Knockdown of Pyruvate Kinase M2 Inhibits Cell Proliferation, Metabolism, and Migration in Renal Cell Carcinoma




 and
and
Abstract
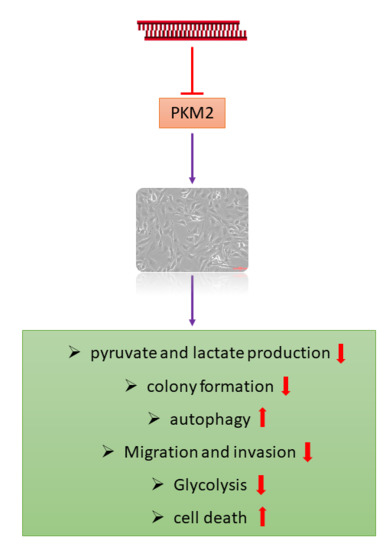
1. Introduction
2. Results
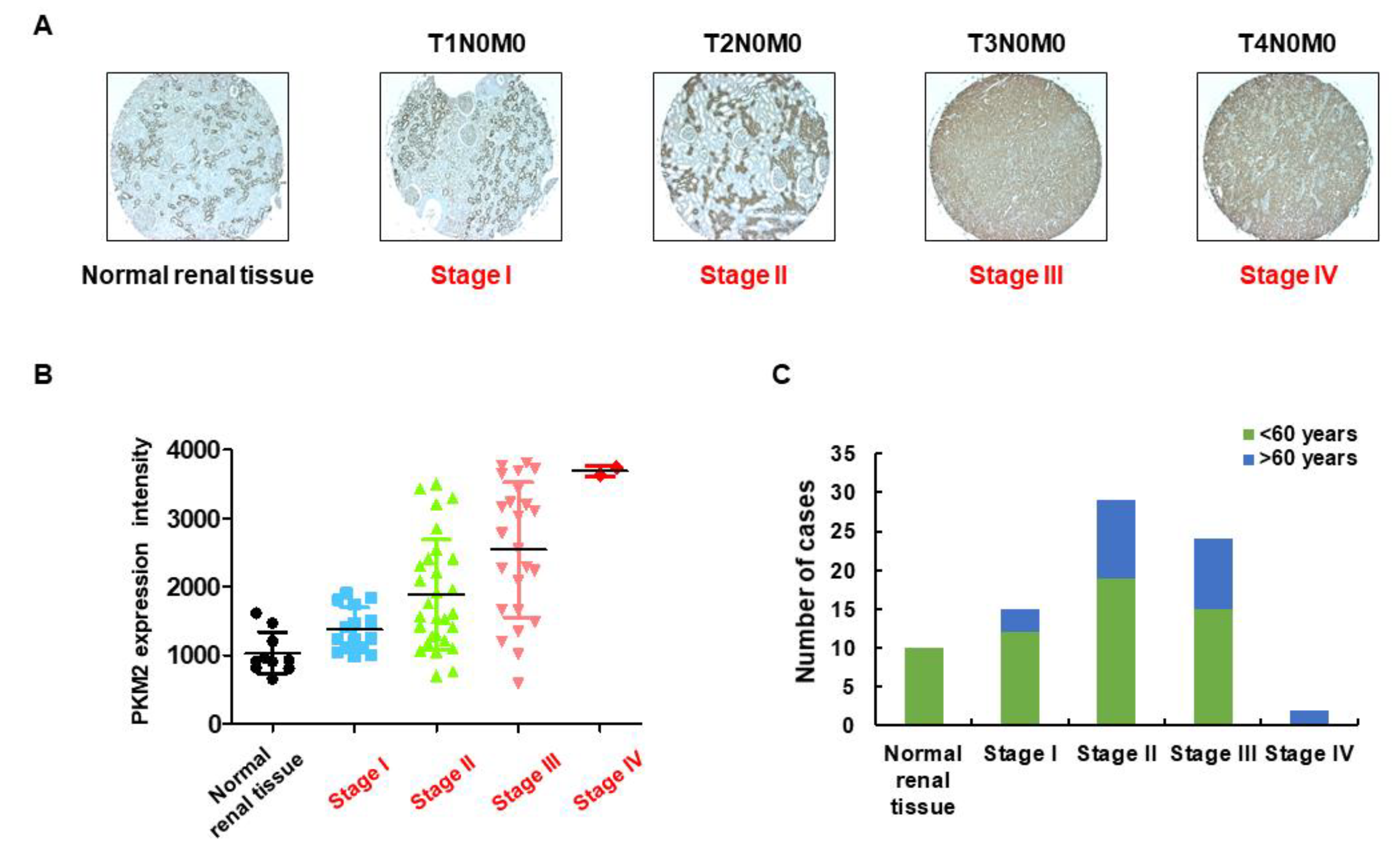
2.1. Identification of PKM2 Expression in RCC
2.2. PKM2 Knockdown Inhibits Tumor Progression of 786-O Cells
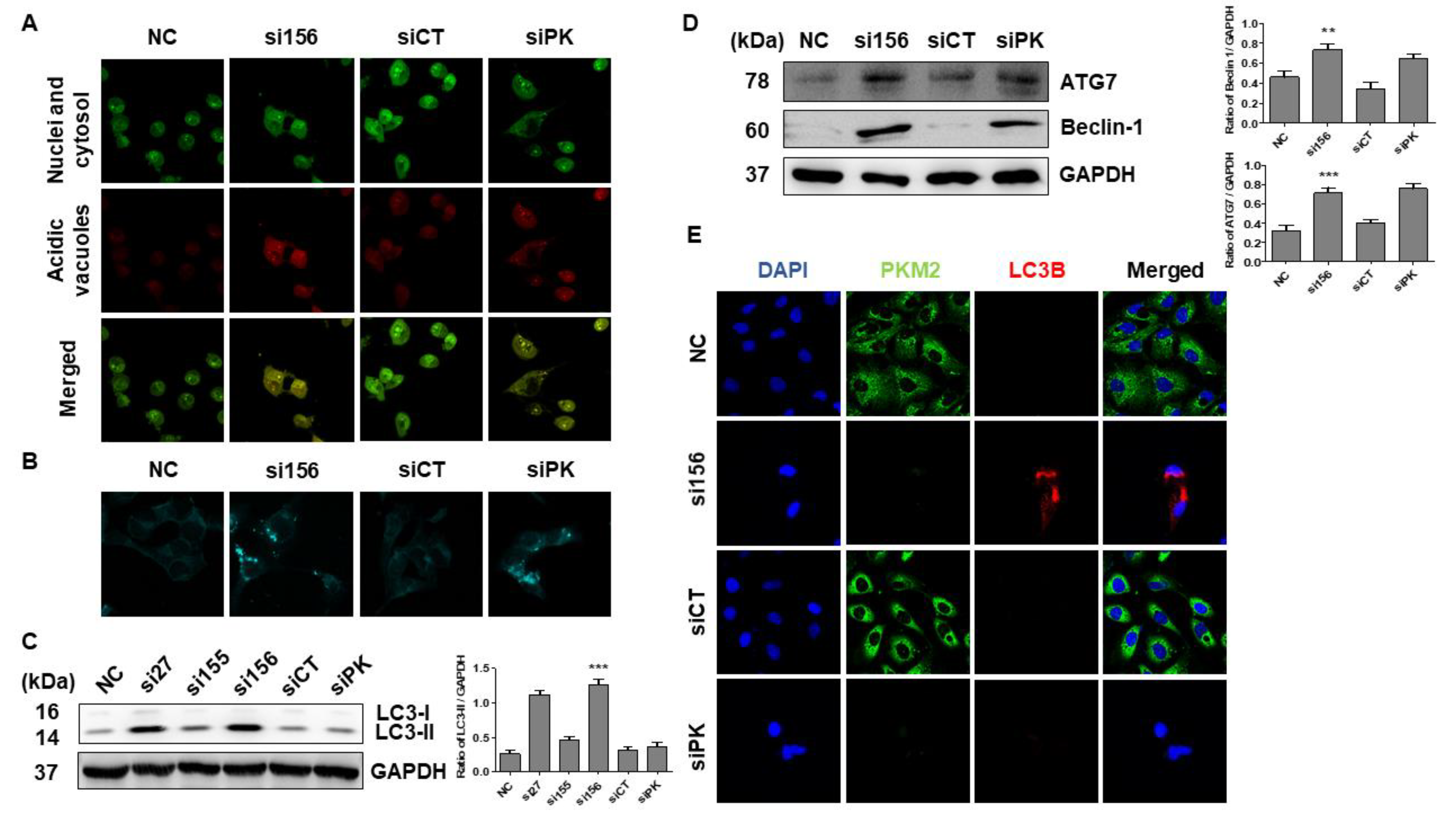
2.3. PKM2 Knockdown Induces Autophagy
2.4. PKM2 Knockdown Impairs Glycolysis, Glycolytic Intermediates, and Mitochondrial Activity
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Kidney Tumor and Normal Tissues
4.3. Immunohistochemical Analysis
4.4. Cell Lines and Cell Culture
4.5. PKM2 Knockdown
4.6. Cell Viability Assay
4.7. Western Blot Analysis
4.8. Immunofluorescence Analysis
4.9. Subcellular Fractionation
4.10. Acridine Orange Staining
4.11. Monodansylcadaverine Staining
4.12. Analysis of Glycolysis, Glycolytic Intermediates, and Mitochondrial Activity
4.13. In Vitro Cell Migration and Invasion Assay
4.14. Colony Formation Assay
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PKM2 | pyruvate kinase M2 |
| RCC | renal cell carcinoma; LDHA |
| LDHA | lactate dehydrogenase A |
| Glut-1 | pyruvate kinase M2 |
| siRNA | small-interfering RNA |
| AKT | protein kinase B |
| mTOR | mechanistic target of rapamycin 1 |
| ccRCC | clear cell renal cell carcinoma |
| TK | tyrosine kinase |
| ERK 1/2 | extracellular signal-regulated kinase 1/2 |
| TMA | tissue microarray |
| HPLC | high-performance liquid chromatography |
| OCR | oxygen consumption rate |
| MMP | matrix metalloproteinase |
| PER | proton efflux rate |
| glycoPER | glycolytic proton efflux rate |
| HIF-1 | hypoxia-inducible factor-1 |
| LC3B | light chain 3B |
| AMPK | AMP-activated protein kinase |
| GSK3β | glycogen synthase kinase 3 beta |
| PI3K | phosphoinositide 3-kinase |
References
- Sanchez, D.J.; Simon, M.C. Genetic and metabolic hallmarks of clear cell renal cell carcinoma. Biochim. Biophys. Acta (BBA) Rev. Cancer 2018, 1870, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Chen, Q.; He, X.; Yao, K.; Li, Z.; Liu, Z.; Chen, J.; Liu, Z.; Guo, C.; Lu, J.; et al. Expression and significance of Cystatin-C in clear cell renal cell carcinoma. Biomed. Pharmacother. 2018, 107, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wu, Y.; Zhang, J.; Fang, Z.; Liu, Z.; Xu, Z.; Fan, Y. Nomograms for predicting long-term overall survival and disease-specific survival of patients with clear cell renal cell carcinoma. OncoTargets Ther. 2018, 11, 5535–5544. [Google Scholar] [CrossRef] [PubMed]
