The Pathogenesis of Endometriosis: Molecular and Cell Biology Insights

 ,
,  ,
,  ,
, 


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
What is Endometriosis?
2. Theories on the Origin of Endometriosis
2.1. The in Situ Theory
2.2. The Transplantation Theory
3. Behind the Origins of Endometriosis
3.1. Comprehensive Models on the Origin of Endometriosis
3.2. Role of Hormones
3.3. The Peritoneal Microenvironment and the Role of Immune Surveillance
3.4. Apoptosis Defects
3.5. Cell-Matrix and Cell-Cell Adhesion
3.6. Extracellular Matrix Remodeling and Matrix Metalloproteinases
3.7. Angiogenesis
3.8. Endometrial Stem Cells
4. The Genetics of Endometriosis
4.1. Candidate Gene Studies
4.2. Linkage and Association Studies
4.3. Genome-Wide Association Studies
5. The Epigenetics of Endometriosis
5.1. Epigenetics in the Eutopic Endometrium
5.2. Epigenetics in Endometriosis
5.3. Implications of Epigenetics in Diagnosis, Prognosis, and Therapy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MeSH | Medical Subject Headings |
| CAM | Chorioallantoic membrane |
| 17β-HSD | 17β-hydroxysteroid dehydrogenase |
| COX-2 | Cyclo-oxygenase type 2 |
| TIAR | Tissue injury and repair |
| TNF | Tumor necrosis factor |
| IL | Interleukin |
| VEGF | Vascular endothelial growth factor |
| Bcl-2 | Antiapoptotic protein B cell lymphoma 2 |
| NK cells | Natural Killer cells |
| sICAM-1 | Soluble form of intercellular adhesion molecule-1 |
| LFA-1 | Leukocyte function antigen-1 |
| MAPK | Mitogen-activated protein kinase |
| ECM | Extracellular matrix |
| HLA-1 | Human leukocyte antigen class I |
| INF | Interferon |
| TGF-β | Transforming growth factor-β |
| E-cadherin | Epithelial cadherin |
| MMPs | Matrix metalloproteinases |
| TIMPs | Tissue inhibitors of MMPs |
| bFGF | Basic fibroblast growth factor |
| PAI | Plasminogen activator inhibitor |
| PR | Progesterone receptor |
| ER | Estrogen receptor |
| SNPs | Single nucleotide polymorphisms |
| LD | Linkage disequilibrium |
| FGF | Fibroblast growth factor |
| FGFR2 | FGF receptor 2 gene |
| GWA | Genome-wide association |
| CDKN2BAS | Cyclin-dependent kinase inhibitor 2B antisense RNA |
| MMTV | Mouse mammary tumor virus |
| WNT4 | Wingless-type mouse mammary tumor virus integration site family 4 |
| rAFS | Revised American Fertility Society |
| FSH | Follicle-stimulating hormone |
| GREB1 | Growth Regulating Estrogen Receptor Binding 1 |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| miRNAs | microRNAs |
| DNMT | DNA methyltransferase |
| HDAC | Histone deacetylase |
| HDACIs | Histone deacetylase inhibitors |
| EDC | Endocrine-disrupting chemicals |
| SF-1 | Steroidogenic factor-1 |
| HOXA10 | Homeobox A transcription factor |
| P-E2 | Prostaglandin E2 |
| E | Estrogen |
| P | Progesterone |
| TSA | Trichostatin A |
| PPAR | Peroxisome proliferator-activated receptor |
| VPA | Valproic acid |
References
- Vercellini, P.; Viganò, P.; Somigliana, E.; Fedele, L. Endometriosis: Pathogenesis and treatment. Nat. Rev. Endocrinol. 2014, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Garai, J.; Molnar, V.; Varga, T.; Koppan, M.; Torok, A.; Bodis, J. Endometriosis: Harmful survival of an ectopic tissue. Front. Biosci. J. Virtual Libr. 2006, 11, 595–619. [Google Scholar] [CrossRef] [PubMed]
- Matalliotakis, M.; Zervou, M.I.; Matalliotaki, C.; Rahmioglu, N.; Koumantakis, G.; Kalogiannidis, I.; Prapas, I.; Zondervan, K.; Spandidos, D.A.; Matalliotakis, I.; et al. The role of gene polymorphisms in endometriosis. Mol. Med. Rep. 2017, 16, 5881–5886. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Wu, M.; Tsai, S. Epigenetic regulation of the pathological process in endometriosis. Reprod. Med. Biol. 2017, 16, 314–319. [Google Scholar] [CrossRef]
- Clement, P.B. The pathology of endometriosis: A survey of the many faces of a common disease emphasizing diagnostic pitfalls and unusual and newly appreciated aspects. Adv. Anat. Pathol. 2007, 14, 241–260. [Google Scholar] [CrossRef]
- Burney, R.O.; Giudice, L.C. Pathogenesis and pathophysiology of endometriosis. Fertil. Steril. 2012, 98, 511–519. [Google Scholar] [CrossRef]
- Laganà, A.S.; Vitale, S.G.; Salmeri, F.M.; Triolo, O.; Ban Frangež, H.; Vrtačnik-Bokal, E.; Stojanovska, L.; Apostolopoulos, V.; Granese, R.; Sofo, V. Unus pro omnibus, omnes pro uno: A novel, evidence-based, unifying theory for the pathogenesis of endometriosis. Med. Hypotheses 2017, 103, 10–20. [Google Scholar] [CrossRef]
- Laganà, A.S.; La Rosa, V.L.; Rapisarda, A.M.; Valenti, G.; Sapia, F.; Chiofalo, B.; Rossetti, D.; Ban Frangež, H.; Vrtačnik Bokal, E.; Vitale, S.G. Anxiety and depression in patients with endometriosis: Impact and management challenges. Int. J. Womens Health 2017, 9, 323–330. [Google Scholar] [CrossRef]
- Laganà, A.S.; Condemi, I.; Retto, G.; Muscatello, M.R.A.; Bruno, A.; Zoccali, R.A.; Triolo, O.; Cedro, C. Analysis of psychopathological comorbidity behind the common symptoms and signs of endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2015, 194, 30–33. [Google Scholar] [CrossRef]
