Druggable Biochemical Pathways and Potential Therapeutic Alternatives to Target Leukemic Stem Cells and Eliminate the Residual Disease in Chronic Myeloid Leukemia


Abstract
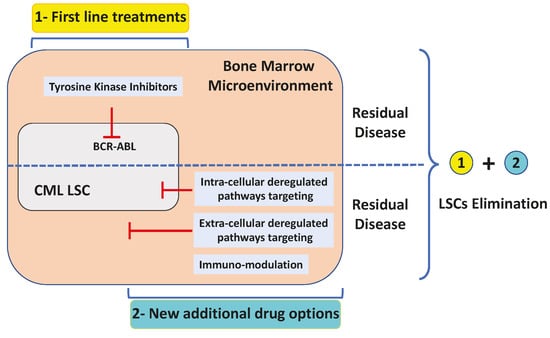
1. Introduction
2. Leukemic Stem Cells: Cannot See the Wood for the Trees
3. BCR-ABL Dependent and Independent Resistances
3.1. BCR-ABL-Dependent Resistances and Alternative Solutions
3.2. BCR-ABL-Independent Resistances
3.2.1. Drug Transporters
3.2.2. Targeting Epigenetic Dysregulation in CML
3.2.3. Apoptosis Defects in Resistance
3.2.4. Role of Autophagy in CML Resistances
3.2.5. Influence of the Microenvironment on CML Resistance
3.2.6. Functional Cross-Talks between the Microenvironment and Immunotherapy in CML
3.2.7. Tumor Microenvironment Impacts CML Cell’s Metabolism
3.2.8. Targeting Signaling Pathways Connected to Key Leukemic Stem Cells Features
4. Conclusions
Funding
Conflicts of Interest
Abbreviations
| 15-LO | 15-Lipoxygenase |
| 4E-BP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| A-loop | Activation-loop |
| AA | Arachidonic acid |
| ABC | ATP-binding cassette |
| ABCB1/MCR1 | Multidrug resistance protein 1/Permeability glycoprotein |
| ABCC1/MRP1 | Multidrug resistance-associated protein 1 |
| ABCG2/BCRP | ATP-binding cassette super-family G member 2 |
| AKT/PKB | Protein kinase B |
| AML | Acute Myeloid Leukemia |
| AMP | Adenosine Monophosphate |
| AMPK | AMP-activated protein kinase |
| AP | Adenomatous Polyposis Coli |
| Ara-C | Cytarabine |
| ATG | Autophagy related genes |
| ATP | Adenosine Tri-Phosphate |
| AXL | Tyrosine-protein kinase receptor UFO |
| BCL-2 | B-cell lymphoma 2 |
| BCL6 | B-cell lymphoma 6 |
| Bcl-xL | B-cell lymphoma-extra large |
| BCR-ABL | Breakpoint Cluster Region-Abelson murine leukemia viral oncogene homolog 1 |
| BET | Bromodomain Extra-Terminal motif |
| BH3 | Bcl-2 homology domain 3 |
| BIM | Bcl-2-like protein 11 |
| BLT2 | Leukotriene B4 receptor 2 |
| BM | Bone Marrow |
| BMI1 | B cell-specific Moloney murine leukemia virus integration site 1 |
| BMM | Bone marrow microenvironment |
| BMP | Bone morphogenetic proteins |
| BP-CML | Blastic phase chronic myeloid leukemia |
| c-ABL | Abelson murine leukemia viral oncogene homolog 1 |
| C/EBPβ | CCAAT/Enhancer-binding Protein beta |
| CBL | Casitas B-lineage Lymphoma |
| CCNG2 | Cyclin G2 |
| CCR | Complete Cytogenetic Remission |
| CHR | Complete Hematological Remission |
| CITED2 | Cbp/p300-interacting transactivator 2 |
| CpN | Cyclophilin |
| CML | Chronic Myeloid Leukemia |
| CQ | Chloroquine |
| CRK | CT10 Regulator of Kinase |
| CRKL | Crk-like protein |
| CsA | Cyclosporine |
| CXCL12 | C-X-C motif chemokine 12 |
| CXCR4 | C-X-C chemokine receptor type 4 |
| Cyt | Cytokines |
| CytR | Cytokine receptors |
| DNA | Deoxyribonucleic acid |
| DVL | Dishevelled |
| ECAR | Extracellular Acidification Rate |
| EIF4E | Eukaryotic translation initiation factor 4E |
| ERK | Extracellular signal-regulated kinases |
| ETC | Electron Transport Chain |
| EZH2 | Enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit |
| FOXO | Forkhead box protein O |
| GAB2 | GRB2-associated-binding protein 2 |
| GAS6 | Growth arrest–specific 6 |
| GLI1 | Glioma-associated oncogene homologue 1 |
| GRB2 | Growth factor receptor-bound protein 2 |
| GSK-3β | Glycogen Synthase Kinase 3 Beta |
| H3K27me3 | Histone H3 trimethylase activity at lysine 27 |
| HCQ | Hydroxychloroquine |
| HIF-2α | Hypoxia-inducible factor-2 alpha |
| HDAC | Histone deacetylase |
| HSC | Hematopoietic Stem Cell |
| IFN | Interferon |
| IL | Interleukin |
| ILR | Interleukin Receptor |
| JAK | Janus-family tyrosine kinase |
| LC3 | microtubule-associated protein 1 light chain 3 |
| LIC | Leukemia-Initiating Cells |
| LRP5/6 | Low density lipoprotein receptor-related protein 5/6 |
| LSC | Leukemic Stem Cell |
| LT | Leukotriene |
| LTB4 | Leukotriene B4 |
| MCL-1 | induced myeloid leukemia cell differentiation protein |
| MCR | Major Cytogenetic Remission |
| MDR | Multidrug Resistance |
| MDM2 | Mouse double minute 2 homolog |
| MEK | Mitogen-activated protein kinase kinase; |
| MHC | Major histocompability complex |
| miRNA | Micro ribo nucleic acid; |
| MNK | MAP kinase-interacting serine/threonine-protein kinase |
| MPT | Mitochondrial protein translation |
| mTOR | mechanistic Target Of Rapamycin |
| NFAT | Nuclear factor of activated T-cells |
| NSG (NOD/SCID) | Nonobese diabetic/severe combined immunodeficient |
| OCR | Oxygen Consumption Rate |
| P-loop | Phosphate-binding loop |
| PAK1 | Serine/threonine-protein kinase 1 |
| PD-1 | Programmed cell death 1 |
| PD-L1 | Programmed death-ligand 1 |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K-III | Phosphoinositide 3-kinase class III |
| PORCN | Porcupine O-acyltransferase |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PRC | Protein regulator of cytokinesis |
| PTCH | Protein patched homolog 1 |
| RAF | Rapidly accelerated fibrosarcoma kinases |
| RPS6 | Ribosomal protein S6 |
| SH2/SH3 | SRC Homology 2/3 Domain |
| SHC | SH2-contaltining collagen-related proteins |
| SHH | Sonic Hedgehog |
| SIRT1 | Sirtuin 1 |
| SMO | Smoothened |
| SOS | Son Of Sevenless guanine nucleotide exchange factor |
| STAT | Signal transducer and activator of transcription; |
| TCR | T cell receptor |
| TGF-β1 | Transforming growth factor beta 1 |
| TGF-βR | abrogated transforming growth factor beta receptor |
| TKI | Tyrosine Kinase Inhibitor |
| TNF | Tumor Necrosis Factor |
| Ub | Ubiquitin |
| ULK1 | unc-51 like autophagy activating kinase 1 |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| WNT | Wingless-related integration site |
References
- Cilloni, D.; Saglio, G. Molecular pathways: BCR-ABL. Clin. Cancer Res. 2012, 18, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M. Optimizing treatment of chronic myeloid leukemia: A rational approach. Oncologist 2004, 9, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Kaleem, B.; Shahab, S.; Ahmed, N.; Shamsi, T.S. Chronic Myeloid Leukemia--Prognostic Value of Mutations. Asian Pac. J. Cancer Prev. 2015, 16, 7415–7423. [Google Scholar] [CrossRef]
- Caldemeyer, L.; Dugan, M.; Edwards, J.; Akard, L. Long-Term Side Effects of Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Curr. Hematol. Malig. Rep. 2016, 11, 71–79. [Google Scholar] [CrossRef]
- Mahon, F.X.; Rea, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Eide, C.A.; Zabriskie, M.S.; Savage Stevens, S.L.; Antelope, O.; Vellore, N.A.; Than, H.; Schultz, A.R.; Clair, P.; Bowler, A.D.; Pomicter, A.D.; et al. Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell 2019, 36, 431–443 e435. [Google Scholar] [CrossRef]
- Scott, M.T.; Korfi, K.; Saffrey, P.; Hopcroft, L.E.; Kinstrie, R.; Pellicano, F.; Guenther, C.; Gallipoli, P.; Cruz, M.; Dunn, K.; et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov. 2016, 6, 1248–1257. [Google Scholar] [CrossRef]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Fiskus, W.; Pranpat, M.; Balasis, M.; Bali, P.; Estrella, V.; Kumaraswamy, S.; Rao, R.; Rocha, K.; Herger, B.; Lee, F.; et al. Cotreatment with vorinostat (suberoylanilide hydroxamic acid) enhances activity of dasatinib (BMS-354825) against imatinib mesylate-sensitive or imatinib mesylate-resistant chronic myelogenous leukemia cells. Clin. Cancer Res. 2006, 12, 5869–5878. [Google Scholar] [CrossRef]