- Xing, T.; He, H. Epigenomics of clear cell renal cell carcinoma: Mechanisms and potential use in molecular pathology. Chin. J. Cancer Res. 2016, 28, 80–91. [Google Scholar] [PubMed]
- Atkins, M.B.; Tannir, N.M. Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treat. Rev. 2018, 70, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Ruan, H.; Wang, K.; Song, Z.; Bao, L.; Xu, T.; Xiao, H.; Wang, C.; Cheng, G.; Tong, J.; et al. Overexpression of PLIN2 is a prognostic marker and attenuates tumor progression in clear cell renal cell carcinoma. Int. J. Oncol. 2018, 53, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Makhov, P.; Joshi, S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol. Cancer Ther. 2018, 17, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Son, J.Y.; Yoon, S.; Tae, I.H.; Park, Y.J.; De, U.; Jeon, Y.; Park, Y.J.; Rhyu, I.J.; Lee, B.M.; Chung, K.H.; et al. Novel therapeutic roles of MC-4 in combination with everolimus against advanced renal cell carcinoma by dual targeting of Akt/pyruvate kinase muscle isozyme M2 and mechanistic target of rapamycin complex 1 pathways. Cancer Med. 2018, 7, 5083–5095. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Energy deregulation: Licensing tumors to grow. Science 2006, 312, 1158–1159. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Hamabe, A.; Konno, M.; Tanuma, N.; Shima, H.; Tsunekuni, K.; Kawamoto, K.; Nishida, N.; Koseki, J.; Mimori, K.; Gotoh, N. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial–mesenchymal transition. Proc. Natl. Acad. Sci. USA 2014, 111, 15526–15531. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Heiden, M.G.V.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef] [PubMed]