- Vitale, S.G.; Petrosino, B.; La Rosa, V.L.; Rapisarda, A.M.C.; Laganà, A.S. A Systematic Review of the Association Between Psychiatric Disturbances and Endometriosis. J. Obstet. Gynaecol. Can. 2016, 38, 1079–1080. [Google Scholar] [CrossRef]
- Laganà, A.S.; La Rosa, V.; Petrosino, B.; Vitale, S.G. Comment on “Risk of developing major depression and anxiety disorders among women with endometriosis: A longitudinal follow-up study”. J. Affect. Disord. 2017, 208, 672–673. [Google Scholar] [CrossRef] [PubMed]
- Raffaelli, R.; Garzon, S.; Baggio, S.; Genna, M.; Pomini, P.; Laganà, A.S.; Ghezzi, F.; Franchi, M. Mesenteric vascular and nerve sparing surgery in laparoscopic segmental intestinal resection for deep infiltrating endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 231, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Baggio, S.; Pomini, P.; Zecchin, A.; Garzon, S.; Bonin, C.; Santi, L.; Festi, A.; Franchi, M.P. Delivery and pregnancy outcome in women with bowel resection for deep endometriosis: A retrospective cohort study. Gynecol. Surg. 2015, 12, 279–285. [Google Scholar] [CrossRef]
- Viganò, P.; Parazzini, F.; Somigliana, E.; Vercellini, P. Endometriosis: Epidemiology and aetiological factors. Best Pract. Res. Clin. Obstet. Gynaecol. 2004, 18, 177–200. [Google Scholar] [CrossRef] [PubMed]
- Buck Louis, G.M.; Hediger, M.L.; Peterson, C.M.; Croughan, M.; Sundaram, R.; Stanford, J.; Chen, Z.; Fujimoto, V.Y.; Varner, M.W.; Trumble, A.; et al. Incidence of endometriosis by study population and diagnostic method: The ENDO study. Fertil. Steril. 2011, 96, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Missmer, S.A.; Cramer, D.W. The epidemiology of endometriosis. Obstet. Gynecol. Clin. North Am. 2003, 30, 1–19. [Google Scholar] [CrossRef]
- Evers, J.L. Endometriosis does not exist; all women have endometriosis. Hum. Reprod. Oxf. Engl. 1994, 9, 2206–2209. [Google Scholar] [CrossRef]
- Evers, J.L. Is adolescent endometriosis a progressive disease that needs to be diagnosed and treated? Hum. Reprod. Oxf. Engl. 2013, 28, 2023. [Google Scholar] [CrossRef]
- Sampson, J.A. Perforating hemorrhage (chocolate) cysts of the ovary: Their importance and especially their relation to pelvic adenomas of endometrial type (“adenomyoma” of the uterus, rectovaginal septum, sigmoid, etc.). Arch. Surg. 1921, 3, 245–323. [Google Scholar] [CrossRef]
- Sampson, J.A. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissue into the peritoneal cavity. Am. J. Obstet. Gynecol. 1927, 14, 422–469. [Google Scholar] [CrossRef]
- Sampson, J.A. The development of the implantation theory for the origin of peritoneal endometriosis. Am. J. Obstet. Gynecol. 1940, 40, 549–557. [Google Scholar] [CrossRef]
- Nisolle, M.; Donnez, J. Peritoneal endometriosis, ovarian endometriosis, and adenomyotic nodules of the rectovaginal septum are three different entities. Fertil. Steril. 1997, 68, 585–596. [Google Scholar] [CrossRef]
- Koninckx, P.R.; Ussia, A.; Adamyan, L.; Wattiez, A.; Donnez, J. Deep endometriosis: Definition, diagnosis, and treatment. Fertil. Steril. 2012, 98, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Koninckx, P.R. Is mild endometriosis a disease?: Is mild endometriosis a condition occurring intermittently in all women? Hum. Reprod. 1994, 9, 2202–2205. [Google Scholar] [CrossRef]
- Donnez, J.; Squifflet, J.; Casanas-Roux, F.; Pirard, C.; Jadoul, P.; Van, A.L. Typical and subtle atypical presentations of endometriosis. Obstet. Gynecol. Clin. North Am. 2003, 30, 83–93. [Google Scholar] [CrossRef]
- Waldeyer, H. “Die epithelialen Eierstockgeschwülste.” Ins besonders die Kystome (The epithelial ovarian tumors, especially the cystic tumors). Arch Gynäkol 1870, 1, 252–316. [Google Scholar] [CrossRef]
- van der Linden, P.J. Theories on the pathogenesis of endometriosis. Hum. Reprod. Oxf. Engl. 1996, 11 (Suppl. 3), 53–65. [Google Scholar] [CrossRef]
- Miyazaki, K.; Dyson, M.T.; Coon V, J.S.; Furukawa, Y.; Yilmaz, B.D.; Maruyama, T.; Bulun, S.E. Generation of Progesterone-Responsive Endometrial Stromal Fibroblasts from Human Induced Pluripotent Stem Cells: Role of the WNT/CTNNB1 Pathway. Stem Cell Rep. 2018, 11, 1136–1155. [Google Scholar] [CrossRef]
- Lauchlan, S.C. THE SECONDARY MÜLLERIAN SYSTEM. Obstet. Gynecol. Surv. 1972, 27, 133. [Google Scholar] [CrossRef]
- Batt, R.E.; Smith, R.A.; Buck, G.L.; Martin, D.C.; Chapron, C.; Koninckx, P.R.; Yeh, J. Müllerianosis. Histol. Histopathol. 2007, 22, 1161–1166. [Google Scholar] [CrossRef]
- Signorile, P.G.; Baldi, F.; Bussani, R.; D’Armiento, M.; De Falco, M.; Baldi, A. Ectopic endometrium in human foetuses is a common event and sustains the theory of müllerianosis in the pathogenesis of endometriosis, a disease that predisposes to cancer. J. Exp. Clin. Cancer Res. 2009, 28, 49. [Google Scholar] [CrossRef] [PubMed]
- Nawroth, F.; Rahimi, G.; Nawroth, C.; Foth, D.; Ludwig, M.; Schmidt, T. Is there an association between septate uterus and endometriosis? Hum. Reprod. 2006, 21, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Mok-Lin, E.Y.; Wolfberg, A.; Hollinquist, H.; Laufer, M.R. Endometriosis in a Patient with Mayer-Rokitansky-Küster-Hauser Syndrome and Complete Uterine Agenesis: Evidence to Support the Theory of Coelomic Metaplasia. J. Pediatr. Adolesc. Gynecol. 2010, 23, e35–e37. [Google Scholar] [CrossRef] [PubMed]
