2014, 65, 1513–1528. [Google Scholar] [CrossRef]
- Low, E.T.; Alias, H.; Boon, S.H.; Shariff, E.M.; Tan, C.Y.; Ooi, L.C.; Cheah, S.C.; Raha, A.R.; Wan, K.L.; Singh, R. Oil palm (Elaeis guineensis Jacq.) tissue culture ESTs: Identifying genes associated with callogenesis and embryogenesis. BMC Plant Biol. 2008, 8, 62. [Google Scholar] [CrossRef]
- Dong, Q.L.; Liu, D.D.; An, X.H.; Hu, D.G.; Yao, Y.X.; Hao, Y.J. MdVHP1 encodes an apple vacuolar H+-PPase and enhances stress tolerance in transgenic apple callus and tomato. J. Plant Physiol. 2011, 168, 2124–2133. [Google Scholar] [CrossRef]
- Miao, Y.; Zentgraf, U. The antagonist function of Arabidopsis WRKY53 and ESR/ESP in leaf senescence is modulated by the jasmonic and salicylic acid equilibrium. Plant Cell 2007, 19, 819–830. [Google Scholar] [CrossRef]
- Murray, S.L.; Ingle, R.A.; Petersen, L.N.; Denby, K.J. Basal resistance against Pseudomonas syringae in Arabidopsis involves WRKY53 and a protein with homology to a nematode resistance protein. Mol. Plant-Microbe Interact. 2007, 20, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Zentgraf, U.; Laun, T.; Miao, Y. The complex regulation of WRKY53 during leaf senescence of Arabidopsis thaliana. Eur. J. Cell Biol. 2010, 89, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, D. Activated expression of AtWRKY53 negatively regulates drought tolerance by mediating stomatal movement. Plant Cell Rep. 2015, 34, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, J.; Lin, G.; Wang, A.; Wang, Z.; Lu, G. Overexpression of AtWRKY28 and AtWRKY75 in Arabidopsis enhances resistance to oxalic acid and Sclerotinia sclerotiorum. Plant Cell Rep. 2013, 32, 1589–1599. [Google Scholar] [CrossRef]
- Choi, C.; Park, Y.H.; Kwon, S.I.; Yun, C.; Ahn, I.; Park, S.R.; Hwang, D.-J. Identification of AtWRKY75 as a transcriptional regulator in the defense response to Pcc through the screening of Arabidopsis activation-tagged lines. Plant Biotechnol. Rep. 2013, 8, 183–192. [Google Scholar] [CrossRef]
- Kunkel, B.N.; Brooks, D.M. Cross talk between signaling pathways in pathogen defense. Curr. Opin. Plant Biol. 2002, 5, 325–331. [Google Scholar] [CrossRef]
- Mur, L.A.; Kenton, P.; Atzorn, R.; Miersch, O.; Wasternack, C. The outcomes of concentration-specific interactions between salicylate and jasmonate signaling include synergy, antagonism, and oxidative stress leading to cell death. Plant Physiol. 2006, 140, 249–262. [Google Scholar] [CrossRef]
- Mur, L.A.; Prats, E.; Pierre, S.; Hall, M.A.; Hebelstrup, K.H. Integrating nitric oxide into salicylic acid and jasmonic acid/ethylene plant defense pathways. Front Plant Sci. 2013, 4, 215. [Google Scholar] [CrossRef]
- Karlidag, H.; Yildirim, E.; Turan, M. Salicylic acid ameliorates the adverse effect of salt stress on strawberry. Sci. Agric. 2009, 66, 180–187. [Google Scholar] [CrossRef]
- Wasti, S.; Mimouni, H.; Smiti, S.; Zid, E.; Ben Ahmed, H. Enhanced salt tolerance of tomatoes by exogenous salicylic acid applied through rooting medium. OMICS 2012, 16, 200–207. [Google Scholar] [CrossRef]
- Qiu, Z.; Guo, J.; Zhu, A.; Zhang, L.; Zhang, M. Exogenous jasmonic acid can enhance tolerance of wheat seedlings to salt stress. Ecotoxicol. Environ. Saf. 2014, 104, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lai, Z.; Shi, J.; Xiao, Y.; Chen, Z.; Xu, X. Roles of arabidopsis WRKY18, WRKY40 and WRKY60 transcription factors in plant responses to abscisic acid and abiotic stress. BMC Plant Biol. 2010, 10, 281. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Raj, S.R.; Jaiswal, P.S.; Patil, V.R.; Punwar, B.S.; Chavda, J.C.; Subhash, N. Effect of plant growth regulators on in vitro plant regeneration of sandalwood (Santalum album L.) via organogenesis. Agrofor. Syst. 2015, 90, 281–288. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- He, C.; Teixeira da Silva, J.A.; Tan, J.; Zhang, J.; Pan, X.; Li, M.; Luo, J.; Duan, J. A genome-wide identification of the WRKY family genes and a survey of potential WRKY target genes in Dendrobium officinale. Sci. Rep. 2017, 7, 9200. [Google Scholar] [CrossRef] [PubMed]