- He, C.L.; Bian, Y.Y.; Xue, Y.; Liu, Z.X.; Zhou, K.Q.; Yao, C.F.; Lin, Y.; Zou, H.F.; Luo, F.X.; Qu, Y.Y.; et al. Pyruvate Kinase M2 Activates mTORC1 by Phosphorylating AKT1S1. Sci. Rep. 2016, 6, 21524. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lu, Z. Pyruvate kinase M2 at a glance. J. Cell Sci. 2015, 128, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.C.; Hung, W.C. Pyruvate kinase M2 fuels multiple aspects of cancer cells: From cellular metabolism, transcriptional regulation to extracellular signaling. Mol. Cancer 2018, 17, 35. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Yung, W.A.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Roessler, S.; Jia, H.L.; Budhu, A.; Forgues, M.; Ye, Q.H.; Lee, J.S.; Thorgeirsson, S.S.; Sun, Z.; Tang, Z.Y.; Qin, L.X.; et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010, 70, 10202–10212. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Leem, S.H.; Lee, S.Y.; Kim, S.C.; Park, E.S.; Kim, S.B.; Kim, S.K.; Kim, Y.J.; Kim, W.J.; Chu, I.S. Expression Signature ofE2F1and Its Associated Genes Predict Superficial to Invasive Progression of Bladder Tumors. J. Clin. Oncol. 2010, 28, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Beer, D.G.; Kardia, S.L.; Huang, C.C.; Giordano, T.J.; Levin, A.M.; Misek, D.E.; Lin, L.; Chen, G.; Gharib, T.G.; Thomas, D.G.; et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 2002, 8, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Hu, N.; Yang, H.H.; Wang, C.; Takikita, M.; Wang, Q.H.; Giffen, C.; Clifford, R.; Hewitt, S.M.; Shou, J.Z.; et al. Global gene expression profiling and validation in esophageal squamous cell carcinoma (ESCC) and its association with clinical phenotypes. Clin. Cancer Res. 2011, 17, 2955–2966. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jiang, J.; Ji, J.; Cai, Q.; Chen, X.; Yu, Y.; Zhu, Z.; Zhang, J. PKM2 promotes cell migration and inhibits autophagy by mediating PI3K/AKT activation and contributes to the malignant development of gastric cancer. Sci. Rep. 2017, 7, 2886. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Kundu, A.; Sachan, R.; Park, J.H.; Ahn, M.Y.; Yoon, K.; Lee, J.; Kim, N.D.; Kim, I.S.; Lee, B.M.; et al. PKM2 Knockdown Induces Autophagic Cell Death via AKT/mTOR Pathway in Human Prostate Cancer Cells. Cell. Physiol. Biochem. 2019, 52, 1535–1552. [Google Scholar] [PubMed]
