Maternal Platelets—Friend or Foe of the Human Placenta?



{kind=link}
{kind=link}
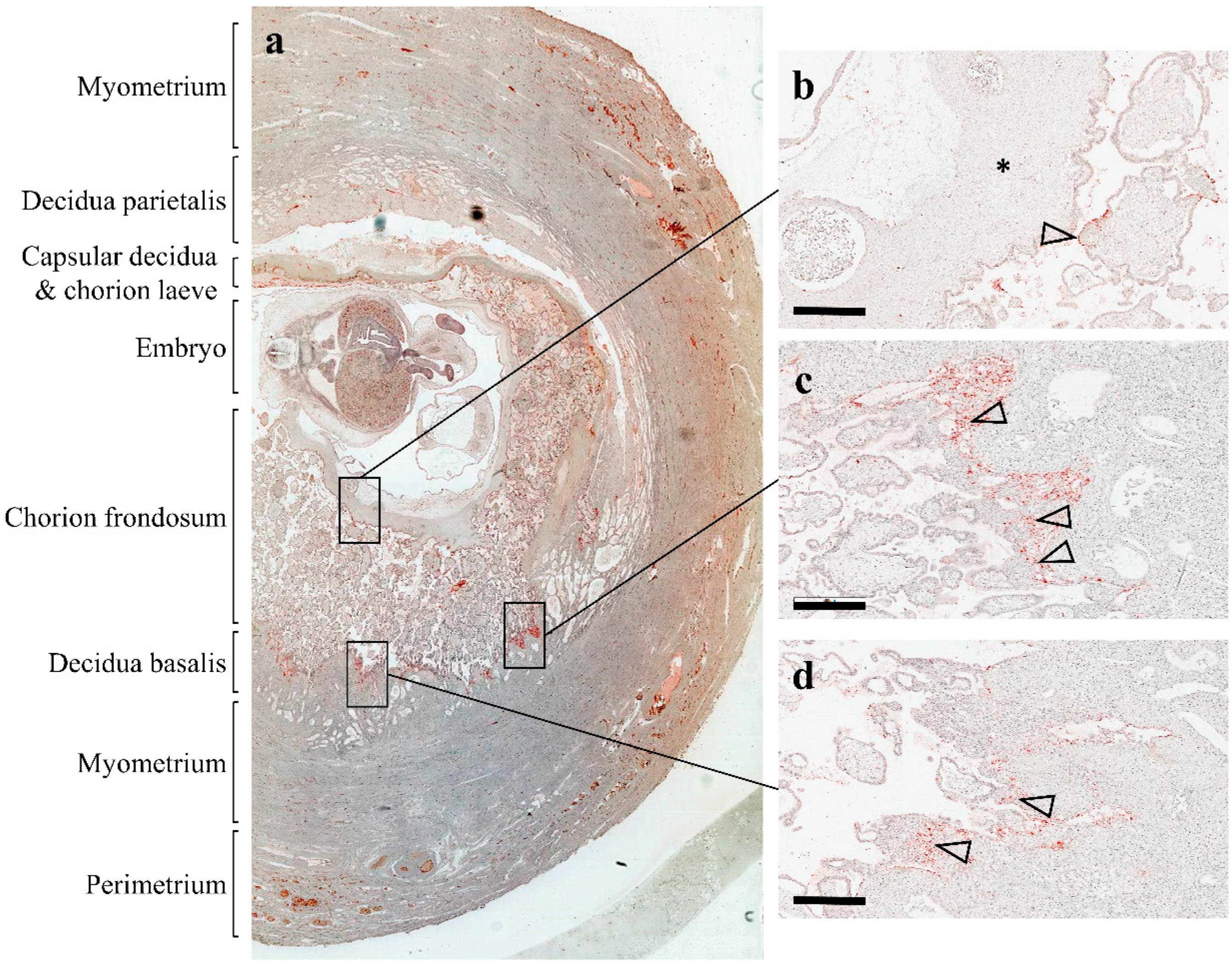{kind=link}
{kind=link}
Abstract
1. Hemochorial Placentation
2. An Effective System for Hemostasis is Key for Hemochorial Placentation
3. Maternal Platelets in Pregnancy
4. Platelets and Their Role in Placenta Development and Function
5. Platelets in Placenta-Associated Pregnancy Pathologies
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| CCL3 | chemokine (C-C motif) ligand 3 (CCL3) |
| CCL5 | chemokine (C-C motif) ligand 5 |
| CCR1 | C-C chemokine receptor type 1 |
| CD146 | cluster of differentiation 146 |
| CD41 | cluster of differentiation 41, integrin alpha-IIb |
| CD42b | cluster of differentiation 42b, platelet glycoprotein Ib alpha chain |
| CX3CL1 | chemokine (C-X3-C motif) ligand 1 |
| CX3CR1 | CX3C chemokine receptor 1 |
| CXCL4 | chemokine (C-X-C motif) ligand 4 |
| EGF | epidermal growth factor |
| EPCR | endothelial protein C receptor |
| EV | extracellular vesicles |
| EVT | extravillous trophoblast |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| HELLP | hemolysis, elevated liver enzymes, low platelet count |
| HLA-G | major histocompatibility complex, class I, G |
| IL-12 | interleukin-12 |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| IL-8 | interleukin-8 |
| IUGR | intrauterine growth restriction |
| MCP-3 | monocyte-chemotactic protein 3 |
| MIP-1α | macrophage inflammatory protein 1-alpha |
| MPV | mean platelet volume |
| p.c. | post conception |
| PAI-1 | plasminogen activator inhibitor-1 |
| PAR | protease activated receptors |
| PDGF | platelet-derived growth factor |
| PE | preeclampsia |
| PSGL-1 | leucocyte P-selectin ligand-1 |
| RANTES | regulated on activation, normal T cell expressed and secreted |
| sFlt-1 | soluble fms-like tyrosine kinase |
| TF | tissue factor |
| TM | thrombomodulin |
| TNF-α | tumor necrosis factor-α |
| TXA2 | thromboxane A2 |
| VEGF | vascular endothelial growth factor |
References
- Benirschke, K.; Burton, G.J.; Baergen, R.N. Pathology of the Human Placenta, 6th ed.; Springer-Verlag: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.P.; Skepper, J.N.; Burton, G.J. Onset of Maternal Arterial Blood Flow and Placental Oxidative Stress. A Possible Factor in Human Early Pregnancy Failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef]
- Jauniaux, E.; Watson, A.; Ozturk, O.; Quick, D.; Burton, G. In-Vivo Measurement of Intrauterine Gases and Acid-Base Values Early in Human Pregnancy. Hum. Reprod. 1999, 14, 2901–2904. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C. Rheological and Physiological Consequences of Conversion of the Maternal Spiral Arteries for Uteroplacental Blood Flow during Human Pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.K. Review: Trophoblast-Vascular Cell Interactions in Early Pregnancy: How to Remodel a Vessel. Placenta 2010, 31, S93–S98. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.D.; Choudhury, R.H.; Matos, P.; Horn, J.A.; Lye, S.J.; Dunk, C.E.; Aplin, J.D.; Jones, R.L.; Harris, L.K. Changes in Vascular Extracellular Matrix Composition during Decidual Spiral Arteriole Remodeling in Early Human Pregnancy. Histol. Histopathol. 2016, 31, 557–571. [Google Scholar] [PubMed]