- Troncon, J.K.; Zani, A.C.T.; Vieira, A.D.D.; Poli-Neto, O.B.; Nogueira, A.A.; Rosa-E-Silva, J.C. Endometriosis in a patient with mayer-rokitansky-küster-hauser syndrome. Case Rep. Obstet. Gynecol. 2014, 2014, 376231. [Google Scholar] [CrossRef] [PubMed]
- Batt, R.E.; Mitwally, M.F.M. Endometriosis from thelarche to midteens: Pathogenesis and prognosis, prevention and pedagogy. J. Pediatr. Adolesc. Gynecol. 2003, 16, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Suginami, H. A reappraisal of the coelomic metaplasia theory by reviewing, endometriosis occurring in unusual sites and instances. Am. J. Obstet. Gynecol. 1991, 165, 214–218. [Google Scholar] [CrossRef]
- Rei, C.; Williams, T.; Feloney, M. Endometriosis in a Man as a Rare Source of Abdominal Pain: A Case Report and Review of the Literature. Case Rep. Obstet. Gynecol. 2018, 2018, 2083121. [Google Scholar] [CrossRef]
- Cullen, T.S. Adeno-myoma uteri diffusum benignum; Johns Hopkins Press: Baltimore, Maryland, 1896. [Google Scholar]
- Meyer, R. Über eine adenomatöse Wucherung der Serosa in einer Bauchnarbe. Zeitschr Geburtsh Gynakol 1903, 49, 32–41. [Google Scholar]
- Novak, E. Pelvic endometriosis: Spontaneous rupture of endometrial cysts, with a report of three cases. Am. J. Obstet. Gynecol. 1931, 22, 826–837. [Google Scholar] [CrossRef]
- Hu, Z.; Mamillapalli, R.; Taylor, H.S. Increased circulating miR-370-3p regulates steroidogenic factor 1 in endometriosis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E373–E382. [Google Scholar] [CrossRef]
- Meyer, R. Uber den Stand der Frage der Adenomyositis und Adenomyome im allgeneinen und insbesondere uber Adenomyositis seroepithelialis und Adenomyometritis sarcomatosa. Zentralbl Gynäkol 1919, 36, 745–750. [Google Scholar]
- Munrós, J.; Martínez-Zamora, M.-A.; Tàssies, D.; Reverter, J.C.; Rius, M.; Gracia, M.; Ros, C.; Carmona, F. Total Circulating Microparticle Levels After Laparoscopic Surgical Treatment for Endometrioma: A Pilot, Prospective, Randomized Study Comparing Stripping with CO2 Laser Vaporization. J. Minim. Invasive Gynecol. 2019, 26, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Levander, G.; Normann, P. The Pathogenesis of Endometriosis. Acta Obstet. Gynecol. Scand. 1955, 34, 366–398. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.A. Endometrial induction of endometriosis across Millipore filters. Am J Obstet Gynecol 1966, 94, 780–790. [Google Scholar] [PubMed]
- Maniglio, P.; Ricciardi, E.; Meli, F.; Vitale, S.G.; Noventa, M.; Vitagliano, A.; Valenti, G.; La Rosa, V.L.; Laganà, A.S.; Caserta, D. Catamenial pneumothorax caused by thoracic endometriosis. Radiol. Case Rep. 2018, 13, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Uğur, M.; Turan, C.; Mungan, T.; Kuşçu, E.; Şenöz, S.; Ağış, H.T.; Gökmen, O. Endometriosis in Association with Müllerian Anomalies. Gynecol. Obstet. Investig. 1995, 40, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Olive, D.L.; Henderson, D.Y. Endometriosis and mullerian anomalies. Obstet. Gynecol. 1987, 69, 412–415. [Google Scholar]
- Javert, C.T. Pathogenesis of endometriosis based on endometrial homeoplasia, direct extension, exfoliation and implantation, lymphatic and hematogenous metastasis. Including five case reports of endometrial tissue in pelvic lymph nodes. Cancer 1949, 2, 399–410. [Google Scholar] [CrossRef]
- Halme, J.; Hammond, M.G.; Hulka, J.F.; Raj, S.G.; Talbert, L.M. Retrograde menstruation in healthy women and in patients with endometriosis. Obstet. Gynecol. 1984, 64, 151–154. [Google Scholar]
- Kruitwagen, R.F.; Poels, L.G.; Willemsen, W.N.; de Ronde, I.J.; Jap, P.H.; Rolland, R. Endometrial epithelial cells in peritoneal fluid during the early follicular phase. Fertil. Steril. 1991, 55, 297–303. [Google Scholar] [CrossRef]
- Koks, C.A.M.; Dunselman, G.A.J.; de Goeij, A.F.P.M.; Arends, J.W.; Evers, J.L.H. Evaluation of a menstrual cup to collect shed endometrium for in vitro studies. Fertil. Steril. 1997, 68, 560–564. [Google Scholar] [CrossRef]
- Vercellini, P.; Aimi, G.; De Giorgi, O.; Maddalena, S.; Carinelli, S.; Crosignani, P.G. Is cystic ovarian endometriosis an asymmetric disease? Br. J. Obstet. Gynaecol. 1998, 105, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Vercellini, P.; Abbiati, A.; Vigano, P.; Somigliana, E.D.; Daguati, R.; Meroni, F.; Crosignani, P.G. Asymmetry in distribution of diaphragmatic endometriotic lesions: Evidence in favour of the menstrual reflux theory. Hum. Reprod. 2007, 22, 2359–2367. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.; Olive, D.L.; Haney, A.F. Endometriosis: Pathogenetic implications of the anatomic distribution. Obstet. Gynecol. 1986, 67, 335–338. [Google Scholar]
- Martin, D.C. Laparoscopic Appearance of Endometriosis. Available online: https://www.danmartinmd.com/files/lae1988.pdf (accessed on 15 September 2019).