- Fan, F.T.; Shen, C.S.; Tao, L.; Tian, C.; Liu, Z.G.; Zhu, Z.J.; Liu, Y.P.; Pei, C.S.; Wu, H.Y.; Zhang, L.; et al. PKM2 Regulates Hepatocellular Carcinoma Cell Epithelial-mesenchymal Transition and Migration upon EGFR Activation. Asian Pac. J. Cancer Prev. 2014, 15, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Yang, G.; Wang, J.; Wang, Y. Knockdown of PKM2 induces apoptosis and autophagy in human A549 alveolar adenocarcinoma cells. Mol. Med. Rep. 2015, 12, 4358–4363. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, U.; Bensalah, K.; Bex, A.; Boorjian, S.A.; Bray, F.; Coleman, J.; Gore, J.L.; Sun, M.; Wood, C.; Russo, P. Epidemiology of Renal Cell Carcinoma. Eur. Urol. 2019, 75, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Dutcher, J.P. Recent developments in the treatment of renal cell carcinoma. Ther. Adv. Urol. 2013, 5, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Kanesvaran, R.; Tan, M.H. Targeted therapy for renal cell carcinoma: The next lap. J. Carcinog. 2014, 13, 3. [Google Scholar] [PubMed]
- Ghatalia, P.; Koenigsberg, R.; Pisarcik, D.; Handorf, E.A.; Geynisman, D.M.; Zibelman, M. The Evolution of Clinical Trials in Renal Cell Carcinoma: A Status Report for 2013–2016 from the ClinicalTrials.gov Website. Kidney Cancer 2017, 1, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Sathianathen, N.J.; Krishna, S.; Anderson, J.K.; Weight, C.J.; Gupta, S.; Konety, B.R.; Griffith, T.S. The current status of immunobased therapies for metastatic renal-cell carcinoma. ImmunoTargets Ther. 2017, 6, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Akins, N.S.; Nielson, T.C.; Le, H.V. Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer. Curr. Top. Med. Chem. 2018, 18, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, J.M. Altered energy metabolism in cancer: A unique opportunity for therapeutic intervention. Cancer Biol. Ther. 2013, 14, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, H.; Lindgren, D.; Forsberg, A.M.; Mulder, H.; Axelson, H.; Johansson, M.E. Primary clear cell renal carcinoma cells display minimal mitochondrial respiratory capacity resulting in pronounced sensitivity to glycolytic inhibition by 3-Bromopyruvate. Cell Death Dis. 2015, 6, e1585. [Google Scholar] [CrossRef] [PubMed]