- Moser, G.; Gauster, M.; Orendi, K.; Glasner, A.; Theuerkauf, R.; Huppertz, B. Endoglandular Trophoblast, an Alternative Route of Trophoblast Invasion? Analysis with Novel Confrontation Co-Culture Models. Hum. Reprod. 2010, 25, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Moser, G.; Weiss, G.; Gauster, M.; Sundl, M.; Huppertz, B. Evidence from the very Beginning: Endoglandular Trophoblasts Penetrate and Replace Uterine Glands in Situ and in Vitro. Hum. Reprod. 2015, 30, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Craven, C.M.; Zhao, L.; Ward, K. Lateral Placental Growth Occurs by Trophoblast Cell Invasion of Decidual Veins. Placenta 2000, 21, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Moser, G.; Huppertz, B. Implantation and Extravillous Trophoblast Invasion: From Rare Archival Specimens to Modern Biobanking. Placenta 2017, 56, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Moser, G.; Drewlo, S.; Huppertz, B.; Armant, D.R. Trophoblast Retrieval and Isolation from the Cervix: Origins of Cervical Trophoblasts and their Potential Value for Risk Assessment of Ongoing Pregnancies. Hum. Reprod. Update 2018, 24, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Moser, G.; Weiss, G.; Sundl, M.; Gauster, M.; Siwetz, M.; Lang-Olip, I.; Huppertz, B. Extravillous Trophoblasts Invade More than Uterine Arteries: Evidence for the Invasion of Uterine Veins. Histochem. Cell Biol. 2017, 147, 353–366. [Google Scholar] [CrossRef] [PubMed]
- He, N.; van Iperen, L.; de Jong, D.; Szuhai, K.; Helmerhorst, F.M.; van der Westerlaken, L.A.; Chuva de Sousa Lopes, S.M. Human Extravillous Trophoblasts Penetrate Decidual Veins and Lymphatics before Remodeling Spiral Arteries during Early Pregnancy. PLoS ONE 2017, 12, e0169849. [Google Scholar] [CrossRef] [PubMed]
- Windsperger, K.; Dekan, S.; Pils, S.; Golletz, C.; Kunihs, V.; Fiala, C.; Kristiansen, G.; Knofler, M.; Pollheimer, J. Extravillous Trophoblast Invasion of Venous as Well as Lymphatic Vessels is Altered in Idiopathic, Recurrent, Spontaneous Abortions. Hum. Reprod. 2017, 32, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fan, X.; Wang, R.; Lu, X.; Dang, Y.L.; Wang, H.; Lin, H.Y.; Zhu, C.; Ge, H.; Cross, J.C.; et al. Single-Cell RNA-Seq Reveals the Diversity of Trophoblast Subtypes and Patterns of Differentiation in the Human Placenta. Cell Res. 2018, 28, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, H.; Morozov, P.; Straus, A.; Sahasrabudhe, N.; Max, K.E.A.; Garzia, A.; Kustagi, M.; Tuschl, T.; Williams, Z. A