- Roman, H.; Hennetier, C.; Darwish, B.; Badescu, A.; Csanyi, M.; Aziz, M.; Tuech, J.-J.; Abo, C. Bowel occult microscopic endometriosis in resection margins in deep colorectal endometriosis specimens has no impact on short-term postoperative outcomes. Fertil. Steril. 2016, 105, 423–429. [Google Scholar] [CrossRef]
- Badescu, A.; Roman, H.; Aziz, M.; Puscasiu, L.; Molnar, C.; Huet, E.; Sabourin, J.-C.; Stolnicu, S. Mapping of bowel occult microscopic endometriosis implants surrounding deep endometriosis nodules infiltrating the bowel. Fertil. Steril. 2016, 105, 430–434. [Google Scholar] [CrossRef]
- Bruner-Tran, K.L.; Mokshagundam, S.; Herington, J.L.; Ding, T.; Osteen, K.G. Rodent Models of Experimental Endometriosis: Identifying Mechanisms of Disease and Therapeutic Targets. Curr. Womens Health Rev. 2018, 14, 173–188. [Google Scholar] [CrossRef]
- Laganà, A.S.; Garzon, S.; Franchi, M.; Casarin, J.; Gullo, G.; Ghezzi, F. Translational animal models for endometriosis research: A long and windy road. Ann. Transl. Med. 2018, 6, 431. [Google Scholar] [CrossRef]
- Klemmt, P.A.B.; Starzinski-Powitz, A. Molecular and Cellular Pathogenesis of Endometriosis. Curr. Womens Health Rev. 2018, 14, 106–116. [Google Scholar] [CrossRef]
- Ota, H.; Igarashi, S. Expression of major histocompatibility complex class II antigen in endometriotic tissue in patients with endometriosis and adenomyosis. Fertil. Steril. 1993, 60, 834–838. [Google Scholar] [CrossRef]
- Chung, H.W.; Wen, Y.; Chun, S.H.; Nezhat, C.; Woo, B.H.; Lake Polan, M. Matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-3 mRNA expression in ectopic and eutopic endometrium in women with endometriosis: A rationale for endometriotic invasiveness. Fertil. Steril. 2001, 75, 152–159. [Google Scholar] [CrossRef]
- Di Carlo, C.; Bonifacio, M.; Tommaselli, G.A.; Bifulco, G.; Guerra, G.; Nappi, C. Metalloproteinases, vascular endothelial growth factor, and angiopoietin 1 and 2 in eutopic and ectopic endometrium. Fertil. Steril. 2009, 91, 2315–2323. [Google Scholar] [CrossRef] [PubMed]
- Redwine, D.B. Was Sampson wrong? Fertil. Steril. 2002, 78, 686–693. [Google Scholar] [CrossRef]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Tamura, R.; Mori, Y.; Yamawaki, K.; Adachi, S.; Takahashi, T.; Kase, H.; et al. Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep. 2018, 24, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- Leyendecker, G.; Wildt, L.; Mall, G. The pathophysiology of endometriosis and adenomyosis: Tissue injury and repair. Arch. Gynecol. Obstet. 2009, 280, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Young, V.J.; Ahmad, S.F.; Duncan, W.C.; Horne, A.W. The role of TGF-β in the pathophysiology of peritoneal endometriosis. Hum. Reprod. Update 2017, 23, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Maas, J.W.; Groothuis, P.G.; Dunselman, G.A.; de Goeij, A.F.; Struijker-Boudier, H.A.; Evers, J.L. Development of endometriosis-like lesions after transplantation of human endometrial fragments onto the chick embryo chorioallantoic membrane. Hum. Reprod. 2001, 16, 627–631. [Google Scholar] [CrossRef]
- Nap, A.W.; Groothuis, P.G.; Demir, A.Y.; Maas, J.W.; Dunselman, G.A.; de Goeij, A.F.; Evers, J.L. Tissue integrity is essential for ectopic implantation of human endometrium in the chicken chorioallantoic membrane. Hum. Reprod. 2003, 18, 30–34. [Google Scholar] [CrossRef]
- Winterhager, E. Role of Steroid Hormones: Estrogen and Endometriosis. In Endometriosis: Science and Practice; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012; pp. 140–144. ISBN 978-1-4443-9851-9. [Google Scholar]