- Leisz, S.; Schulz, K.; Erb, S.; Oefner, P.; Dettmer, K.; Mougiakakos, D.; Wang, E.; Marincola, F.M.; Stehle, F.; Seliger, B. Distinct von Hippel-Lindau gene and hypoxia-regulated alterations in gene and protein expression patterns of renal cell carcinoma and their effects on metabolism. Oncotarget 2015, 6, 11395–11406. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Toschi, A.; Lee, E.; Thompson, S.; Gadir, N.; Yellen, P.; Drain, C.M.; Ohh, M.; Foster, D.A. Phospholipase D-mTOR requirement for the Warburg effect in human cancer cells. Cancer Lett. 2010, 299, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Gui, D.Y.; Lewis, C.A.; Heiden, M.G.V. Allosteric Regulation of PKM2 Allows Cellular Adaptation to Different Physiological States. Sci. Signal. 2013, 6, pe7. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genome Res. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Facompre, N.D.; Sinha, I.; El-Bayoumy, K.; Pinto, J.T.; Sinha, R. Remarkable inhibition of mTOR signaling by the combination of rapamycin and 1,4-phenylenebis(methylene)selenocyanate in human prostate cancer cells. Int. J. Cancer 2012, 131, 2134–2142. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Robb, V.A.; Karbowniczek, M.; Klein-Szanto, A.J.; Henske, E.P. Activation of the mTOR Signaling Pathway in Renal Clear Cell Carcinoma. J. Urol. 2007, 177, 346–352. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Sun, X.; Yang, H.; Li, X.; Yang, D. TOFA induces cell cycle arrest and apoptosis in ACHN and 786-O cells through inhibiting PI3K/Akt/mTOR pathway. J. Cancer 2018, 9, 2734–2742. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; German, P.; Bai, S.; Barnes, S.; Guo, W.; Qi, X.; Lou, H.; Liang, J.; Jonasch, E.; Mills, G.B.; et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. J. Genet. Genom. 2015, 42, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Cui, S.P.; Ke, Y. MiR-144 inhibits cell proliferation of renal cell carcinoma by targeting MTOR. J. Huazhong Univ. Sci. Technol. Med. Sci. 2016, 36, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Koshikawa, N.; Mochizuki, S.; Takenaga, K. 3-Methyladenine suppresses cell migration and invasion of HT1080 fibrosarcoma cells through inhibiting phosphoinositide 3-kinases independently of autophagy inhibition. Int. J. Oncol. 2007, 31, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, W.; Wang, K.; Fan, Y.; Chen, J.; Ma, J.; Wang, X.; He, D.; Zeng, J.; Li, L. Tetrandrine inhibits migration and invasion of human renal cell carcinoma by regulating Akt/NF-κB/MMP-9 signaling. PLoS ONE 2017, 12, e0173725. [Google Scholar] [CrossRef] [PubMed]