- Naka, K.; Ishihara, K.; Jomen, Y.; Jin, C.H.; Kim, D.H.; Gu, Y.K.; Jeong, E.S.; Li, S.; Krause, D.S.; Kim, D.W.; et al. Novel oral transforming growth factor-beta signaling inhibitor EW-7197 eradicates CML-initiating cells. Cancer Sci. 2016, 107, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Cervera, E.; Candelaria, M.; Lopez-Navarro, O.; Labardini, J.; Gonzalez-Fierro, A.; Taja-Chayeb, L.; Cortes, J.; Gordillo-Bastidas, D.; Duenas-Gonzalez, A. Epigenetic therapy with hydralazine and magnesium valproate reverses imatinib resistance in patients with chronic myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2012, 12, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, L.; Li, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.; Bhatia, R. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 2012, 21, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Qiu, S.; Chacko, B.K.; Li, H.; Paterson, A.; He, J.; Agarwal, P.; Shah, M.; Welner, R.; Darley-Usmar, V.M.; et al. SIRT1 regulates metabolism and leukemogenic potential in CML stem cells. J. Clin. Investig. 2019, 130, 2685–2701. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yang, L.; Zhao, M.; Zhu, S.; Kang, R.; Vernon, P.; Tang, D.; Cao, L. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia 2012, 26, 1752–1760. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, X.; Feng, J.; Zhang, X. Overexpression of miR-202 resensitizes imatinib resistant chronic myeloid leukemia cells through targetting Hexokinase 2. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.S.; Li, L.; Li, L.; Chu, S.; Shiang, K.D.; Li, M.; Sun, H.Y.; Xu, J.; Xiao, F.J.; Sun, G.; et al. MicroRNA-486 regulates normal erythropoiesis and enhances growth and modulates drug response in CML progenitors. Blood 2015, 125, 1302–1313. [Google Scholar] [CrossRef]
- Tu, Y.X.; Wang, S.B.; Fu, L.Q.; Li, S.S.; Guo, Q.P.; Wu, Y.; Mou, X.Z.; Tong, X.M. Ovatodiolide targets chronic myeloid leukemia stem cells by epigenetically upregulating hsa-miR-155, suppressing the BCR-ABL fusion gene and dysregulating the PI3K/AKT/mTOR pathway. Oncotarget 2018, 9, 3267–3277. [Google Scholar] [CrossRef]
- Hurtz, C.; Hatzi, K.; Cerchietti, L.; Braig, M.; Park, E.; Kim, Y.M.; Herzog, S.; Ramezani-Rad, P.; Jumaa, H.; Muller, M.C.; et al. BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J. Exp. Med. 2011, 208, 2163–2174. [Google Scholar] [CrossRef]
- Madapura, H.S.; Nagy, N.; Ujvari, D.; Kallas, T.; Krohnke, M.C.L.; Amu, S.; Bjorkholm, M.; Stenke, L.; Mandal, P.K.; McMurray, J.S.; et al. Interferon gamma is a STAT1-dependent direct inducer of BCL6 expression in imatinib-treated chronic myeloid leukemia cells. Oncogene 2017, 36, 4619–4628. [Google Scholar] [CrossRef]
- Goff, D.J.; Court Recart, A.; Sadarangani, A.; Chun, H.J.; Barrett, C.L.; Krajewska, M.; Leu, H.; Low-Marchelli, J.; Ma, W.; Shih, A.Y.; et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 2013, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Mak, D.H.; Wang, R.Y.; Schober, W.D.; Konopleva, M.; Cortes, J.; Kantarjian, H.; Andreeff, M.; Carter, B.Z. Activation of apoptosis signaling eliminates CD34+ progenitor cells in blast crisis CML independent of response to tyrosine kinase inhibitors. Leukemia 2012, 26, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Airiau, K.; Mahon, F.X.; Josselin, M.; Jeanneteau, M.; Turcq, B.; Belloc, F. ABT-737 increases tyrosine kinase inhibitor-induced apoptosis in chronic myeloid leukemia cells through XIAP downregulation and sensitizes CD34+ CD38(-) population to imatinib. Exp. Hematol. 2012, 40, 367–378.e362. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Mak, P.Y.; Mak, D.H.; Ruvolo, V.R.; Schober, W.; McQueen, T.; Cortes, J.; Kantarjian, H.M.; Champlin, R.E.; Konopleva, M.; et al. Synergistic effects of p53 activation via MDM2 inhibition in combination with inhibition of Bcl-2 or Bcr-Abl in CD34+ proliferating and quiescent chronic myeloid leukemia blast crisis cells. Oncotarget 2015, 6, 30487–30499. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Mak, P.Y.; Mu, H.; Wang, X.; Tao, W.; Mak, D.H.; Dettman, E.J.; Cardone, M.; Zernovak, O.; Seki, T.; et al. Combined inhibition of MDM2 and Bcr-Abl tyrosine kinase targets chronic myeloid leukemia stem/progenitor cells in a murine model. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.K.; Chuah, C.T.; Huang, J.W.; Ng, K.P.; Ong, S.T. The BCL2 inhibitor ABT-199 significantly enhances imatinib-induced cell death in chronic myeloid leukemia progenitors. Oncotarget 2014, 5, 9033–9038. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Mak, P.Y.; Mu, H.; Zhou, H.; Mak, D.H.; Schober, W.; Leverson, J.D.; Zhang, B.; Bhatia, R.; Huang, X.; et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci. Transl. Med. 2016, 8, 355ra117. [Google Scholar] [CrossRef]
- Calabretta, B.; Salomoni, P. Inhibition of autophagy: A new strategy to enhance sensitivity of chronic myeloid leukemia stem cells to tyrosine kinase inhibitors. Leuk. Lymphoma 2011, 52, 54–59. [Google Scholar] [CrossRef]
- Pena, M.C.R.; Pardini, M.I.M.C.; Colturato, V.A.R.; Pinheiro, N.A. Methylation status of the SOCS 1 and JUNB genes in chronic myeloid leukemia patients. Rev. Bras. Hematol. Hemoter. 2009, 31, 147–152. [Google Scholar] [CrossRef]
- Carella, A.M.; Beltrami, G.; Catania, G.; Pica, G.; Ghiggi, C.; Garuti, A.; Carella, A. Inhibition of autophagy with clarithromycin: A new strategy to enhance sensitivity of CML stem cells to tyrosine kinase inhibitors. Leuk. Suppl. 2012, 1, S49–S50. [Google Scholar] [CrossRef][Green Version]
- Shao, S.; Li, S.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Tauchi, T.; Okabe, S.; Minami, Y.; Kimura, S.; Maekawa, T.; Naoe, T.; Ohyashiki, K. Combination of ponatinib with Hedgehog antagonist vismodegib for therapy-resistant BCR-ABL1-positive leukemia. Clin. Cancer Res. 2013, 19, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Vianello, F.; Villanova, F.; Tisato, V.; Lymperi, S.; Ho, K.K.; Gomes, A.R.; Marin, D.; Bonnet, D.; Apperley, J.; Lam, E.W.; et al. Bone marrow mesenchymal stromal cells non-selectively protect chronic myeloid leukemia cells from imatinib-induced apoptosis via the CXCR4/CXCL12 axis. Haematologica 2010, 95, 1081–1089. [Google Scholar] [CrossRef]
- Agarwal, P.; Zhang, B.; Ho, Y.; Cook, A.; Li, L.; Mikhail, F.M.; Wang, Y.; McLaughlin, M.E.; Bhatia, R. Enhanced targeting of CML stem and progenitor cells by inhibition of porcupine acyltransferase in combination with TKI. Blood 2017, 129, 1008–1020. [Google Scholar] [CrossRef]
- Zhou, H.; Mak, P.Y.; Mu, H.; Mak, D.H.; Zeng, Z.; Cortes, J.; Liu, Q.; Andreeff, M.; Carter, B.Z. Combined inhibition of beta-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 2017, 31, 2065–2074. [Google Scholar] [CrossRef]
- Jin, Y.; Nie, D.; Li, J.; Du, X.; Lu, Y.; Li, Y.; Liu, C.; Zhou, J.; Pan, J. Gas6/AXL Signaling Regulates Self-Renewal of Chronic Myelogenous Leukemia Stem Cells by Stabilizing beta-Catenin. Clin. Cancer Res. 2017, 23, 2842–2855. [Google Scholar] [CrossRef]
- Dillmann, F.; Veldwijk, M.R.; Laufs, S.; Sperandio, M.; Calandra, G.; Wenz, F.; Zeller, J.; Fruehauf, S. Plerixafor inhibits chemotaxis toward SDF-1 and CXCR4-mediated stroma contact in a dose-dependent manner resulting in increased susceptibility of BCR-ABL+ cell to Imatinib and Nilotinib. Leuk. Lymphoma 2009, 50, 1676–1686. [Google Scholar] [CrossRef]
- Helgason, G.V.; Karvela, M.; Holyoake, T.L. Kill one bird with two stones: Potential efficacy of BCR-ABL and autophagy inhibition in CML. Blood 2011, 118, 2035–2043. [Google Scholar] [CrossRef]
- Beider, K.; Darash-Yahana, M.; Blaier, O.; Koren-Michowitz, M.; Abraham, M.; Wald, H.; Wald, O.; Galun, E.; Eizenberg, O.; Peled, A.; et al. Combination of imatinib with CXCR4 antagonist BKT140 overcomes the protective effect of stroma and targets CML in vitro and in vivo. Mol. Cancer 2014, 13, 1155–1169. [Google Scholar] [CrossRef]
- Kuepper, M.K.; Butow, M.; Herrmann, O.; Ziemons, J.; Chatain, N.; Maurer, A.; Kirschner, M.; Maie, T.; Costa, I.G.; Eschweiler, J.; et al. Stem cell persistence in CML is mediated by extrinsically activated JAK1-STAT3 signaling. Leukemia 2019, 33, 1964–1977. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, X.; Liu, Y.; Yang, L.; Huang, S.; Lu, L.; Wang, S.; Guo, Q.; Zhao, L. BM microenvironmental protection of CML cells from imatinib through Stat5/NF-kappaB signaling and reversal by Wogonin. Oncotarget 2016, 7, 24436–24454. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Y.; Wan, H.; Hu, J. Antibiotic ivermectin selectively induces apoptosis in chronic myeloid leukemia through inducing mitochondrial dysfunction and oxidative stress. Biochem. Biophys. Res. Commun. 2018, 497, 241–247. [Google Scholar] [CrossRef]
- Riether, C.; Schurch, C.M.; Flury, C.; Hinterbrandner, M.; Druck, L.; Huguenin, A.L.; Baerlocher, G.M.; Radpour, R.; Ochsenbein, A.F. Tyrosine kinase inhibitor-induced CD70 expression mediates drug resistance in leukemia stem cells by activating Wnt signaling. Sci. Transl. Med. 2015, 7, 298ra119. [Google Scholar] [CrossRef]