Single-Cell Survey of the Human First-Trimester Placenta and Decidua. Sci. Adv. 2018, 4, eaau4788. [Google Scholar] [CrossRef] [PubMed]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polanski, K.; Goncalves, A.; et al. Single-Cell Reconstruction of the Early Maternal-Fetal Interface in Humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Sundl, M.; Glasner, A.; Huppertz, B.; Moser, G. The Trophoblast Plug during Early Pregnancy: A Deeper Insight. Histochem. Cell Biol. 2016, 146, 749–756. [Google Scholar] [CrossRef] [PubMed]
- James, J.L.; Saghian, R.; Perwick, R.; Clark, A.R. Trophoblast Plugs: Impact on Utero-Placental Haemodynamics and Spiral Artery Remodelling. Hum. Reprod. 2018. [Google Scholar] [CrossRef] [PubMed]
- Roberts, V.H.J.; Morgan, T.K.; Bednarek, P.; Morita, M.; Burton, G.J.; Lo, J.O.; Frias, A.E. Early First Trimester Uteroplacental Flow and the Progressive Disintegration of Spiral Artery Plugs: New Insights from Contrast-Enhanced Ultrasound and Tissue Histopathology. Hum. Reprod. 2017, 32, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Saghian, R.; Bogle, G.; James, J.L.; Clark, A.R. Establishment of Maternal Blood Supply to the Placenta: Insights into Plugging, Unplugging and Trophoblast Behaviour from an Agent-Based Model. Interface Focus. 2019, 9, 20190019. [Google Scholar] [CrossRef] [PubMed]
- Roberts, V.H.; Lo, J.O.; Salati, J.A.; Lewandowski, K.S.; Lindner, J.R.; Morgan, T.K.; Frias, A.E. Quantitative Assessment of Placental Perfusion by Contrast-Enhanced Ultrasound in Macaques and Human Subjects. Am. J. Obstet. Gynecol. 2016, 214, e1–e369. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Fujiwara, H.; Zeng, B.X.; Higuchi, T.; Yoshioka, S.; Fujii, S. Platelet-Derived Soluble Factors Induce Human Extravillous Trophoblast Migration and Differentiation: Platelets are a Possible Regulator of Trophoblast Infiltration into Maternal Spiral Arteries. Blood 2005, 106, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Blaschitz, A.; Siwetz, M.; Schlenke, P.; Gauster, M. Adhering Maternal Platelets can Contribute to the Cytokine and Chemokine Cocktail Released by Human First Trimester Villous Placenta. Placenta 2015, 36, 1333–1336. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.F.; Wagner, G.P. The Origin of Platelets Enabled the Evolution of Eutherian Placentation. Biol. Lett. 2019, 15, 20190374. [Google Scholar] [CrossRef] [PubMed]
- Claver, J.A.; Quaglia, A.I. Comparative Morphology, Development, and Function of Blood Cells in Nonmammalian Vertebrates. J. Exot. Pet. Med. 2009, 18, 87–97. [Google Scholar] [CrossRef]
- Theopold, U.; Schmidt, O.; Soderhall, K.; Dushay, M.S. Coagulation in Arthropods: Defence, Wound Closure and Healing. Trends Immunol. 2004, 25, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Michelson, A.D. Platelets, 3rd ed.; Academic Press, Elsevier: London, UK, 2013; p. 1353. [Google Scholar]