- Dollé, L.; Depypere, H.; Bracke, M. Anti-Invasive and Anti-Metastasis Strategies: New Roads, New Tools and New Hopes. Curr. Cancer Drug Targets 2006, 6, 729–751. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, W.; Zhang, X.; Lin, S.; Chen, Z. Overexpression of KiSS-1 reduces colorectal cancer cell invasion by downregulating MMP-9 via blocking PI3K/Akt/NF-κB signal pathway. Int. J. Oncol. 2016, 48, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Chou, P.Y.; Chien, Y.C.; Wu, C.H.; Wu, T.S.; Sheu, M.J. Ethanol extracts of fruiting bodies of Antrodia cinnamomea exhibit anti-migration action in human adenocarcinoma CL1-0 cells through the MAPK and PI3K/AKT signaling pathways. Phytomedicine 2012, 19, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Sharp, P.A. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J. Exp. Med. 2012, 209, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 2016, 7, 13280. [Google Scholar] [CrossRef] [PubMed]
- Tae, I.H.; Park, E.Y.; Dey, P.; Son, J.Y.; Lee, S.Y.; Jung, J.H.; Saloni, S.; Kim, M.H.; Kim, H.S. Novel SIRT1 inhibitor 15-deoxy-Δ12,14-prostaglandin J2 and its derivatives exhibit anticancer activity through apoptotic or autophagic cell death pathways in SKOV3 cells. Int. J. Oncol. 2018, 53, 2518–2530. [Google Scholar] [CrossRef] [PubMed]
- Iansante, V.; Choy, P.M.; Fung, S.W.; Liu, Y.; Chai, J.G.; Dyson, J.; Del Rio, A.; D’Santos, C.; Williams, R.; Chokshi, S. PARP14 promotes the Warburg effect in hepatocellular carcinoma by inhibiting JNK1-dependent PKM2 phosphorylation and activation. Nat. Commun. 2015, 6, 7882. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.K.; Rao, K.V.K.; Aalinkeel, R.; Mahajan, S.; Chawda, R.; Schwartz, S.A. Inhibition of Prostate Cancer Cell Colony Formation by the Flavonoid Quercetin Correlates with Modulation of Specific Regulatory Genes. Clin. Diagn. Lab. Immunol. 2004, 11, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Stage and Grade | Variable | No. of Samples | Healthy Subject (%) | Cancer Patient (%) | ||
|---|---|---|---|---|---|---|
| <60 | >60 | <60 | >60 | |||
| Patients (Male and Female) | 70 Cancer Patients, 10 Healthy Subjects | 10 (100%) | - | 6 (60%) | 4 (40%) | |
| Tumor stage (%) | I | 15 | - | - | 12 (17.14%) | 3 (4.29%) |