- Kolosova, N.; Miller, B.; Ralph, S.; Ellis, B.E.; Douglas, C.; Ritland, K.; Bohlmann, J. Isolation of high-quality RNA from gymnosperm and angiosperm trees. BioTechniques 2004, 36, 821–824. [Google Scholar] [CrossRef]
- Yan, H.; Zhang, Y.; Xiong, Y.; Chen, Q.; Liang, H.; Niu, M.; Guo, B.; Li, M.; Zhang, X.; Li, Y.; et al. Selection and validation of novel RT-qPCR reference genes under hormonal stimuli and in different tissues of Santalum album. Sci. Rep. 2018, 8, 17511. [Google Scholar] [CrossRef]
- Yoo, S.D.; Cho, Y.H.; Sheen, J. Arabidopsis mesophyll protoplasts: A versatile cell system for transient gene expression analysis. Nat. Protoc. 2007, 2, 1565–1572. [Google Scholar] [CrossRef]
- Weigel, D.; Glazebrook, J. Transformation of Agrobacterium using the freeze-thaw method. CSH Protoc. 2006, 7, 1031–1036. [Google Scholar] [CrossRef]
- Zhang, X.; Henriques, R.; Lin, S.S.; Niu, Q.W.; Chua, N.H. Agrobacterium-mediated transformation of Arabidopsis thaliana using the floral dip method. Nat. Protoc. 2006, 1, 641–646. [Google Scholar] [CrossRef]
- Porra, R.J.; Thompson, W.A.; Kriedemann, P.E. Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents: Verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. Biochim. Biophys. Acta 1989, 975, 384–394. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | ORF (aa) | Conserved Motif | Zinc-Finger Type | Subcellular Location | Group |
|---|---|---|---|---|---|
| SaWRKY5 | 549 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY8 | 273 | WRKYGQK | C-X4-C-X22-HXH | Golgi | I |
| SaWRKY12 | 764 | WRKYGQK/WRKYGQK | C-X5-C-X23-HXT/C-X4-C-X22-HXH/ C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY18 | 700 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY20 | 507 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY25 | 342 | WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH/ C-X7-C-X23-HXL | Nucleus | I |
| SaWRKY30 | 427 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY31 | 443 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY34 | 580 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY36 | 440 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY38 | 511 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY39 | 521 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY41 | 621 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X5-C-X23-HXH | Nucleus | I |
| SaWRKY44 | 627 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY55 | 577 | WRKYGQK/WRKYGQK | C-X4-C-X22-HXH/C-X4-C-X23-HXH | Nucleus | I |
| SaWRKY1 | 333 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIa |
| SaWRKY3 | 320 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIa |
| SaWRKY49 | 369 | WRKYGQK | C-X5-C-X23-HXH | Chloroplast | IIa |
| SaWRKY10 | 555 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIb |
| SaWRKY11 | 331 | WRKYGQK | C-X6-CX23-HXH | Nucleus | IIb |
| SaWRKY17 | 639 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIb |
| SaWRKY24 | 545 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIb |
| SaWRKY35 | 435 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIb |
| SaWRKY40 | 631 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIb |
| SaWRKY2 | 179 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY4 | 329 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY13 | 295 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY15 | 306 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY16 | 199 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY19 | 296 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY23 | 234 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY27 | 266 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY29 | 181 | WRKYGKK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY32 | 311 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY33 | 343 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY37 | 294 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY45 | 121 | WRKYGHK | C-X4-C-X23-HXY | cytoplasm | IIc |
| SaWRKY46 | 177 | WRKYGQK | C-X4-C-X23-HXH | peroxisome | IIc |
| SaWRKY54 | 589 | WRKYGKK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY62 | 341 | WRKYGQK | C-X4-C-X23-HXH | Nucleus | IIc |
| SaWRKY22 | 356 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IId |
| SaWRKY42 | 335 | WRKYGQK | C-X4-C-X22-HXV/C-X5-C-X23-HXH | Nucleus | IId |
| SaWRKY43 | 310 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IId |
| SaWRKY50 | 375 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IId |
| SaWRKY52 | 347 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IId |
| SaWRKY59 | 314 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IId |
| SaWRKY60 | 314 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IId |
| SaWRKY61 | 315 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IId |
| SaWRKY64 | 336 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IId |
| SaWRKY6 | 394 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIe |
| SaWRKY14 | 282 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIe |
| SaWRKY21 | 316 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIe |
| SaWRKY26 | 300 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIe |
| SaWRKY48 | 242 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIe |
| SaWRKY51 | 369 | WRKYGQK | C-X3-C-X22-HXL/C-X5-C-X23-HXH | Nucleus | IIe |
| SaWRKY56 | 422 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIe |
| SaWRKY58 | 319 | WRKYGQK | C-X5-C-X22-HXS | Nucleus | IIe |
| SaWRKY63 | 334 | WRKYGQK | C-X5-C-X23-HXH | Nucleus | IIe |
| SaWRKY7 | 367 | WRKYGQK | C-X7-C-X23-HXC | Nucleus | III |
| SaWRKY9 | 357 | WRKYGQK | C-X7-C-X23-HXC | Nucleus | III |
| SaWRKY28 | 366 | WRKYGQK | C-X7-C-X23-HXC | Nucleus | III |
| SaWRKY47 | 290 | WRKYGQK | C-X7-C-X23-HXC | Nucleus | III |
| SaWRKY53 | 362 | WRKYGQK | C-X7-C-X22-HXT | Nucleus | III |
| SaWRKY57 | 330 | WRKYGQK | C-X7-C-X23-HXC | Nucleus | III |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, H.; Li, M.; Xiong, Y.; Wu, J.; Teixeira da Silva, J.A.; Ma, G. Genome-Wide Characterization, Expression Profile Analysis of WRKY Family Genes in Santalum album and Functional Identification of Their Role in Abiotic Stress. Int. J. Mol. Sci. 2019, 20, 5676. https://doi.org/10.3390/ijms20225676
Yan H, Li M, Xiong Y, Wu J, Teixeira da Silva JA, Ma G. Genome-Wide Characterization, Expression Profile Analysis of WRKY Family Genes in Santalum album and Functional Identification of Their Role in Abiotic Stress. International Journal of Molecular Sciences. 2019; 20(22):5676. https://doi.org/10.3390/ijms20225676
Chicago/Turabian StyleYan, Haifeng, Mingzhi Li, Yuping Xiong, Jianming Wu, Jaime A. Teixeira da Silva, and Guohua Ma. 2019. "Genome-Wide Characterization, Expression Profile Analysis of WRKY Family Genes in Santalum album and Functional Identification of Their Role in Abiotic Stress" International Journal of Molecular Sciences 20, no. 22: 5676. https://doi.org/10.3390/ijms20225676
APA StyleYan, H., Li, M., Xiong, Y., Wu, J., Teixeira da Silva, J. A., & Ma, G. (2019). Genome-Wide Characterization, Expression Profile Analysis of WRKY Family Genes in Santalum album and Functional Identification of Their Role in Abiotic Stress. International Journal of Molecular Sciences, 20(22), 5676. https://doi.org/10.3390/ijms20225676