- Liang, Y.; Xie, H.; Wu, J.; Liu, D.; Yao, S. Villainous role of estrogen in macrophage-nerve interaction in endometriosis. Reprod. Biol. Endocrinol. 2018, 16, 122. [Google Scholar] [CrossRef]
- Rier, S.E.; Martin, D.C.; Bowman, R.E.; Becker, J.L. Immunoresponsiveness in endometriosis: Implications of estrogenic toxicants. Environ. Health Perspect. 1995, 103, 151–156. [Google Scholar]
- Maruyama, T.; Yoshimura, Y. Molecular and cellular mechanisms for differentiation and regeneration of the uterine endometrium. Endocr. J. 2008, 55, 795–810. [Google Scholar] [CrossRef] [PubMed]
- de Almeida Asencio, F.; Ribeiro, H.A.; Ayrosa Ribeiro, P.; Malzoni, M.; Adamyan, L.; Ussia, A.; Gomel, V.; Martin, D.C.; Koninckx, P.R. Symptomatic endometriosis developing several years after menopause in the absence of increased circulating estrogen concentrations: A systematic review and seven case reports. Gynecol. Surg. 2019, 16, 3. [Google Scholar] [CrossRef]
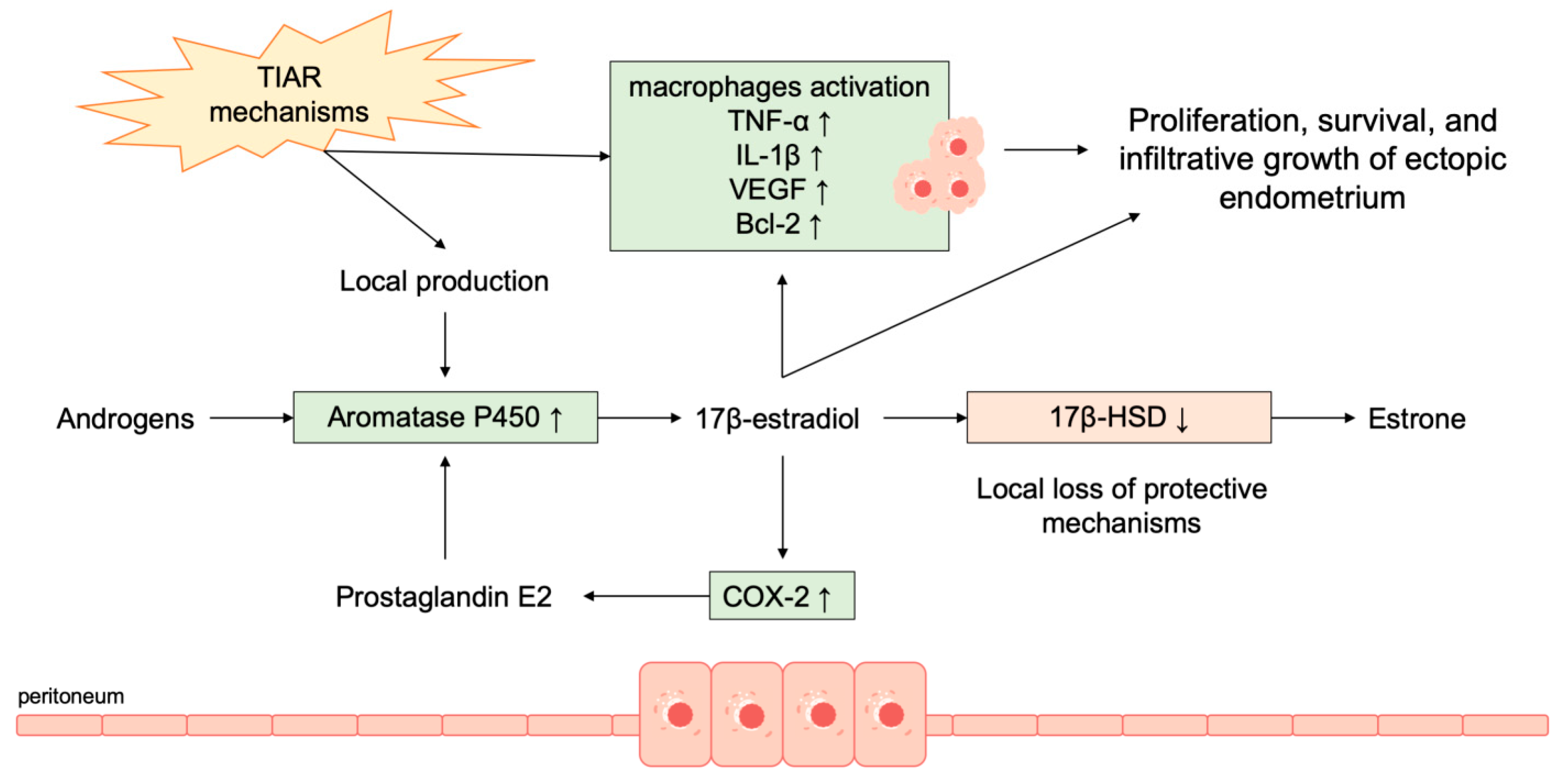
- Kitawaki, J.; Noguchi, T.; Amatsu, T.; Maeda, K.; Tsukamoto, K.; Yamamoto, T.; Fushiki, S.; Osawa, Y.; Honjo, H. Expression of aromatase cytochrome P450 protein and messenger ribonucleic acid in human endometriotic and adenomyotic tissues but not in normal endometrium. Biol. Reprod. 1997, 57, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Noble, L.S.; Simpson, E.R.; Johns, A.; Bulun, S.E. Aromatase expression in endometriosis. J. Clin. Endocrinol. Metab. 1996, 81, 174–179. [Google Scholar] [PubMed]
- Noble, L.S.; Takayama, K.; Zeitoun, K.M.; Putman, J.M.; Johns, D.A.; Hinshelwood, M.M.; Agarwal, V.R.; Zhao, Y.; Carr, B.R.; Bulun, S.E. Prostaglandin E2 stimulates aromatase expression in endometriosis-derived stromal cells. J. Clin. Endocrinol. Metab. 1997, 82, 600–606. [Google Scholar] [CrossRef]
- Zeitoun, K.; Takayama, K.; Sasano, H.; Suzuki, T.; Moghrabi, N.; Andersson, S.; Johns, A.; Meng, L.; Putman, M.; Carr, B. Deficient 17β-hydroxysteroid dehydrogenase type 2 expression in endometriosis: Failure to metabolize 17β-estradiol. J. Clin. Endocrinol. Metab. 1998, 83, 4474–4480. [Google Scholar] [CrossRef]
- TAKAHAsHI, K.; Nagata, H.; Kitao, M. Clinical usefulness of determination of estradiol level in the menstrual blood for patients with endometriosis. Nippon Sanka Fujinka Gakkai Zasshi 1989, 41, 1849–1850. [Google Scholar]
- Bulun, S.E.; Yang, S.; Fang, Z.; Gurates, B.; Tamura, M.; Zhou, J.; Sebastian, S. Role of aromatase in endometrial disease. J. Steroid Biochem. Mol. Biol. 2001, 79, 19–25. [Google Scholar] [CrossRef]
- Götte, M.; Wolf, M.; Staebler, A.; Buchweitz, O.; Kelsch, R.; Schüring, A.N.; Kiesel, L. Increased expression of the adult stem cell marker Musashi-1 in endometriosis and endometrial carcinoma. J. Pathol. 2008, 215, 317–329. [Google Scholar] [CrossRef]
- Valentijn, A.J.; Palial, K.; Al-Lamee, H.; Tempest, N.; Drury, J.; Von Zglinicki, T.; Saretzki, G.; Murray, P.; Gargett, C.E.; Hapangama, D.K. SSEA-1 isolates human endometrial basal glandular epithelial cells: Phenotypic and functional characterization and implications in the pathogenesis of endometriosis. Hum. Reprod. Oxf. Engl. 2013, 28, 2695–2708. [Google Scholar] [CrossRef]