- Irvine, D.A.; Zhang, B.; Kinstrie, R.; Tarafdar, A.; Morrison, H.; Campbell, V.L.; Moka, H.A.; Ho, Y.; Nixon, C.; Manley, P.W.; et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Sci. Rep. 2016, 6, 25476. [Google Scholar] [CrossRef]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef]
- Lin, H.; Chen, M.; Rothe, K.; Lorenzi, M.V.; Woolfson, A.; Jiang, X. Selective JAK2/ABL dual inhibition therapy effectively eliminates TKI-insensitive CML stem/progenitor cells. Oncotarget 2014, 5, 8637–8650. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Lv, C.; Wang, L.; Song, H. Anthelmintic drug niclosamide enhances the sensitivity of chronic myeloid leukemia cells to dasatinib through inhibiting Erk/Mnk1/eIF4E pathway. Biochem. Biophys. Res. Commun. 2016, 478, 893–899. [Google Scholar] [CrossRef]
- Gallipoli, P.; Cook, A.; Rhodes, S.; Hopcroft, L.; Wheadon, H.; Whetton, A.D.; Jorgensen, H.G.; Bhatia, R.; Holyoake, T.L. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood 2014, 124, 1492–1501. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, C.; Abraham, S.A.; Shan, Y.; Guo, Z.; Desouza, N.; Cheloni, G.; Li, D.; Holyoake, T.L.; Li, S. Arachidonate 15-lipoxygenase is required for chronic myeloid leukemia stem cell survival. J. Clin. Investig. 2014, 124, 3847–3862. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Peng, C.; Huang, J.; Li, B.E.; Kim, W.; Smith, E.C.; Fujiwara, Y.; Qi, J.; Cheloni, G.; Das, P.P.; et al. Chronic Myelogenous Leukemia- Initiating Cells Require Polycomb Group Protein EZH2. Cancer Discov. 2016, 6, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Rauzan, M.; Chuah, C.T.; Ko, T.K.; Ong, S.T. The HDAC inhibitor SB939 overcomes resistance to BCR-ABL kinase Inhibitors conferred by the BIM deletion polymorphism in chronic myeloid leukemia. PLoS ONE 2017, 12, e0174107. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhou, J.; Xu, F.; Jin, B.; Cui, L.; Wang, Y.; Du, X.; Li, J.; Li, P.; Ren, R.; et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J. Clin. Investig. 2016, 126, 3961–3980. [Google Scholar] [CrossRef]
- Mitchell, R.; Hopcroft, L.E.M.; Baquero, P.; Allan, E.K.; Hewit, K.; James, D.; Hamilton, G.; Mukhopadhyay, A.; O’Prey, J.; Hair, A.; et al. Targeting BCR-ABL-Independent TKI Resistance in Chronic Myeloid Leukemia by mTOR and Autophagy Inhibition. J. Natl. Cancer Inst. 2018, 110, 467–478. [Google Scholar] [CrossRef]
- Zeng, X.; Zhao, H.; Li, Y.; Fan, J.; Sun, Y.; Wang, S.; Wang, Z.; Song, P.; Ju, D. Targeting Hedgehog signaling pathway and autophagy overcomes drug resistance of BCR-ABL-positive chronic myeloid leukemia. Autophagy 2015, 11, 355–372. [Google Scholar] [CrossRef]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef]
- Tarafdar, A.; Hopcroft, L.E.; Gallipoli, P.; Pellicano, F.; Cassels, J.; Hair, A.; Korfi, K.; Jorgensen, H.G.; Vetrie, D.; Holyoake, T.L.; et al. CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood 2017, 129, 199–208. [Google Scholar] [CrossRef]
- Karvela, M.; Baquero, P.; Kuntz, E.M.; Mukhopadhyay, A.; Mitchell, R.; Allan, E.K.; Chan, E.; Kranc, K.R.; Calabretta, B.; Salomoni, P.; et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy 2016, 12, 936–948. [Google Scholar] [CrossRef]
- Babashah, S.; Sadeghizadeh, M.; Hajifathali, A.; Tavirani, M.R.; Zomorod, M.S.; Ghadiani, M.; Soleimani, M. Targeting of the signal transducer Smo links microRNA-326 to the oncogenic Hedgehog pathway in CD34+ CML stem/progenitor cells. Int. J. Cancer 2013, 133, 579–589. [Google Scholar] [CrossRef]
- Pellicano, F.; Simara, P.; Sinclair, A.; Helgason, G.V.; Copland, M.; Grant, S.; Holyoake, T.L. The MEK inhibitor PD184352 enhances BMS-214662-induced apoptosis in CD34+ CML stem/progenitor cells. Leukemia 2011, 25, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Saw, T.Y.; Zhang, M.; Janes, M.R.; Nacro, K.; Hill, J.; Lim, A.Q.; Chang, C.T.; Fruman, D.A.; Rizzieri, D.A.; et al. Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proc. Natl. Acad. Sci. USA 2013, 110, E2298–E2307. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Ai, H.; Li, T.; Rajoria, P.; Shahu, P.; Li, X. Targeting of the BLT2 in chronic myeloid leukemia inhibits leukemia stem/progenitor cell function. Biochem. Biophys. Res. Commun. 2016, 472, 610–616. [Google Scholar] [CrossRef]
- Abraham, S.A.; Hopcroft, L.E.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 2016, 534, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Holyoake, T.; Jiang, X.; Eaves, C.; Eaves, A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 1999, 94, 2056–2064. [Google Scholar] [CrossRef]
- Uckun, F.M.; Sather, H.; Reaman, G.; Shuster, J.; Land, V.; Trigg, M.; Gunther, R.; Chelstrom, L.; Bleyer, A.; Gaynon, P.; et al. Leukemic cell growth in SCID mice as a predictor of relapse in high-risk B-lineage acute lymphoblastic leukemia. Blood 1995, 85, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Muller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.J.; Holyoake, T.L. Preclinical approaches in chronic myeloid leukemia: From cells to systems. Exp. Hematol. 2017, 47, 13–23. [Google Scholar] [CrossRef]
- Milojkovic, D.; Apperley, J. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin. Cancer Res. 2009, 15, 7519–7527. [Google Scholar] [CrossRef] [PubMed]
- Zabriskie, M.S.; Eide, C.A.; Tantravahi, S.K.; Vellore, N.A.; Estrada, J.; Nicolini, F.E.; Khoury, H.J.; Larson, R.A.; Konopleva, M.; Cortes, J.E.; et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell 2014, 26, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.J.; Palaiologou, D.; Panousopoulou, E.; Schultheis, B.; Yong, A.S.; Wong, A.; Pattacini, L.; Goldman, J.M.; Melo, J.V. Bcr-Abl expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res. 2005, 65, 8912–8919. [Google Scholar] [CrossRef]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem. 2000, 275, 24273–24278. [Google Scholar] [CrossRef]
- Kim, J.H.; Chu, S.C.; Gramlich, J.L.; Pride, Y.B.; Babendreier, E.; Chauhan, D.; Salgia, R.; Podar, K.; Griffin, J.D.; Sattler, M. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood 2005, 105, 1717–1723. [Google Scholar] [CrossRef]
- Burger, H.; van Tol, H.; Boersma, A.W.; Brok, M.; Wiemer, E.A.; Stoter, G.; Nooter, K. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 2004, 104, 2940–2942. [Google Scholar] [CrossRef]
- Breedveld, P.; Pluim, D.; Cipriani, G.; Wielinga, P.; van Tellingen, O.; Schinkel, A.H.; Schellens, J.H. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): Implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res. 2005, 65, 2577–2582. [Google Scholar] [CrossRef] [PubMed]
- Dohse, M.; Scharenberg, C.; Shukla, S.; Robey, R.W.; Volkmann, T.; Deeken, J.F.; Brendel, C.; Ambudkar, S.V.; Neubauer, A.; Bates, S.E. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab. Dispos. 2010, 38, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Strauss, A.C.; Chu, S.; Li, M.; Ho, Y.; Shiang, K.D.; Snyder, D.S.; Huettner, C.S.; Shultz, L.; Holyoake, T.; et al. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell 2010, 17, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Kinstrie, R.; Karamitros, D.; Goardon, N.; Morrison, H.; Hamblin, M.; Robinson, L.; Clark, R.E.; Copland, M.; Vyas, P. Heterogeneous leukemia stem cells in myeloid blast phase chronic myeloid leukemia. Blood Adv. 2016, 1, 160–169. [Google Scholar] [CrossRef]
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017, 129, 1595–1606. [Google Scholar] [CrossRef]
- Koschmieder, S.; Vetrie, D. Epigenetic dysregulation in chronic myeloid leukaemia: A myriad of mechanisms and therapeutic options. Semin. Cancer Biol. 2018, 51, 180–197. [Google Scholar] [CrossRef]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef]
- Laugesen, A.; Helin, K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell 2014, 14, 735–751. [Google Scholar] [CrossRef]
- Lessard, J.; Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003, 423, 255–260. [Google Scholar] [CrossRef]
- Rizo, A.; Horton, S.J.; Olthof, S.; Dontje, B.; Ausema, A.; van Os, R.; van den Boom, V.; Vellenga, E.; de Haan, G.; Schuringa, J.J. BMI1 collaborates with BCR-ABL in leukemic transformation of human CD34+ cells. Blood 2010, 116, 4621–4630. [Google Scholar] [CrossRef]