- Reese, J.A.; Peck, J.D.; McIntosh, J.J.; Vesely, S.K.; George, J.N. Platelet Counts in Women with Normal Pregnancies: A Systematic Review. Am. J. Hematol. 2017, 92, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.A.; Peck, J.D.; Yu, Z.; Scordino, T.A.; Deschamps, D.R.; McIntosh, J.J.; Terrell, D.R.; Vesely, S.K.; George, J.N. Platelet Sequestration and Consumption in the Placental Intervillous Space Contribute to Lower Platelet Counts during Pregnancy. Am. J. Hematol. 2019, 94, E8–E11. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, V.; Bock, J.E.; Nielsen, J.B. Significance of Plasma Skimming and Plasma Volume Expansion. J. Appl. Physiol. 1992, 72, 2047–2051. [Google Scholar] [CrossRef] [PubMed]
- Aster, R.H. Pooling of Platelets in the Spleen: Role in the Pathogenesis of “Hypersplenic” Thrombocytopenia. J. Clin. Investig. 1966, 45, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Boehlen, F.; Hohlfeld, P.; Extermann, P.; Perneger, T.V.; de Moerloose, P. Platelet Count at Term Pregnancy: A Reappraisal of the Threshold. Obstet. Gynecol. 2000, 95, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Shehata, N.; Burrows, R.; Kelton, J.G. Gestational Thrombocytopenia. Clin. Obstet. Gynecol. 1999, 42, 327–334. [Google Scholar] [CrossRef] [PubMed]
- McCrae, K.R. Thrombocytopenia in Pregnancy. Hematology Am. Soc. Hematol. Educ. Program. 2010, 2010, 397–402. [Google Scholar]
- Gardiner, C.; Vatish, M. Impact of Haemostatic Mechanisms on Pathophysiology of Preeclampsia. Thromb. Res. 2017, 151, S48–S52. [Google Scholar] [CrossRef]
- Lockwood, C.J.; Huang, S.J.; Krikun, G.; Caze, R.; Rahman, M.; Buchwalder, L.F.; Schatz, F. Decidual Hemostasis, Inflammation, and Angiogenesis in Pre-Eclampsia. Semin. Thromb. Hemost. 2011, 37, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Erlich, J.; Parry, G.C.; Fearns, C.; Muller, M.; Carmeliet, P.; Luther, T.; Mackman, N. Tissue Factor is Required for Uterine Hemostasis and Maintenance of the Placental Labyrinth during Gestation. Proc. Natl. Acad. Sci. USA 1999, 96, 8138–8143. [Google Scholar] [CrossRef] [PubMed]
- Isermann, B.; Kashif, M.; Nawroth, P.P. Platelet Activation Impairs Placental Function. Thromb. Res. 2009, 123, S85–S87. [Google Scholar] [CrossRef]
- Sood, R.; Sholl, L.; Isermann, B.; Zogg, M.; Coughlin, S.R.; Weiler, H. Maternal Par4 and Platelets Contribute to Defective Placenta Formation in Mouse Embryos Lacking Thrombomodulin. Blood 2008, 112, 585–591. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaufmann, P.; Huppertz, B.; Frank, H.G. The Fibrinoids of the Human Placenta: Origin, Composition and Functional Relevance. Ann. Anat. 1996, 178, 485–501. [Google Scholar] [CrossRef]
- Pierleoni, C.; Castellucci, M.; Kaufmann, P.; Lund, L.R.; Schnack Nielsen, B. Urokinase Receptor is Up-Regulated in Endothelial Cells and Macrophages Associated with Fibrinoid Deposits in the Human Placenta. Placenta 2003, 24, 677–685. [Google Scholar] [CrossRef]