- Leyendecker, G.; Herbertz, M.; Kunz, G.; Mall, G. Endometriosis results from the dislocation of basal endometrium. Hum. Reprod. 2002, 17, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Agic, A.; Djalali, S.; Diedrich, K.; Hornung, D. Apoptosis in endometriosis. Gynecol. Obstet. Investig. 2009, 68, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gong, P.; Chen, Y.; Nwachukwu, J.C.; Srinivasan, S.; Ko, C.; Bagchi, M.K.; Taylor, R.N.; Korach, K.S.; Nettles, K.W.; et al. Dual suppression of estrogenic and inflammatory activities for targeting of endometriosis. Sci. Transl. Med. 2015, 7, 271ra9. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.A.; Rodriguez, K.F.; Hewitt, S.C.; Janardhan, K.S.; Young, S.L.; Korach, K.S. Role of estrogen receptor signaling required for endometriosis-like lesion establishment in a mouse model. Endocrinology 2012, 153, 3960–3971. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Jung, S.Y.; Wu, S.-P.; Hawkins, S.M.; Park, M.J.; Kyo, S.; Qin, J.; Lydon, J.P.; Tsai, S.Y.; Tsai, M.-J.; et al. Estrogen Receptor β Modulates Apoptosis Complexes and the Inflammasome to Drive the Pathogenesis of Endometriosis. Cell 2015, 163, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Simmen, R.C.M.; Kelley, A.S. Reversal of Fortune: Estrogen Receptor-Beta in Endometriosis. J. Mol. Endocrinol. 2016, 57, F23–F27. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, K.J.; Arao, Y.; Korach, K.S. Estrogen hormone physiology: Reproductive findings from estrogen receptor mutant mice. Reprod. Biol. 2014, 14, 3–8. [Google Scholar] [CrossRef]
- Bulun, S.E.; Monsavais, D.; Pavone, M.E.; Dyson, M.; Xue, Q.; Attar, E.; Tokunaga, H.; Su, E.J. Role of Estrogen Receptor-β in Endometriosis. Semin. Reprod. Med. 2012, 30, 39–45. [Google Scholar] [CrossRef]
- Han, S.J.; Lee, J.E.; Cho, Y.J.; Park, M.J.; O’Malley, B.W. Genomic Function of Estrogen Receptor β in Endometriosis. Endocrinology 2019, 160, 2495–2516. [Google Scholar] [CrossRef]
- McKinnon, B.; Mueller, M.; Montgomery, G. Progesterone Resistance in Endometriosis: An Acquired Property? Trends Endocrinol. Metab. 2018, 29, 535–548. [Google Scholar] [CrossRef]
- Patel, B.G.; Rudnicki, M.; Yu, J.; Shu, Y.; Taylor, R.N. Progesterone resistance in endometriosis: Origins, consequences and interventions. Acta Obstet. Gynecol. Scand. 2017, 96, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Cheng, Y.-H.; Yin, P.; Imir, G.; Utsunomiya, H.; Attar, E.; Innes, J.; Julie Kim, J. Progesterone resistance in endometriosis: Link to failure to metabolize estradiol. Mol. Cell. Endocrinol. 2006, 248, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Grandi, G.; Mueller, M.D.; Bersinger, N.A.; Facchinetti, F.; McKinnon, B.D. The association between progestins, nuclear receptors expression and inflammation in endometrial stromal cells from women with endometriosis. Gynecol. Endocrinol. Off. J. Int. Soc. Gynecol. Endocrinol. 2017, 33, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Allport, V.C.; Pieber, D.; Slater, D.M.; Newton, R.; White, J.O.; Bennett, P.R. Human labour is associated with nuclear factor-kappaB activity which mediates cyclo-oxygenase-2 expression and is involved with the “functional progesterone withdrawal”. Mol. Hum. Reprod. 2001, 7, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Cinar, O.; Seval, Y.; Uz, Y.H.; Cakmak, H.; Ulukus, M.; Kayisli, U.A.; Arici, A. Differential regulation of Akt phosphorylation in endometriosis. Reprod. Biomed. Online 2009, 19, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Treloar, S.A.; Zhao, Z.Z.; Armitage, T.; Duffy, D.L.; Wicks, J.; O’Connor, D.T.; Martin, N.G.; Montgomery, G.W. Association between polymorphisms in the progesterone receptor gene and endometriosis. Mol. Hum. Reprod. 2005, 11, 641–647. [Google Scholar] [CrossRef][Green Version]
- Ren, Y.; Liu, X.; Ma, D.; Feng, Y.; Zhong, N. Down-regulation of the progesterone receptor by the methylation of progesterone receptor gene in endometrial cancer cells. Cancer Genet. Cytogenet. 2007, 175, 107–116. [Google Scholar] [CrossRef]