- Saudy, N.S.; Fawzy, I.M.; Azmy, E.; Goda, E.F.; Eneen, A.; Abdul Salam, E.M. BMI1 gene expression in myeloid leukemias and its impact on prognosis. Blood Cells Mol. Dis. 2014, 53, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Mourgues, L.; Imbert, V.; Nebout, M.; Colosetti, P.; Neffati, Z.; Lagadec, P.; Verhoeyen, E.; Peng, C.; Duprez, E.; Legros, L.; et al. The BMI1 polycomb protein represses cyclin G2-induced autophagy to support proliferation in chronic myeloid leukemia cells. Leukemia 2015, 29, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Mohty, M.; Yong, A.S.; Szydlo, R.M.; Apperley, J.F.; Melo, J.V. The polycomb group BMI1 gene is a molecular marker for predicting prognosis of chronic myeloid leukemia. Blood 2007, 110, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Branford, S.; Wang, P.; Yeung, D.T.; Thomson, D.; Purins, A.; Wadham, C.; Shahrin, N.H.; Marum, J.E.; Nataren, N.; Parker, W.T.; et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood 2018, 132, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Boultwood, J.; Perry, J.; Zaman, R.; Fernandez-Santamaria, C.; Littlewood, T.; Kusec, R.; Pellagatti, A.; Wang, L.; Clark, R.E.; Wainscoat, J.S. High-density single nucleotide polymorphism array analysis and ASXL1 gene mutation screening in chronic myeloid leukemia during disease progression. Leukemia 2010, 24, 1139–1145. [Google Scholar] [CrossRef]
- Menezes, J.; Salgado, R.N.; Acquadro, F.; Gomez-Lopez, G.; Carralero, M.C.; Barroso, A.; Mercadillo, F.; Espinosa-Hevia, L.; Talavera-Casanas, J.G.; Pisano, D.G.; et al. ASXL1, TP53 and IKZF3 mutations are present in the chronic phase and blast crisis of chronic myeloid leukemia. Blood Cancer J. 2013, 3, e157. [Google Scholar] [CrossRef]
- Yang, H.; Kurtenbach, S.; Guo, Y.; Lohse, I.; Durante, M.A.; Li, J.; Li, Z.; Al-Ali, H.; Li, L.; Chen, Z.; et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 2018, 131, 328–341. [Google Scholar] [CrossRef]
- Nimmanapalli, R.; Fuino, L.; Stobaugh, C.; Richon, V.; Bhalla, K. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cells. Blood 2003, 101, 3236–3239. [Google Scholar] [CrossRef]
- Nimmanapalli, R.; Fuino, L.; Bali, P.; Gasparetto, M.; Glozak, M.; Tao, J.; Moscinski, L.; Smith, C.; Wu, J.; Jove, R.; et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003, 63, 5126–5135. [Google Scholar]
- Fiskus, W.; Pranpat, M.; Bali, P.; Balasis, M.; Kumaraswamy, S.; Boyapalle, S.; Rocha, K.; Wu, J.; Giles, F.; Manley, P.W.; et al. Combined effects of novel tyrosine kinase inhibitor AMN107 and histone deacetylase inhibitor LBH589 against Bcr-Abl-expressing human leukemia cells. Blood 2006, 108, 645–652. [Google Scholar] [CrossRef]
- Wang, Z.; Yuan, H.; Roth, M.; Stark, J.M.; Bhatia, R.; Chen, W.Y. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene 2013, 32, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Wang, Z.; Li, L.; Zhang, H.; Modi, H.; Horne, D.; Bhatia, R.; Chen, W. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 2012, 119, 1904–1914. [Google Scholar] [CrossRef]
- Roman-Gomez, J.; Castillejo, J.A.; Jimenez, A.; Cervantes, F.; Boque, C.; Hermosin, L.; Leon, A.; Granena, A.; Colomer, D.; Heiniger, A.; et al. Cadherin-13, a mediator of calcium-dependent cell-cell adhesion, is silenced by methylation in chronic myeloid leukemia and correlates with pretreatment risk profile and cytogenetic response to interferon alfa. J. Clin. Oncol. 2003, 21, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- San Jose-Eneriz, E.; Agirre, X.; Jimenez-Velasco, A.; Cordeu, L.; Martin, V.; Arqueros, V.; Garate, L.; Fresquet, V.; Cervantes, F.; Martinez-Climent, J.A.; et al. Epigenetic down-regulation of BIM expression is associated with reduced optimal responses to imatinib treatment in chronic myeloid leukaemia. Eur. J. Cancer 2009, 45, 1877–1889. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.; Kim, D.Y.; Park, S.H.; Byun, H.M.; Kim, I.; Yoon, S.S.; Kim, B.K.; Park, E.; Yang, A.S.; Park, S. Increased BCR promoter DNA methylation status strongly correlates with favorable response to imatinib in chronic myeloid leukemia patients. Oncol. Lett. 2011, 2, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.C.; Lin, S.F.; Chang, J.G.; Yang, M.Y.; Hung, S.Y.; Chang, C.S. Epigenetic alteration of the SOCS1 gene in chronic myeloid leukaemia. Br. J. Haematol. 2003, 123, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lin, J.; Qian, J.; Deng, Z.Q.; Qian, W.; Yang, J.; Li, Y.; Chen, X.X.; Ma, Y.J.; Ma, J.C.; et al. The methylation status of the DDX43 promoter in Chinese patients with chronic myeloid leukemia. Genet. Test. Mol. Biomark. 2013, 17, 508–511. [Google Scholar] [CrossRef]
- Ahmad, I.; Mir, R.; Javid, J.; Farooq, S.; Yadav, P.; Zuberi, M.; Masroor, M.; Gupta, N.; Ray, P.C.; Saxena, A. Epigenetic Silencing of P16 (INK4a) Gene by Promoter Hypermethylation in Chronic Myelogenous Leukemia. Clin. Lymphoma Myeloma Leuk. 2014, 14, S139. [Google Scholar] [CrossRef]
- Nagy, E.; Beck, Z.; Kiss, A.; Csoma, E.; Telek, B.; Konya, J.; Olah, E.; Rak, K.; Toth, F.D. Frequent methylation of p16INK4A and p14ARF genes implicated in the evolution of chronic myeloid leukaemia from its chronic to accelerated phase. Eur. J. Cancer 2003, 39, 2298–2305. [Google Scholar] [CrossRef]
- Bodoor, K.; Haddad, Y.; Alkhateeb, A.; Al-Abbadi, A.; Dowairi, M.; Magableh, A.; Bsoul, N.; Ghabkari, A. DNA hypermethylation of cell cycle (p15 and p16) and apoptotic (p14, p53, DAPK and TMS1) genes in peripheral blood of leukemia patients. Asian Pac. J. Cancer Prev. 2014, 15, 75–84. [Google Scholar] [CrossRef]
- Qian, J.; Wang, Y.L.; Lin, J.; Yao, D.M.; Xu, W.R.; Wu, C.Y. Aberrant methylation of the death-associated protein kinase 1 (DAPK1) CpG island in chronic myeloid leukemia. Eur. J. Haematol. 2009, 82, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Pehlivan, M.; Sercan, Z.; Sercan, H.O. sFRP1 promoter methylation is associated with persistent Philadelphia chromosome in chronic myeloid leukemia. Leuk. Res. 2009, 33, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, J.; Gharibyan, V.; Estecio, M.R.; Kondo, K.; He, R.; Chung, W.; Lu, Y.; Zhang, N.; Liang, S.; Kantarjian, H.M.; et al. Aberrant DNA methylation is associated with disease progression, resistance to imatinib and shortened survival in chronic myelogenous leukemia. PLoS ONE 2011, 6, e22110. [Google Scholar] [CrossRef] [PubMed]
- Maupetit-Mehouas, S.; Court, F.; Bourgne, C.; Guerci-Bresler, A.; Cony-Makhoul, P.; Johnson, H.; Etienne, G.; Rousselot, P.; Guyotat, D.; Janel, A.; et al. DNA methylation profiling reveals a pathological signature that contributes to transcriptional defects of CD34(+) CD15(-) cells in early chronic-phase chronic myeloid leukemia. Mol. Oncol. 2018, 12, 814–829. [Google Scholar] [CrossRef]
- Janssen, J.J.; Denkers, F.; Valk, P.; Cornelissen, J.J.; Schuurhuis, G.J.; Ossenkoppele, G.J. Methylation patterns in CD34 positive chronic myeloid leukemia blast crisis cells. Haematologica 2010, 95, 1036–1037. [Google Scholar] [CrossRef][Green Version]
- Oki, Y.; Kantarjian, H.M.; Gharibyan, V.; Jones, D.; O’Brien, S.; Verstovsek, S.; Cortes, J.; Morris, G.M.; Garcia-Manero, G.; Issa, J.P. Phase II study of low-dose decitabine in combination with imatinib mesylate in patients with accelerated or myeloid blastic phase of chronic myelogenous leukemia. Cancer 2007, 109, 899–906. [Google Scholar] [CrossRef]
- Pelechano, V.; Steinmetz, L.M. Gene regulation by antisense transcription. Nat. Rev. Genet. 2013, 14, 880–893. [Google Scholar] [CrossRef]
- Lu, Y.; Li, Y.; Chai, X.; Kang, Q.; Zhao, P.; Xiong, J.; Wang, J. Long noncoding RNA HULC promotes cell proliferation by regulating PI3K/AKT signaling pathway in chronic myeloid leukemia. Gene 2017, 607, 41–46. [Google Scholar] [CrossRef]
- Sun, C.; Luan, S.; Zhang, G.; Wang, N.; Shao, H.; Luan, C. CEBPA-mediated upregulation of the lncRNA PLIN2 promotes the development of chronic myelogenous leukemia via the GSK3 and Wnt/beta-catenin signaling pathways. Am. J. Cancer Res. 2017, 7, 1054–1067. [Google Scholar]
- Guo, G.; Kang, Q.; Zhu, X.; Chen, Q.; Wang, X.; Chen, Y.; Ouyang, J.; Zhang, L.; Tan, H.; Chen, R.; et al. A long noncoding RNA critically regulates Bcr-Abl-mediated cellular transformation by acting as a competitive endogenous RNA. Oncogene 2015, 34, 1768–1779. [Google Scholar] [CrossRef]