- Benirschke, K.; Kaufmann, P.; Baergen, R.N. Pathology of the Human Placenta, 5th ed.; Springer: New York, NY, USA, 2006; pp. 42–49. [Google Scholar]
- Nelson, D.M.; Crouch, E.C.; Curran, E.M.; Farmer, D.R. Trophoblast Interaction with Fibrin Matrix. Epithelialization of Perivillous Fibrin Deposits as a Mechanism for Villous Repair in the Human Placenta. Am. J. Pathol. 1990, 136, 855–865. [Google Scholar] [PubMed]
- Maloney, S.; Smith, A.; Furst, D.E.; Myerson, D.; Rupert, K.; Evans, P.C.; Nelson, J.L. Microchimerism of Maternal Origin Persists into Adult Life. J. Clin. Investig. 1999, 104, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Dawe, G.S.; Tan, X.W.; Xiao, Z.C. Cell Migration from Baby to Mother. Cell. Adh Migr. 2007, 1, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Srivatsa, B.; Srivatsa, S.; Johnson, K.L.; Bianchi, D.W. Maternal Cell Microchimerism in Newborn Tissues. J. Pediatr. 2003, 142, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, J.S.; Zogg, M.; Talmage, K.E.; Degen, J.L.; Weiler, H.; Isermann, B.H. Role of Fibrinogen- and Platelet-Mediated Hemostasis in Mouse Embryogenesis and Reproduction. J. Thromb. Haemost. 2004, 2, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.L.; Zheng, Y.W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V., Jr.; Tam, C.; Coughlin, S.R. A Dual Thrombin Receptor System for Platelet Activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Sambrano, G.R.; Weiss, E.J.; Zheng, Y.W.; Huang, W.; Coughlin, S.R. Role of Thrombin Signalling in Platelets in Haemostasis and Thrombosis. Nature 2001, 413, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Sherer, D.M.; Lerner, R. Glanzmann’s Thrombasthenia in Pregnancy: A Case and Review of the Literature. Am. J. Perinatol. 1999, 16, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Seoud, M.; Tannous, R.; Usta, I.; Shamseddine, A. Bernard-Soulier Syndrome in Pregnancy: Case Report and Review of the Literature. Clin. Lab. Haematol. 1998, 20, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Peitsidis, P.; Datta, T.; Pafilis, I.; Otomewo, O.; Tuddenham, E.G.; Kadir, R.A. Bernard Soulier Syndrome in Pregnancy: A Systematic Review. Haemophilia 2010, 16, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.J.; Spencer, R.A.; Miyazawa, K. Successful Cesarean Section in a Gravida with the Thrombocytopenia with Absent Radius Syndrome. A Case Report. J. Reprod. Med. 1986, 31, 260–262. [Google Scholar] [PubMed]
- Kohli, S.; Isermann, B. Placental Hemostasis and Sterile Inflammation: New Insights into Gestational Vascular Disease. Thromb. Res. 2017, 151, S30–S33. [Google Scholar] [CrossRef]