| II | 29 | - | - | 19 (27.14%) | 10 (14.29%) | |
| III | 24 | - | - | 15 (21.43%) | 9 (12.86%) | |
| IV | 2 | - | - | - | 2 (2.86%) | |
| TNM grade | T1N0M0 | - | - | - | 12 (17.14%) | 3 (4.29%) |
| T2N0M0 | - | - | - | 19 (27.14%) | 10 (14.29%) | |
| T3N0M0 | - | - | - | 7 (10%) | 4 (5.71%) | |
| T3NxM0 | - | - | - | 1 (1.43%) | 1 (1.43%) | |
| T3aN0M0 | - | - | - | 5 (7.14%) | 4 (5.71%) | |
| T3bN1M0 | - | - | - | 1 (1.43%) | - | |
| T3aNxM0 | - | - | - | 1 (1.43%) | - | |
| T4N0M0 | - | - | - | - | 1 (1.43%) | |
| T4N1M1 | - | - | - | - | 1 (1.43%) | |
| siRNAs | Sense Stand Sequences | Source |
|---|---|---|
| si27 | AGGCAGAGGCUGCCAUCUA | Bioneer Corporation |
| si155 | GCCAUAAUCGUCCUCACCA | |
| si156 | CCAUAAUCGUCCUCACCAA | |
| siCT (negative control siRNA targeting the firefly luciferase gene) | CUUACGCUGAGUACUUCGA | |
| siPK (positive control siRNA, commercially available, targeting both PKM1 and PKM2 mRNAs) | GGACCUGAGAUCCGAACUG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dey, P.; Son, J.Y.; Kundu, A.; Kim, K.S.; Lee, Y.; Yoon, K.; Yoon, S.; Lee, B.M.; Nam, K.T.; Kim, H.S. Knockdown of Pyruvate Kinase M2 Inhibits Cell Proliferation, Metabolism, and Migration in Renal Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 5622. https://doi.org/10.3390/ijms20225622
Dey P, Son JY, Kundu A, Kim KS, Lee Y, Yoon K, Yoon S, Lee BM, Nam KT, Kim HS. Knockdown of Pyruvate Kinase M2 Inhibits Cell Proliferation, Metabolism, and Migration in Renal Cell Carcinoma. International Journal of Molecular Sciences. 2019; 20(22):5622. https://doi.org/10.3390/ijms20225622
Chicago/Turabian StyleDey, Prasanta, Ji Yeon Son, Amit Kundu, Kyeong Seok Kim, Yura Lee, Kyungsil Yoon, Sungpil Yoon, Byung Mu Lee, Ki Taek Nam, and Hyung Sik Kim. 2019. "Knockdown of Pyruvate Kinase M2 Inhibits Cell Proliferation, Metabolism, and Migration in Renal Cell Carcinoma" International Journal of Molecular Sciences 20, no. 22: 5622. https://doi.org/10.3390/ijms20225622
APA StyleDey, P., Son, J. Y., Kundu, A., Kim, K. S., Lee, Y., Yoon, K., Yoon, S., Lee, B. M., Nam, K. T., & Kim, H. S. (2019). Knockdown of Pyruvate Kinase M2 Inhibits Cell Proliferation, Metabolism, and Migration in Renal Cell Carcinoma. International Journal of Molecular Sciences, 20(22), 5622. https://doi.org/10.3390/ijms20225622