- Li, Z.; Yang, L.; Liu, X.; Nie, Z.; Luo, J. Long noncoding RNA MEG3 inhibits proliferation of chronic myeloid leukemia cells by sponging microRNA21. Biomed. Pharm. 2018, 104, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Agirre, X.; Jimenez-Velasco, A.; San Jose-Eneriz, E.; Garate, L.; Bandres, E.; Cordeu, L.; Aparicio, O.; Saez, B.; Navarro, G.; Vilas-Zornoza, A.; et al. Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol. Cancer Res. 2008, 6, 1830–1840. [Google Scholar] [CrossRef] [PubMed]
- Flamant, S.; Ritchie, W.; Guilhot, J.; Holst, J.; Bonnet, M.L.; Chomel, J.C.; Guilhot, F.; Turhan, A.G.; Rasko, J.E. Micro-RNA response to imatinib mesylate in patients with chronic myeloid leukemia. Haematologica 2010, 95, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Machova Polakova, K.; Lopotova, T.; Klamova, H.; Burda, P.; Trneny, M.; Stopka, T.; Moravcova, J. Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol. Cancer 2011, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lin, Z.; Du, J.; Zhou, X.; Yang, L.; Liu, G. Studies on microRNAs that are correlated with the cancer stem cells in chronic myeloid leukemia. Mol. Cell Biochem. 2014, 390, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Kotagama, K.; Chang, Y.; Mangone, M. miRNAs as Biomarkers in Chronic Myelogenous Leukemia. Drug Dev. Res. 2015, 76, 278–285. [Google Scholar] [CrossRef] [PubMed]
- San Jose-Eneriz, E.; Roman-Gomez, J.; Jimenez-Velasco, A.; Garate, L.; Martin, V.; Cordeu, L.; Vilas-Zornoza, A.; Rodriguez-Otero, P.; Calasanz, M.J.; Prosper, F.; et al. MicroRNA expression profiling in Imatinib-resistant Chronic Myeloid Leukemia patients without clinically significant ABL1-mutations. Mol. Cancer 2009, 8, 69. [Google Scholar] [CrossRef]
- Salati, S.; Salvestrini, V.; Carretta, C.; Genovese, E.; Rontauroli, S.; Zini, R.; Rossi, C.; Ruberti, S.; Bianchi, E.; Barbieri, G.; et al. Deregulated expression of miR-29a-3p, miR-494-3p and miR-660-5p affects sensitivity to tyrosine kinase inhibitors in CML leukemic stem cells. Oncotarget 2017, 8, 49451–49469. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, Y.; Tao, K.; Wang, X.; Xiao, Q.; Huang, Z.; Zhong, L.; Cao, W.; Wen, J.; Feng, W. Inhibition of BCR/ABL protein expression by miR-203 sensitizes for imatinib mesylate. PLoS ONE 2013, 8, e61858. [Google Scholar] [CrossRef]
- Zhang, B.; Nguyen, L.X.T.; Li, L.; Zhao, D.; Kumar, B.; Wu, H.; Lin, A.; Pellicano, F.; Hopcroft, L.; Su, Y.L.; et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat. Med. 2018, 24, 450–462. [Google Scholar] [CrossRef]
- Di Bacco, A.; Keeshan, K.; McKenna, S.L.; Cotter, T.G. Molecular abnormalities in chronic myeloid leukemia: Deregulation of cell growth and apoptosis. Oncologist 2000, 5, 405–415. [Google Scholar] [CrossRef][Green Version]
- Fernandez-Luna, J.L. Bcr-Abl and inhibition of apoptosis in chronic myelogenous leukemia cells. Apoptosis 2000, 5, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Tzifi, F.; Economopoulou, C.; Gourgiotis, D.; Ardavanis, A.; Papageorgiou, S.; Scorilas, A. The Role of BCL2 Family of Apoptosis Regulator Proteins in Acute and Chronic Leukemias. Adv. Hematol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Guillem, V.; Amat, P.; Collado, M.; Cervantes, F.; Alvarez-Larran, A.; Martinez, J.; Tormo, E.; Eroles, P.; Solano, C.; Hernandez-Boluda, J.C. BCL2 gene polymorphisms and splicing variants in chronic myeloid leukemia. Leuk. Res. 2015, 39, 1278–1284. [Google Scholar] [CrossRef]
- Kim, D.H.; Xu, W.; Ma, C.; Liu, X.; Siminovitch, K.; Messner, H.A.; Lipton, J.H. Genetic variants in the candidate genes of the apoptosis pathway and susceptibility to chronic myeloid leukemia. Blood 2009, 113, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- Deming, P.B.; Schafer, Z.T.; Tashker, J.S.; Potts, M.B.; Deshmukh, M.; Kornbluth, S. Bcr-Abl-mediated protection from apoptosis downstream of mitochondrial cytochrome c release. Mol. Cell Biol. 2004, 24, 10289–10299. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Rothe, K.; Porter, V.; Jiang, X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. Int. J. Mol. Sci. 2019, 20, 461. [Google Scholar] [CrossRef]
- Rothe, K.; Lin, H.; Lin, K.B.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, D.L.; Gorski, S.M.; Jiang, X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef]
- Sheng, Z.; Ma, L.; Sun, J.E.; Zhu, L.J.; Green, M.R. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood 2011, 118, 2840–2848. [Google Scholar] [CrossRef]
- Martinelli, G.; Oehler, V.G.; Papayannidis, C.; Courtney, R.; Shaik, M.N.; Zhang, X.; O’Connell, A.; McLachlan, K.R.; Zheng, X.; Radich, J.; et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: A phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015, 2, e339–e346. [Google Scholar] [CrossRef]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Ianniciello, A.; Dumas, P.Y.; Drullion, C.; Guitart, A.; Villacreces, A.; Peytour, Y.; Chevaleyre, J.; Brunet de la Grange, P.; Vigon, I.; Desplat, V.; et al. Chronic myeloid leukemia progenitor cells require autophagy when leaving hypoxia-induced quiescence. Oncotarget 2017, 8, 96984–96992. [Google Scholar] [CrossRef] [PubMed]
- Schepers, K.; Campbell, T.B.; Passegue, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef]
- Shah, M.; Bhatia, R. Preservation of Quiescent Chronic Myelogenous Leukemia Stem Cells by the Bone Marrow Microenvironment. Adv. Exp. Med. Biol. 2018, 1100, 97–110. [Google Scholar] [CrossRef]
- Zhang, B.; Ho, Y.W.; Huang, Q.; Maeda, T.; Lin, A.; Lee, S.U.; Hair, A.; Holyoake, T.L.; Huettner, C.; Bhatia, R. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 2012, 21, 577–592. [Google Scholar] [CrossRef]
- Bhatia, R.; Wayner, E.A.; McGlave, P.B.; Verfaillie, C.M. Interferon-alpha restores normal adhesion of chronic myelogenous leukemia hematopoietic progenitors to bone marrow stroma by correcting impaired beta 1 integrin receptor function. J. Clin. Investig. 1994, 94, 384–391. [Google Scholar] [CrossRef]
- Bhatia, R.; McCarthy, J.B.; Verfaillie, C.M. Interferon-alpha restores normal beta 1 integrin-mediated inhibition of hematopoietic progenitor proliferation by the marrow microenvironment in chronic myelogenous leukemia. Blood 1996, 87, 3883–3891. [Google Scholar] [CrossRef]
- Krause, D.S.; Lazarides, K.; von Andrian, U.H.; Van Etten, R.A. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat. Med. 2006, 12, 1175–1180. [Google Scholar] [CrossRef]
- Krause, D.S.; Lazarides, K.; Lewis, J.B.; von Andrian, U.H.; Van Etten, R.A. Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood 2014, 123, 1361–1371. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, Y.; Douglas, L.; Li, S. beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia 2009, 23, 109–116. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, B.; Li, M.; McDonald, T.; Holyoake, T.L.; Moon, R.T.; Campana, D.; Shultz, L.; Bhatia, R. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood 2013, 121, 1824–1838. [Google Scholar] [CrossRef] [PubMed]
- Laperrousaz, B.; Jeanpierre, S.; Sagorny, K.; Voeltzel, T.; Ramas, S.; Kaniewski, B.; Ffrench, M.; Salesse, S.; Nicolini, F.E.; Maguer-Satta, V. Primitive CML cell expansion relies on abnormal levels of BMPs provided by the niche and on BMPRIb overexpression. Blood 2013, 122, 3767–3777. [Google Scholar] [CrossRef] [PubMed]
- Grockowiak, E.; Laperrousaz, B.; Jeanpierre, S.; Voeltzel, T.; Guyot, B.; Gobert, S.; Nicolini, F.E.; Maguer-Satta, V. Immature CML cells implement a BMP autocrine loop to escape TKI treatment. Blood 2017, 130, 2860–2871. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Isringhausen, S.; Li, H.; Paterson, A.J.; He, J.; Gomariz, A.; Nagasawa, T.; Nombela-Arrieta, C.; Bhatia, R. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell 2019, 24, 769–784.e766. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Azab, A.K.; Manley, P.W.; Kung, A.L.; Christie, A.L.; Bronson, R.; Ghobrial, I.M.; Griffin, J.D. Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia 2012, 26, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Sadovnik, I.; Cerny-Reiterer, S.; Rulicke, T.; Stefanzl, G.; Willmann, M.; Hoermann, G.; Bilban, M.; Blatt, K.; Herndlhofer, S.; et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 2014, 123, 3951–3962. [Google Scholar] [CrossRef]