- Isermann, B.; Nawroth, P.P. The Role of Platelets during Reproduction. Pathophysiol. Haemost. Thromb. 2006, 35, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Bass, K.E.; Morrish, D.; Roth, I.; Bhardwaj, D.; Taylor, R.; Zhou, Y.; Fisher, S.J. Human Cytotrophoblast Invasion is Up-Regulated by Epidermal Growth Factor: Evidence that Paracrine Factors Modify this Process. Dev. Biol. 1994, 164, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Lash, G.E.; Warren, A.Y.; Underwood, S.; Baker, P.N. Vascular Endothelial Growth Factor is a Chemoattractant for Trophoblast Cells. Placenta 2003, 24, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Fujiwara, H.; Konishi, I. Role of Platelets in Placentation. Med. Mol. Morphol. 2010, 43, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Kohli, S.; Ranjan, S.; Hoffmann, J.; Kashif, M.; Daniel, E.A.; Al-Dabet, M.M.; Bock, F.; Nazir, S.; Huebner, H.; Mertens, P.R.; et al. Maternal Extracellular Vesicles and Platelets Promote Preeclampsia Via Inflammasome Activation in Trophoblasts. Blood 2016, 128, 2153–2164. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.J. Pre-Eclampsia. Lancet 2000, 356, 1260–1265. [Google Scholar] [CrossRef]
- Tranquilli, A.L.; Dekker, G.; Magee, L.; Roberts, J.; Sibai, B.M.; Steyn, W.; Zeeman, G.G.; Brown, M.A. The Classification, Diagnosis and Management of the Hypertensive Disorders of Pregnancy: A Revised Statement from the ISSHP. Pregnancy Hypertens. 2014, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Haram, K.; Mortensen, J.H.; Nagy, B. Genetic Aspects of Preeclampsia and the HELLP Syndrome. J. Pregnancy 2014, 2014, 910751. [Google Scholar] [CrossRef] [PubMed]
- Socol, M.L.; Weiner, C.P.; Louis, G.; Rehnberg, K.; Rossi, E.C. Platelet Activation in Preeclampsia. Am. J. Obstet. Gynecol. 1985, 151, 494–497. [Google Scholar] [CrossRef]
- Redman, C.W.; Bonnar, J.; Beilin, L. Early Platelet Consumption in Pre-Eclampsia. Br. Med. J. 1978, 1, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Janes, S.L.; Kyle, P.M.; Redman, C.; Goodall, A.H. Flow Cytometric Detection of Activated Platelets in Pregnant Women Prior to the Development of Pre-Eclampsia. Thromb. Haemost. 1995, 74, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Kanat-Pektas, M.; Yesildager, U.; Tuncer, N.; Arioz, D.T.; Nadirgil-Koken, G.; Yilmazer, M. Could Mean Platelet Volume in Late First Trimester of Pregnancy Predict Intrauterine Growth Restriction and Pre-Eclampsia? J. Obstet. Gynaecol. Res. 2014, 40, 1840–1845. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, C.; Larsen, J.B.; Fuglsang, J.; Hvas, A.M. Platelet Function in Preeclampsia - a Systematic Review and Meta-Analysis. Platelets 2019, 30, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Dundar, O.; Yoruk, P.; Tutuncu, L.; Erikci, A.A.; Muhcu, M.; Ergur, A.R.; Atay, V.; Mungen, E. Longitudinal Study of Platelet Size Changes in Gestation and Predictive Power of Elevated MPV in Development of Pre-Eclampsia. Prenat. Diagn. 