- Teague, E.M.C.O.; Print, C.G.; Hull, M.L. The role of microRNAs in endometriosis and associated reproductive conditions. Hum. Reprod. Update 2010, 16, 142–165. [Google Scholar] [CrossRef]
- Rižner, T.L. Diagnostic potential of peritoneal fluid biomarkers of endometriosis. Expert Rev. Mol. Diagn. 2015, 15, 557–580. [Google Scholar] [CrossRef]
- Haney, A.F.; Muscato, J.J.; Weinberg, J.B. Peritoneal fluid cell populations in infertility patients. Fertil. Steril. 1981, 35, 696–698. [Google Scholar] [CrossRef]
- Hill, J.A.; Faris, H.M.; Schiff, I.; Anderson, D.J. Characterization of leukocyte subpopulations in the peritoneal fluid of women with endometriosis. Fertil. Steril. 1988, 50, 216–222. [Google Scholar] [CrossRef]
- Jones, R.K.; Bulmer, J.N.; Searle, R.F. Phenotypic and functional studies of leukocytes in human endometrium and endometriosis. Hum. Reprod. Update 1998, 4, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Darrow, S.L.; Vena, J.E.; Batt, R.E.; Zielezny, M.A.; Michalek, A.M.; Selman, S. Menstrual cycle characteristics and the risk of endometriosis. Epidemiology 1993, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Vercellini, P.; De Giorgi, O.; Aimi, G.; Panazza, S.; Uglietti, A.; Crosignani, P.G. Menstrual characteristics in women with and without endometriosis. Obstet. Gynecol. 1997, 90, 264–268. [Google Scholar] [CrossRef]
- Salamanca, A.; Beltrán, E. Subendometrial contractility in menstrual phase visualized by transvaginal sonography in patients with endometriosis. Fertil. Steril. 1995, 64, 193–195. [Google Scholar] [CrossRef]
- Sanfilippo, J.S.; Wakim, N.G.; Schikler, K.N.; Yussman, M.A. Endometriosis in association with uterine anomaly. Am. J. Obstet. Gynecol. 1986, 154, 39–43. [Google Scholar] [CrossRef]
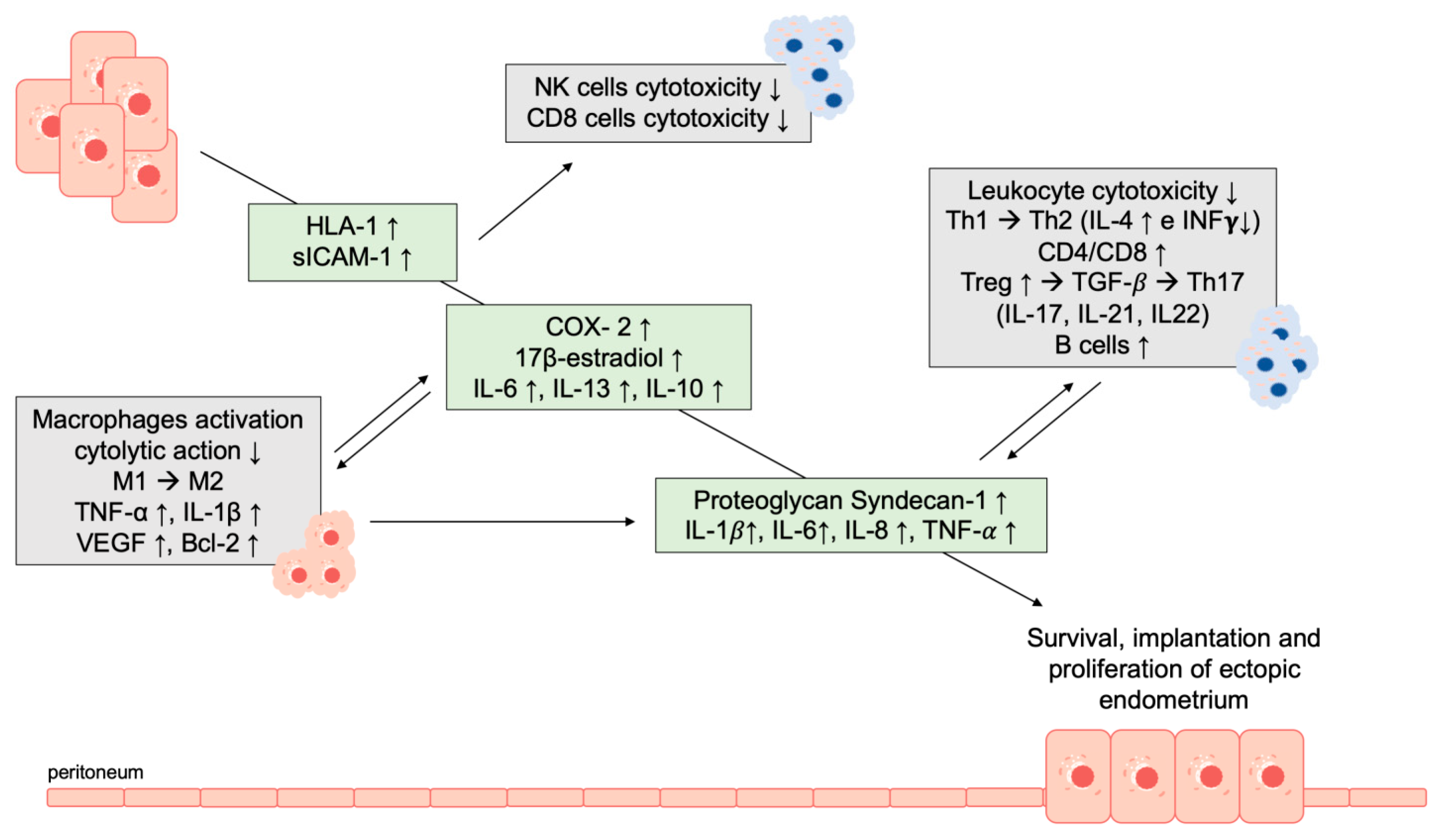
- Oosterlynck, D.J.; Cornillie, F.J.; Waer, M.; Vandeputte, M.; Koninckx, P.R. Women with endometriosis show a defect in natural killer activity resulting in a decreased cytotoxicity to autologous endometrium. Fertil. Steril. 1991, 56, 45–51. [Google Scholar] [CrossRef]
- Laganà, A.S.; Triolo, O.; Salmeri, F.M.; Granese, R.; Palmara, V.I.; Ban Frangež, H.; Vrtčnik Bokal, E.; Sofo, V. Natural Killer T cell subsets in eutopic and ectopic endometrium: A fresh look to a busy corner. Arch. Gynecol. Obstet. 2016, 293, 941–949. [Google Scholar] [CrossRef]
- Semino, C.; Semino, A.; Pietra, G.; Mingari, M.C.; Barocci, S.; Venturini, P.L.; Ragni, N.; Melioli, G. Role of major histocompatibility complex class I expression and natural killer-like T cells in the genetic control of endometriosis*. Fertil. Steril. 1995, 64, 909–916. [Google Scholar] [CrossRef]
- Somigliana, E.; Viganò, P.; Gaffuri, B.; Guarneri, D.; Busacca, M.; Vignali, M. Human endometrial stromal cells as a source of soluble intercellular adhesion molecule (ICAM)-1 molecules. Hum. Reprod. Oxf. Engl. 1996, 11, 1190–1194. [Google Scholar] [CrossRef]
- Viganò, P.; Gaffuri, B.; Somigliana, E.; Busacca, M.; Di Blasio, A.M.; Vignali, M. Expression of intercellular adhesion molecule (ICAM)-1 mRNA and protein is enhanced in endometriosis versus endometrial stromal cells in culture. Mol. Hum. Reprod. 1998, 4, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Králíčková, M.; Vetvicka, V. Immunological aspects of endometriosis: A review. Ann. Transl. Med. 2015, 3, 153. [Google Scholar] [PubMed]
- Riccio, L.D.G.C.; Santulli, P.; Marcellin, L.; Abrão, M.S.; Batteux, F.; Chapron, C. Immunology of endometriosis. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 50, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Santulli, P.; Marcellin, L.; Tosti, C.; Chouzenoux, S.; Cerles, O.; Borghese, B.; Batteux, F.; Chapron, C. MAP kinases and the inflammatory signaling cascade as targets for the treatment of endometriosis? Expert Opin. Ther. Targets 2015, 19, 1465–1483. [Google Scholar] [CrossRef]