- Willmann, M.; Sadovnik, I.; Eisenwort, G.; Entner, M.; Bernthaler, T.; Stefanzl, G.; Hadzijusufovic, E.; Berger, D.; Herrmann, H.; Hoermann, G.; et al. Evaluation of cooperative antileukemic effects of nilotinib and vildagliptin in Ph(+) chronic myeloid leukemia. Exp. Hematol. 2018, 57, 50–59.e56. [Google Scholar] [CrossRef]
- Reynaud, D.; Pietras, E.; Barry-Holson, K.; Mir, A.; Binnewies, M.; Jeanne, M.; Sala-Torra, O.; Radich, J.P.; Passegue, E. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell 2011, 20, 661–673. [Google Scholar] [CrossRef]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegue, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef]
- Welner, R.S.; Amabile, G.; Bararia, D.; Czibere, A.; Yang, H.; Zhang, H.; Pontes, L.L.; Ye, M.; Levantini, E.; Di Ruscio, A.; et al. Treatment of chronic myelogenous leukemia by blocking cytokine alterations found in normal stem and progenitor cells. Cancer Cell 2015, 27, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tu, H.; Yang, Y.; Jiang, X.; Hu, X.; Luo, Q.; Li, J. Bone marrow-derived mesenchymal stromal cells promote resistance to tyrosine kinase inhibitors in chronic myeloid leukemia via the IL-7/JAK1/STAT5 pathway. J. Biol. Chem. 2019, 294, 12167–12179. [Google Scholar] [CrossRef]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef]
- Glodkowska-Mrowka, E.; Manda-Handzlik, A.; Stelmaszczyk-Emmel, A.; Seferynska, I.; Stoklosa, T.; Przybylski, J.; Mrowka, P. PPARgamma ligands increase antileukemic activity of second- and third-generation tyrosine kinase inhibitors in chronic myeloid leukemia cells. Blood Cancer J. 2016, 6, e377. [Google Scholar] [CrossRef]
- Zhou, H.S.; Carter, B.Z.; Andreeff, M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1alpha is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef]
- Ng, K.P.; Manjeri, A.; Lee, K.L.; Huang, W.; Tan, S.Y.; Chuah, C.T.; Poellinger, L.; Ong, S.T. Physiologic hypoxia promotes maintenance of CML stem cells despite effective BCR-ABL1 inhibition. Blood 2014, 123, 3316–3326. [Google Scholar] [CrossRef] [PubMed]
- Ilander, M.; Kreutzman, A.; Mustjoki, S. IFNalpha induces prolonged remissions modeling curative immunologic responses in chronic myeloid leukemia. Oncoimmunology 2014, 3, e28781. [Google Scholar] [CrossRef][Green Version]
- Bruck, O.; Blom, S.; Dufva, O.; Turkki, R.; Chheda, H.; Ribeiro, A.; Kovanen, P.; Aittokallio, T.; Koskenvesa, P.; Kallioniemi, O.; et al. Immune cell contexture in the bone marrow tumor microenvironment impacts therapy response in CML. Leukemia 2018, 32, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Bachy, E.; Bernaud, J.; Roy, P.; Rigal, D.; Nicolini, F.E. Quantitative and functional analyses of CD4(+) CD25(+) FoxP3(+) regulatory T cells in chronic phase chronic myeloid leukaemia patients at diagnosis and on imatinib mesylate. Br. J. Haematol. 2011, 153, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Christiansson, L.; Soderlund, S.; Svensson, E.; Mustjoki, S.; Bengtsson, M.; Simonsson, B.; Olsson-Stromberg, U.; Loskog, A.S. Increased level of myeloid-derived suppressor cells, programmed death receptor ligand 1/programmed death receptor 1, and soluble CD25 in Sokal high risk chronic myeloid leukemia. PLoS ONE 2013, 8, e55818. [Google Scholar] [CrossRef]
- Riether, C.; Gschwend, T.; Huguenin, A.L.; Schurch, C.M.; Ochsenbein, A.F. Blocking programmed cell death 1 in combination with adoptive cytotoxic T-cell transfer eradicates chronic myelogenous leukemia stem cells. Leukemia 2015, 29, 1781–1785. [Google Scholar] [CrossRef] [PubMed]
- Sweet, K.; Hazlehurst, L.; Sahakian, E.; Powers, J.; Nodzon, L.; Kayali, F.; Hyland, K.; Nelson, A.; Pinilla-Ibarz, J. A phase I clinical trial of ruxolitinib in combination with nilotinib in chronic myeloid leukemia patients with molecular evidence of disease. Leuk. Res. 2018, 74, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G.; Da Silva, P.; Max, H.; Kalbacher, H.; Schmidt, H.; Bruserud, O.; Zugel, U.; Baier, W.; Rehbein, A.; Pohla, H. Relative roles of natural killer- and T cell-mediated anti-leukemia effects in chronic myelogenous leukemia patients treated with interferon-alpha. Leuk. Lymphoma 1995, 18, 471–478. [Google Scholar] [CrossRef] [PubMed]
- de Castro, F.A.; Palma, P.V.; Morais, F.R.; Simoes, B.P.; Carvalho, P.V.; Ismael, S.J.; Lima, C.P.; Voltarelli, J.C. Immunological effects of interferon-alpha on chronic myelogenous leukemia. Leuk. Lymphoma 2003, 44, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Burchert, A.; Muller, M.C.; Kostrewa, P.; Erben, P.; Bostel, T.; Liebler, S.; Hehlmann, R.; Neubauer, A.; Hochhaus, A. Sustained molecular response with interferon alfa maintenance after induction therapy with imatinib plus interferon alfa in patients with chronic myeloid leukemia. J. Clin. Oncol. 2010, 28, 1429–1435. [Google Scholar] [CrossRef]
- Preudhomme, C.; Guilhot, J.; Nicolini, F.E.; Guerci-Bresler, A.; Rigal-Huguet, F.; Maloisel, F.; Coiteux, V.; Gardembas, M.; Berthou, C.; Vekhoff, A.; et al. Imatinib plus peginterferon alfa-2a in chronic myeloid leukemia. N. Engl. J. Med. 2010, 363, 2511–2521. [Google Scholar] [CrossRef]
- Simonsson, B.; Gedde-Dahl, T.; Markevarn, B.; Remes, K.; Stentoft, J.; Almqvist, A.; Bjoreman, M.; Flogegard, M.; Koskenvesa, P.; Lindblom, A.; et al. Combination of pegylated IFN-alpha2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood 2011, 118, 3228–3235. [Google Scholar] [CrossRef]
- Nemkov, T.; D’Alessandro, A.; Reisz, J.A. Metabolic underpinnings of leukemia pathology and treatment. Cancer Rep. 2019, 2, e1139. [Google Scholar] [CrossRef]
- A, J.; Qian, S.; Wang, G.; Yan, B.; Zhang, S.; Huang, Q.; Ni, L.; Zha, W.; Liu, L.; Cao, B.; et al. Chronic myeloid leukemia patients sensitive and resistant to imatinib treatment show different metabolic responses. PLoS ONE 2010, 5, e13186. [Google Scholar] [CrossRef]
- Karlikova, R.; Siroka, J.; Friedecky, D.; Faber, E.; Hrda, M.; Micova, K.; Fikarova, I.; Gardlo, A.; Janeckova, H.; Vrobel, I.; et al. Metabolite Profiling of the Plasma and Leukocytes of Chronic Myeloid Leukemia Patients. J. Proteome Res. 2016, 15, 3158–3166. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A.; et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740 e724. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.; Chen, H.N.; Wang, K.; Yuan, K.; Lei, Y.; Li, K.; Lan, J.; Chen, Y.; Huang, Z.; Xie, N.; et al. Ivermectin Induces Cytostatic Autophagy by Blocking the PAK1/Akt Axis in Breast Cancer. Cancer Res. 2016, 76, 4457–4469. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Feng, M.; Liu, Z.L.; Liu, Y.; Huang, Z.L.; Li, H.; Feng, W.L. Potential role of Wnt/beta-catenin signaling in blastic transformation of chronic myeloid leukemia: Cross talk between beta-catenin and BCR-ABL. Tumour Biol. 2016, 39. [Google Scholar] [CrossRef]
- Gregory, M.A.; Phang, T.L.; Neviani, P.; Alvarez-Calderon, F.; Eide, C.A.; O’Hare, T.; Zaberezhnyy, V.; Williams, R.T.; Druker, B.J.; Perrotti, D.; et al. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell 2010, 18, 74–87. [Google Scholar] [CrossRef]
- Zhao, C.; Blum, J.; Chen, A.; Kwon, H.Y.; Jung, S.H.; Cook, J.M.; Lagoo, A.; Reya, T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007, 12, 528–541. [Google Scholar] [CrossRef]
- Schurch, C.; Riether, C.; Matter, M.S.; Tzankov, A.; Ochsenbein, A.F. CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J. Clin. Investig. 2012, 122, 624–638. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Langdon, W.Y.; Varticovski, L. Tyrosine phosphorylation of p120cbl in BCR/abl transformed hematopoietic cells mediates enhanced association with phosphatidylinositol 3-kinase. Oncogene 1997, 14, 2217–2228. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chai, S.K.; Nichols, G.L.; Rothman, P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J. Immunol. 1997, 159, 4720–4728. [Google Scholar]
- de Groot, R.P.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L. STAT5-Dependent CyclinD1 and Bcl-xL expression in Bcr-Abl-transformed cells. Mol. Cell Biol. Res. Commun. 2000, 3, 299–305. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Bertrand, F.E.; Ludwig, D.E.; Basecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia 2008, 22, 708–722. [Google Scholar] [CrossRef]