2008, 28, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Pickel, K.; Stern, C.; Eberhard, K.; Lang, U.; Obermayer-Pietsch, B.; Cervar-Zivkovic, M. Comparison of Mean Platelet Volume (MPV) and sFlt-1/PlGF Ratio as Predictive Markers for Preeclampsia. J. Matern. Fetal. Neonatal Med. 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Schoene, N.; Harris, W. Mean Platelet Volume as an Indicator of Platelet Activation: Methodological Issues. Platelets 2002, 13, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Morrison, R.; Crawford, J.; MacPherson, M.; Heptinstall, S. Platelet Behaviour in Normal Pregnancy, Pregnancy Complicated by Essential Hypertension and Pregnancy-Induced Hypertension. Thromb. Haemost. 1985, 54, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Rabini, R.A.; Salvolini, E.; Staffolani, R.; Pugnaloni, A.; Simonelli, L.; Biagini, G.; Cester, N.; Garzetti, G.G.; Mazzanti, L. Biochemical-Morphological Modifications of Platelet Membranes during Pregnancy-Induced Hypertension. Exp. Mol. Pathol. 1995, 63, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Kiumi, F.; Mitsuya, K. Changes in Platelet ATP Secretion and Aggregation during Pregnancy and in Preeclampsia. Am. J. Med. Sci. 1999, 318, 115–121. [Google Scholar] [CrossRef]
- Germain, S.J.; Sacks, G.P.; Sooranna, S.R.; Sargent, I.L.; Redman, C.W. Systemic Inflammatory Priming in Normal Pregnancy and Preeclampsia: The Role of Circulating Syncytiotrophoblast Microparticles. J. Immunol. 2007, 178, 5949–5956. [Google Scholar] [CrossRef] [PubMed]
- Tannetta, D.S.; Hunt, K.; Jones, C.I.; Davidson, N.; Coxon, C.H.; Ferguson, D.; Redman, C.W.; Gibbins, J.M.; Sargent, I.L.; Tucker, K.L. Syncytiotrophoblast Extracellular Vesicles from Pre-Eclampsia Placentas Differentially Affect Platelet Function. PLoS ONE 2015, 10, e0142538. [Google Scholar] [CrossRef] [PubMed]
- Bujold, E.; Morency, A.M.; Roberge, S.; Lacasse, Y.; Forest, J.C.; Giguere, Y. Acetylsalicylic Acid for the Prevention of Preeclampsia and Intra-Uterine Growth Restriction in Women with Abnormal Uterine Artery Doppler: A Systematic Review and Meta-Analysis. J. Obstet. Gynaecol. Can. 2009, 31, 818–826. [Google Scholar] [CrossRef]
- Roberge, S.; Bujold, E.; Nicolaides, K.H. Aspirin for the Prevention of Preterm and Term Preeclampsia: Systematic Review and Metaanalysis. Am. J. Obstet. Gynecol. 2018, 218, 287–293.e1. [Google Scholar] [CrossRef] [PubMed]
- Reilly, C.R.; Cuesta-Fernandez, A.; Kayaleh, O.R. Successful Gestation and Delivery using Clopidogrel for Secondary Stroke Prophylaxis: A Case Report and Literature Review. Arch. Gynecol. Obstet. 2014, 290, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Schrottmaier, W.C.; Kral, J.B.; Badrnya, S.; Assinger, A. Aspirin and P2Y12 Inhibitors in Platelet-Mediated Activation of Neutrophils and Monocytes. Thromb. Haemost. 2015, 114, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Siwetz, M.; Blaschitz, A.; El-Heliebi, A.; Hiden, U.; Desoye, G.; Huppertz, B.; Gauster, M. TNF-Alpha Alters the Inflammatory Secretion Profile of Human First Trimester Placenta. Lab. Investig. 2016, 96, 428. [Google Scholar] [CrossRef] [PubMed]