- Danisz, K.; Blasiak, J. Role of anti-apoptotic pathways activated by BCR/ABL in the resistance of chronic myeloid leukemia cells to tyrosine kinase inhibitors. Acta Biochim. Pol. 2013, 60, 503–514. [Google Scholar] [CrossRef]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef]
- Burchert, A.; Wang, Y.; Cai, D.; von Bubnoff, N.; Paschka, P.; Muller-Brusselbach, S.; Ottmann, O.G.; Duyster, J.; Hochhaus, A.; Neubauer, A. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia 2005, 19, 1774–1782. [Google Scholar] [CrossRef]
- Samanta, A.; Perazzona, B.; Chakraborty, S.; Sun, X.; Modi, H.; Bhatia, R.; Priebe, W.; Arlinghaus, R. Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia 2011, 25, 463–472. [Google Scholar] [CrossRef]
- Modi, H.; Li, L.; Chu, S.; Rossi, J.; Yee, J.K.; Bhatia, R. Inhibition of Grb2 expression demonstrates an important role in BCR-ABL-mediated MAPK activation and transformation of primary human hematopoietic cells. Leukemia 2011, 25, 305–312. [Google Scholar] [CrossRef]
- Ohanian, M.; Tari Ashizawa, A.; Garcia-Manero, G.; Pemmaraju, N.; Kadia, T.; Jabbour, E.; Ravandi, F.; Borthakur, G.; Andreeff, M.; Konopleva, M.; et al. Liposomal Grb2 antisense oligodeoxynucleotide (BP1001) in patients with refractory or relapsed haematological malignancies: A single-centre, open-label, dose-escalation, phase 1/1b trial. Lancet Haematol. 2018, 5, e136–e146. [Google Scholar] [CrossRef]
- Reddiconto, G.; Toto, C.; Palama, I.; De Leo, S.; de Luca, E.; De Matteis, S.; Dini, L.; Passerini, C.G.; Di Renzo, N.; Maffia, M.; et al. Targeting of GSK3beta promotes imatinib-mediated apoptosis in quiescent CD34+ chronic myeloid leukemia progenitors, preserving normal stem cells. Blood 2012, 119, 2335–2345. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Li, S.A. Mitotic kinases: The key to duplication, segregation, and cytokinesis errors, chromosomal instability, and oncogenesis. Pharmacol. Ther. 2006, 111, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Cortes, J.; Jones, D.; Bergstrom, D.; Kantarjian, H.; Freedman, S.J. MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood 2007, 109, 500–502. [Google Scholar] [CrossRef]
- Giles, F.J.; Swords, R.T.; Nagler, A.; Hochhaus, A.; Ottmann, O.G.; Rizzieri, D.A.; Talpaz, M.; Clark, J.; Watson, P.; Xiao, A.; et al. MK-0457, an Aurora kinase and BCR-ABL inhibitor, is active in patients with BCR-ABL T315I leukemia. Leukemia 2013, 27, 113–117. [Google Scholar] [CrossRef]
- Seymour, J.F.; Kim, D.W.; Rubin, E.; Haregewoin, A.; Clark, J.; Watson, P.; Hughes, T.; Dufva, I.; Jimenez, J.L.; Mahon, F.X.; et al. A phase 2 study of MK-0457 in patients with BCR-ABL T315I mutant chronic myelogenous leukemia and philadelphia chromosome-positive acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e238. [Google Scholar] [CrossRef]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef]
- Kizilors, A.; Crisa, E.; Lea, N.; Passera, R.; Mian, S.; Anwar, J.; Best, S.; Nicolini, F.E.; Ireland, R.; Aldouri, M.; et al. Effect of low-level BCR-ABL1 kinase domain mutations identified by next-generation sequencing in patients with chronic myeloid leukaemia: A population-based study. Lancet Haematol. 2019, 6, e276–e284. [Google Scholar] [CrossRef]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef]


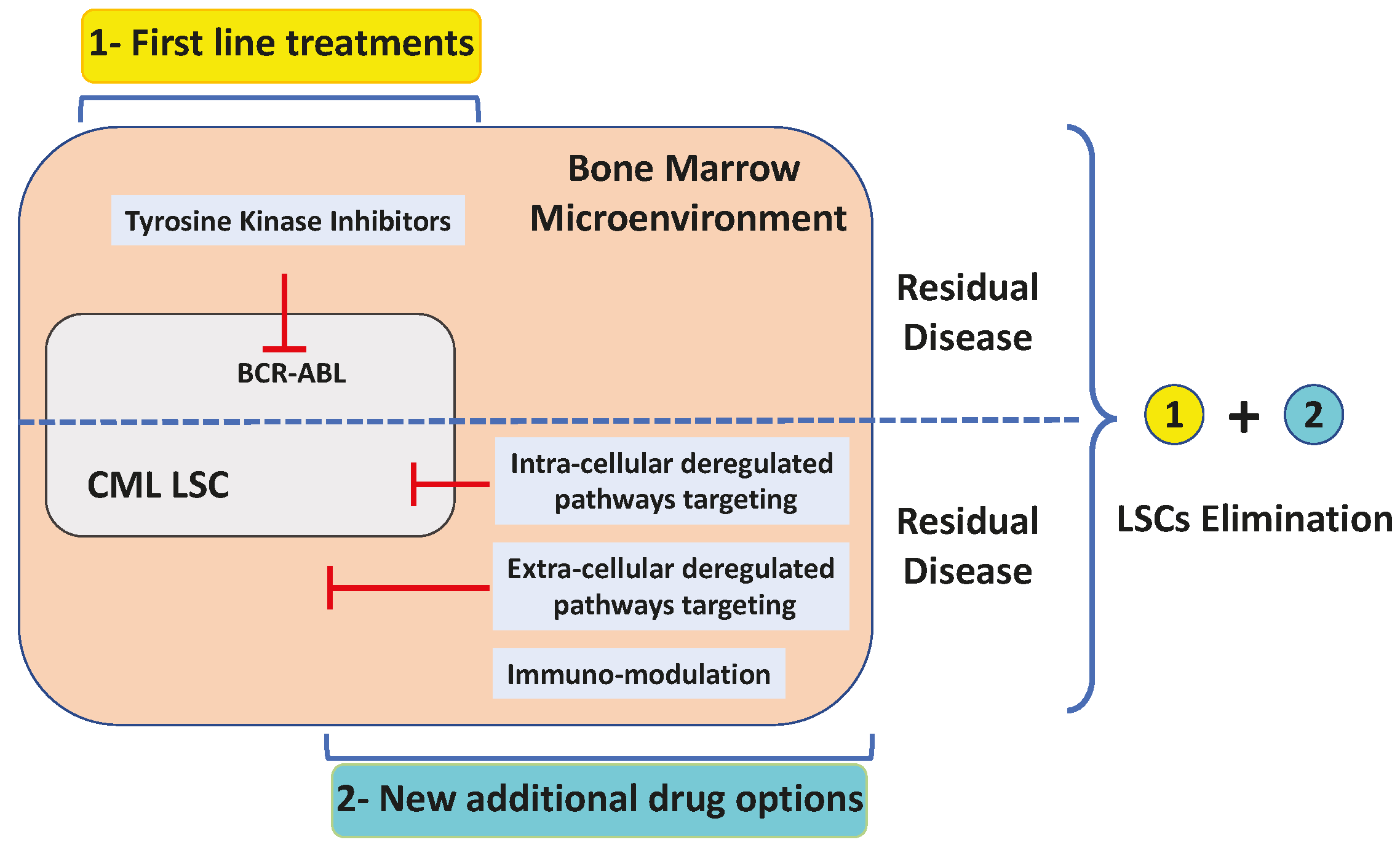{kind=link}
{kind=link}
| Scheme Number | Treatment 1 | Treatment 2 | Pathway | References |
|---|---|---|---|---|
| 1 | Ponatinib | Asciminib | BCR-ABL1 allosteric inhibitor | [7] |
| 2 | Imatinib Dasatinib Nilotinib | GSK343 | EZH2 inhibitor | [8] |
| 3 | Imatinib | LBH589 | HDAC inhibitor | [9] |
| 4 | Nilotinib | LBH589 | HDAC inhibitor | [10] |
| 5 | Imatinib | LAQ824 | HDAC inhibitor | [11] |
| 6 | Imatinib | Hydralazine and Magnesium Valporate | HDAC inhibitors | [12] |
| 7 | Imatinib | Tenovin-6 | SIRT1 inhibitor | [13] |
| 8 | Nilotinib | TV39OH | SIRT1 inhibitor | [14] |
| 9 | Imatinib | miR30a inhibition | Genetic repressor/activator | [15] |
| 10 | Imatinib | miR-202 overexpression | Inhibition of hexokinase 2 (HK2) and glycolysis | [16] |
| 11 | Imatinib | miR-486 | Genetic repressor/activator | [17] |
| 12 | Imatinib | Ovatodiolide | miR-155 upregulation/PI3K/mTOR inhibition | [18] |
| 13 | Imatinib | RI-BPI | BCL6 inhibitor | [19] |
| 14 | Imatinib | FX1 | BCL6 inhibitor | [20] |
| 15 | Imatinib | A-1210477 | MCL-1 inhibitor | [20] |
| 16 | Dasatinib | Sabutoclax | Pan-BCL2 inhibitor | [21] |
| 17 | Imatinib | ABT-737 | BCL2 and Bcl-xL inhibitor | [22,23] |
| 18 | Nilotinib | Nutlin3a ABT-737 | MDM2 inhibitor BCL2 and Bcl-xL inhibitor | [24] |
| 19 | Imatinib | DS-5272 | MDM2 inhibitor | [25] |
| 20 | Imatinib | ABT-199 (Venetoclax) | BCL2 inhibitor | [26] |
| 21 | Nilotinib | ABT-199 (Venetoclax) | BCL2 inhibitor | [27] |
| 22 | Imatinib Nilotinib Dasatinib | Chloroquine Bafilomycin A1 | Autophagy inhibitors | [9,28] |
| 23 | Imatinib | Chloroquine Bafilomycin A1 | Autophagy inhibitors | [29] |
| 24 | Imatinib Nilotinib Dasatinib | Clarithromycin | Autophagy inhibitor | [30] |
| 25 | Imatinib | Spautin-1 | Autophagy inhibitor | [31] |
| 26 | Ponatinib | Vismodegib (GDC-0449) | Smo antagonist Hedgehog pathway | [32] |
| 27 | Nilotinib | Lys05 | Autophagy inhibitor | [33] |
| 28 | Nilotinib | PIK-III | VPS34 inhibitor Autophagy | [33] |
| 29 | Imatinib | PTC-209 | BMI1 inhibitor | [34] |
| 30 | Nilotinib | WNT974 | PORCN selective inhibitor Wnt pathway | [35] |
| 31 | Nilotinib | PRI-724 | CBP inhibitor Wnt/β-catenin inhibition | [36] |
| 32 | Nilotinib | XL880 R428 | AXL inhibitors | [37] |
| 33 | Imatinib | Plerifaxor (ADM3100) | CXCR4 antagonist | [34] |
| 34 | Imatinib Nilotinib | Plerifaxor (ADM3100) | CXCR4 antagonist | [38] |
| 35 | Nilotinib | Plerifaxor (ADM3100) | CXCR4 antagonist | [39] |
| 36 | Imatinib | BTK140 | CXCR4 antagonist | [40] |
| 37 | Imatinib | Filgotinib Itacitinib | JAK1 specific inhibitors, JAK1/STAT3 pathway | [41] |
| 38 | Imatinib | Wogonin | STAT5 pathway inhibitor | [42] |
| 39 | Imatinib | Tigecycline | Mitochondrial ribosome protein translation and respiratory chain | [43] |
| 40 | Nilotinib | SR18292 | PGC-1α inhibitor/SIRT1 pathway inhibition | [14] |
| 41 | Imatinib Dasatinib | Ivermectin | mTOR inhibitor | [44] |
| 42 | Imatinib | Anti-CD70 | Wnt/β-catenin inhibition | [45] |
| 43 | Imatinib | Sonidegib (LDE225) | Smo antagonist Hedgehog pathway inhibition | [46] |
| 44 | Imatinib | Ly364947 | TGF-βRI inhibitor TGFβ/Activin/NODAL pathway | [47] |
| 45 | Imatinib Dasatinib Ponatinib | EW-7197 | TGF-β signaling inhibitor | [11] |
| 46 | Dasatinib | BMS-911543 | JAK2 inhibitor | [48] |
| 47 | Dasatinib | Niclosamide | ERK/MNK1/eIF4E pathway inhibition | [49] |
| 48 | Imatinib | SB216763 | GSK-3 specific inhibitor | [50] |
| 49 | Imatinib Nilotinib | PD146176 | 15-LO inhibition/ arachidonic acid -leukotriene pathway inhibition | [51] |
| 50 | none | EZH2 CRISPR/Cas9 invalidation | EZH2 inhibition | [52] |
| 51 | none | Pracinostat | HDAC inhibitor | [53] |
| 52 | none | PJ-68 | Methyltransferase inhibitor (PRMT5) Wnt/β-catenin pathway inhibition | [54] |
| 53 | none | Dactolisib (BEZ235) Chloroquine | PI3K/mTOR inhibitor Autophagy inhibitor | [55] |
| 54 | none | Vismodegib (GDC-0449) | Smo antagonistHH pathway inhibition | [56] |
| 55 | none | Resveratrol | Autophagic cell death induction | [57] |
| 56 | none | IFN-α Ruxolitinib | MHC II increased expression JAK1/2 inhibitor | [58] |
| 57 | none | shATG7 | Mitochondrial ROS and oxidative phosphorylation increase Autophagy inhibition | [59] |
| 58 | none | miR-326 overexpression | Downregulation of SmoHH pathway inhibition | [60] |
| 59 | none | PD184352 BMS-21462 | MEK specific inhibitor Farnesyl transferase inhibitor | [61] |
| 60 | none | CGP57380 | MNK1 inhibitor ERK/MNK1/eIF4E pathway inhibition | [62] |
| 61 | none | LY255283 | BLT2 inhibitor/arachidonic acid -leukotriene pathway inhibition | [63] |
| 62 | none | RG7112/7388 CPI-203/0610 | HMD2 inhibitors (p53 inhibition) BET inhibitors (c-Myc inhibition) | [64] |
| Scheme Number | Treatment 1 | Treatment 2 | Pathway | Investigation | Phase | Identifier | First Posted | Status |
|---|---|---|---|---|---|---|---|---|
| 1 | Imatinib Nilotinib Dasatinib | Asciminib | BCR-ABL1 allosteric inhibitor | Frontline combination in CP- CML | II | NCT03906292 | 2019 | Recruiting |
| 2 | none | Asciminib | BCR-ABL1 allosteric inhibitor | Efficacy of ABL001 versus Bosutinib in CP-CML patients previously treated with TKIs | III | NCT03106779 | 2017 | Recruiting |
| 3 | Dasatinib | Asciminib | BCR-ABL1 allosteric inhibitor | Combination in CML in lymphoid blast crisis | I | NCT03595917 | 2018 | Recruiting |
| 4 | Imatinib | Asciminib | BCR-ABL1 allosteric inhibitor | Efficacy and safety of combination in patients with CP-CML | II | NCT03578367 | 2018 | Recruiting |
| 5 | Imatinib Nilotinib Dasatinib | Asciminib | BCR-ABL1 allosteric inhibitor | Oral ABL001 in CML Patients | I | NCT02081378 | 2014 | Recruiting |
| 6 | Imatinib | Cytarabine | DNA synthesis inhibitor | Combination in patients with CML | II | NCT00022490 | 2003 | Terminated |
| 7 | Imatinib | Cytarabine | DNA synthesis inhibitor | Combination in patients with CML | I/II | NCT00015834 | 2003 | Completed |
| 8 | Dasatinib | SAHA (Vorinostat) | HDAC inhibitor | Combination in treating patients with accelerated phase or BP- CML | I | NCT00816283 | 2009 | Completed |
| 9 | Imatinib | Panobinostat (LBH589) | HDAC inhibitor | Safety and tolerability of LBH589 combined with imatinib in CML patients in MCR | I | NCT00686218 | 2008 | Completed |
| 10 | Imatinib | Decitabine | DNA methyltransferase inhibitor | Combination in patients with CML | II | NCT00054431 | 2003 | Completed |
| 11 | Dasatinib | Venetoclax | BCL2 inhibitor | Combination in treating patients with BCR-ABL1 positive early chronic phase | II | NCT02689440 | 2016 | Recruiting |
| 12 | Ponatinib | Venetoclax Dexamethasone | BCL2 inhibitor Anti-inflammatory | Triple combination in BCR-ABL positive relapsed CML | I/II | NCT03576547 | 2018 | Recruiting |
| 13 | Imatinib | Oblimersen | bcl-2 antisense oligodeoxynucleotide | Oblimersen and Imatinib in treating patients with CML | II | NCT00049192 | 2003 | Completed |
| 14 | Imatinib | Hydroxychloroquine | Autophagy inhibitor | Effectiveness of combination on BCR/ABL levels in CML patients in MCR | II | NCT01227135 | 2010 | Unknown |
| 15 | Imatinib | Everolimus (RAD001) | mTOR inhibitor | Combination in patients in CP-CML who are not in CCR after previous Imatinib | I/II | NCT00093639 | 2004 | Completed |
| 16 | Imatinib | Temsirolimus | mTOR inhibitor | Temsirolimus and Imatinib in treating patients with CML | I | NCT00101088 | 2005 | Terminated |
| 17 | Dasatinib | PF04449913 (Glasdegib) | Smo antagonist HH inhibition | Study of PF-04449913 in select hematologic malignancies | I | NCT00953758 | 2009 | Completed |
| 18 | Imatinib | Interferon-α | Immunomodulatory effect | INF-α and Imatinib in CML patients | II | NCT00045422 | 2003 | Completed |
| 19 | Imatinib | Pioglitazone | PPARγ agonist STAT5 inhibition | Combination in patients with relapsed CML | II | NCT02767063 | 2016 | Terminated |
| 20 | Imatinib | Pioglitazone | PPARγ agonist STAT5 inhibition | Efficiency of combination to treat CML | II | NCT02687425 | 2016 | Unknown |
| 21 | TKIs | Pioglitazone | PPARγ agonist STAT5 inhibition | Pioglitazone and TKI in patients with relapsed CML | II | NCT02730195 | 2019 | Terminated |
| 22 | Dasatinib | Cyclosporine | IL-2 inhibitor | Combination in patients with CML refractory or intolerant to imatinib | I | NCT01426334 | 2011 | Terminated |
| 23 | Pioglitazone | Avelumab (Anti-PD-L1) | PPARγ agonist STAT5 inhibition PD-1/PD-L1 inhibition | Therapies in combination or sequentially with TKIs in CP-CML patients in CCR (ACTIW) | I/II | NCT02767063 | 2016 | Recruiting |
| 24 | Imatinib Nilotinib Dasatinib | Pembrolizumab (Anti-PD-1) | PD-1/PD-L1 inhibition | Pembrolizumab and TKIs in CML patients with persistently detectable MRD | II | NCT03516279 | 2018 | Recruiting |
| 25 | Dasatinib Nilotinib | Ruxolitinib | JAK1/2 selective inhibitor | Ruxolitinib phosphate and Dasatinib or Nilotinib in treating CML patients | II | NCT03654768 | 2018 | Recruiting |
| 26 | Nilotinib | Ruxolitinib | JAK1/2 selective inhibitor | Ruxolitinib in treating participants with CML with MRD after TKIs | I/II | NCT01751425 | 2012 | Active, not recruiting |
| 27 | Nilotinib | Ruxolitinib | JAK1/2 selective inhibitor | Nilotinib/Ruxolitinb therapy for TKI resistant Ph-Leukemia | I//II | NCT01914484 | 2013 | Unknown |
| 28 | Nilotinib | Ruxolitinib | JAK1/2 selective inhibitor | Combination in CP-CML Patients | I | NCT01702064 | 2012 | Completed |
| 29 | Nilotinib | Peginterferon α2b | Immunomodulatory effect | Evaluation of TKI and INF-α | III | NCT01657604 | 2012 | Active, not recruiting |
| 30 | Imatinib Nilotinib | Peginterferon α2b | Immunomodulatory effect | Imatinib or Nilotinib with Pegylated Interferon-α2b in CML | II | NCT00573378 | 2007 | Withdrawn |
| 31 | Dasatinib | Peginterferon α2b | Immunomodulatory effect | Safety and efficacy of combination in newly diagnosed CML (NordCML007) | II | NCT01725204 | 2012 | Completed |
| 32 | Bosutinib | Ropeginterferon | Immunomodulatory effect | Long-acting low dose Ropeginterferon for CML treated with Bosutinib from diagnosis | II | NCT03831776 | 2019 | Recruiting |
| 33 | Nilotinib | Sonidegib (LDE225) | Smo antagonist HH inhibition | Combination in CML patients who developed resistance to prior therapy | I | NCT01456676 | 2011 | Completed |
| 34 | Dasatinib | BMS-833923 | Smo antagonist HH inhibition | Combination therapy in CML | I/II | NCT01218477 | 2010 | Completed |
| 35 | Dasatinib | BMS833923 | Smo antagonist HH inhibition | Dasatinib combo with Smo antagonist | II | NCT01357655 | 2011 | Terminated |
| 36 | Dasatinib | PF04449913 | Smo antagonist HH inhibition | Study in select hematologic malignancies | I | NCT00953758 | 2009 | Completed |
| 37 | Imatinib | Zarnestra | Farnesyltransferase inhibitor | Zarnestra and Gleevec in CP- CML | I | NCT00040105 | 2002 | Completed |
| 38 | Imatinib | Lonafarnib | Farnesyltransferase inhibitor | Lonafarnib and Gleevec in CML | I | NCT00047502 | 2002 | Completed |
| 39 | none | Sorafenib (BAY43-9006) | Raf/VEGFR/PDGFR inhibitor | Raf kinase inhibitor BAY 43-9006 in CML patients resistant to Gleevec | II | NCT00661180 | 2008 | Completed |
| 40 | Imatinib | Vatalanib (PTK 787) | VEGFR inhibitor | Combination in patients with BP-CML | I/II | NCT00088231 | 2004 | Completed |
| 41 | Dasatinib | BP1001 | Grb2 inhibitor | Combination of liposomal Grb2 antisense oligonucleotide with Dasatinib in CML patients | I/II | NCT02923986 | 2016 | Recruiting |
| 42 | none | MK0457 | Aurora kinase inhibition | MK0457 in CML patients (0457-003) | I | NCT00111683 | 2005 | Completed |
| 43 | Dasatinib | MK0457 | Aurora kinase inhibition | Evaluation of efficacy and safety in patients with CML | I | NCT00500006 | 2007 | Terminated |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muselli, F.; Peyron, J.-F.; Mary, D. Druggable Biochemical Pathways and Potential Therapeutic Alternatives to Target Leukemic Stem Cells and Eliminate the Residual Disease in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2019, 20, 5616. https://doi.org/10.3390/ijms20225616
Muselli F, Peyron J-F, Mary D. Druggable Biochemical Pathways and Potential Therapeutic Alternatives to Target Leukemic Stem Cells and Eliminate the Residual Disease in Chronic Myeloid Leukemia. International Journal of Molecular Sciences. 2019; 20(22):5616. https://doi.org/10.3390/ijms20225616
Chicago/Turabian StyleMuselli, Fabien, Jean-François Peyron, and Didier Mary. 2019. "Druggable Biochemical Pathways and Potential Therapeutic Alternatives to Target Leukemic Stem Cells and Eliminate the Residual Disease in Chronic Myeloid Leukemia" International Journal of Molecular Sciences 20, no. 22: 5616. https://doi.org/10.3390/ijms20225616
APA StyleMuselli, F., Peyron, J.-F., & Mary, D. (2019). Druggable Biochemical Pathways and Potential Therapeutic Alternatives to Target Leukemic Stem Cells and Eliminate the Residual Disease in Chronic Myeloid Leukemia. International Journal of Molecular Sciences, 20(22), 5616. https://doi.org/10.3390/ijms20225616