- Flierl, U.; Bauersachs, J.; Schafer, A. Modulation of Platelet and Monocyte Function by the Chemokine Fractalkine (CX3 CL1) in Cardiovascular Disease. Eur. J. Clin. Investig. 2015, 45, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Nonn, O.; Guttler, J.; Forstner, D.; Maninger, S.; Zadora, J.; Balogh, A.; Frolova, A.; Glasner, A.; Herse, F.; Gauster, M. Placental CX3CL1 is Deregulated by Angiotensin II and Contributes to a Pro-Inflammatory Trophoblast-Monocyte Interaction. Int. J. Mol. Sci. 2019, 20, 461. [Google Scholar] [CrossRef] [PubMed]
- Siwetz, M.; Sundl, M.; Kolb, D.; Hiden, U.; Herse, F.; Huppertz, B.; Gauster, M. Placental Fractalkine Mediates Adhesion of THP-1 Monocytes to Villous Trophoblast. Histochem. Cell Biol. 2015, 143, 565–574. [Google Scholar]
- Siwetz, M.; Dieber-Rotheneder, M.; Cervar-Zivkovic, M.; Kummer, D.; Kremshofer, J.; Weiss, G.; Herse, F.; Huppertz, B.; Gauster, M. Placental Fractalkine is Up-Regulated in Severe Early-Onset Preeclampsia. Am. J. Pathol. 2015, 185, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Macey, M.G.; Bevan, S.; Alam, S.; Verghese, L.; Agrawal, S.; Beski, S.; Thuraisingham, R.; MacCallum, P.K. Platelet Activation and Endogenous Thrombin Potential in Pre-Eclampsia. Thromb. Res. 2010, 125, e76–e81. [Google Scholar] [CrossRef] [PubMed]
- Major, H.D.; Campbell, R.A.; Silver, R.M.; Branch, D.W.; Weyrich, A.S. Synthesis of sFlt-1 by Platelet-Monocyte Aggregates Contributes to the Pathogenesis of Preeclampsia. Am. J. Obstet. Gynecol. 2014, 210, e1–e547. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble Endoglin and Other Circulating Antiangiogenic Factors in Preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Malassine, A.; Frendo, J.L.; Evain-Brion, D. A Comparison of Placental Development and Endocrine Functions between the Human and Mouse Model. Hum. Reprod. Update 2003, 9, 531–539. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moser, G.; Guettler, J.; Forstner, D.; Gauster, M. Maternal Platelets—Friend or Foe of the Human Placenta? Int. J. Mol. Sci. 2019, 20, 5639. https://doi.org/10.3390/ijms20225639
Moser G, Guettler J, Forstner D, Gauster M. Maternal Platelets—Friend or Foe of the Human Placenta? International Journal of Molecular Sciences. 2019; 20(22):5639. https://doi.org/10.3390/ijms20225639
Chicago/Turabian StyleMoser, Gerit, Jacqueline Guettler, Désirée Forstner, and Martin Gauster. 2019. "Maternal Platelets—Friend or Foe of the Human Placenta?" International Journal of Molecular Sciences 20, no. 22: 5639. https://doi.org/10.3390/ijms20225639
APA StyleMoser, G., Guettler, J., Forstner, D., & Gauster, M. (2019). Maternal Platelets—Friend or Foe of the Human Placenta? International Journal of Molecular Sciences, 20(22), 5639. https://doi.org/10.3390/ijms20225639