- Martínez-Román, S.; Balasch, J.; Creus, M.; Fábregues, F.; Carmona, F.; Vilella, R.; Vanrell, J.A. Transferrin Receptor (CD71) Expression in Peritoneal Macrophages from Fertile and Infertile Women With and Without Endometriosis. Am. J. Reprod. Immunol. 1997, 38, 413–417. [Google Scholar] [CrossRef]
- Raiter-Tenenbaum, A.; Barañao, R.I.; Etchepareborda, J.J.; Meresman, G.F.; Rumi, L.S. Functional and phenotypic alterations in peritoneal macrophages from patients with early and advanced endometriosis. Arch. Gynecol. Obstet. 1998, 261, 147–157. [Google Scholar] [CrossRef]
- Halme, J.; Becker, S.; Wing, R. Accentuated cyclic activation of peritoneal macrophages in patients with endometriosis. Am. J. Obstet. Gynecol. 1984, 148, 85–90. [Google Scholar] [CrossRef]
- McLaren, J.; Prentice, A.; Charnock-Jones, D.S.; Sharkey, A.M.; Smith, S.K. Immunolocalization of the apoptosis regulating proteins Bcl-2 and Bax in human endometrium and isolated peritoneal fluid macrophages in endometriosis. Hum. Reprod. 1997, 12, 146–152. [Google Scholar] [CrossRef][Green Version]
- Bacci, M.; Capobianco, A.; Monno, A.; Cottone, L.; Di Puppo, F.; Camisa, B.; Mariani, M.; Brignole, C.; Ponzoni, M.; Ferrari, S.; et al. Macrophages Are Alternatively Activated in Patients with Endometriosis and Required for Growth and Vascularization of Lesions in a Mouse Model of Disease. Am. J. Pathol. 2009, 175, 547–556. [Google Scholar] [CrossRef]
- McLaren, J.; Dealtry, G.; Prentice, A.; Charnock-Jones, D.S.; Smith, S.K. Decreased levels of the potent regulator of monocyte/macrophage activation, interleukin-13, in the peritoneal fluid of patients with endometriosis. Hum. Reprod. Oxf. Engl. 1997, 12, 1307–1310. [Google Scholar] [CrossRef][Green Version]
- Schneider, C.; Kässens, N.; Greve, B.; Hassan, H.; Schüring, A.N.; Starzinski-Powitz, A.; Kiesel, L.; Seidler, D.G.; Götte, M. Targeting of syndecan-1 by micro-ribonucleic acid miR-10b modulates invasiveness of endometriotic cells via dysregulation of the proteolytic milieu and interleukin-6 secretion. Fertil. Steril. 2013, 99, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Joussen, A.M.; Klein, C.; Andre, P.; Wagner, D.D.; Hinkes, M.T.; Kirchhof, B.; Adamis, A.P.; Bernfield, M. Role of syndecan-1 in leukocyte-endothelial interactions in the ocular vasculature. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1135–1141. [Google Scholar] [PubMed]
- Averbeck, M.; Kuhn, S.; Bühligen, J.; Götte, M.; Simon, J.C.; Polte, T. Syndecan-1 regulates dendritic cell migration in cutaneous hypersensitivity to haptens. Exp. Dermatol. 2017, 26, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, A.; Salmeri, F.M.; Ardita, F.V.; Sofo, V.; Tripepi, M.; Marsico, S. Behaviour of cytokine levels in serum and peritoneal fluid of women with endometriosis. Gynecol. Obstet. Investig. 2002, 54, 82–87. [Google Scholar] [CrossRef]
- de Barros, I.B.L.; Malvezzi, H.; Gueuvoghlanian-Silva, B.Y.; Piccinato, C.A.; Rizzo, L.V.; Podgaec, S. “What do we know about regulatory T cells and endometriosis? A systematic review”. J. Reprod. Immunol. 2017, 120, 48–55. [Google Scholar] [CrossRef]
- Riccio, L.G.C.; Baracat, E.C.; Chapron, C.; Batteux, F.; Abrão, M.S. The role of the B lymphocytes in endometriosis: A systematic review. J. Reprod. Immunol. 2017, 123, 29–34. [Google Scholar] [CrossRef]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef]
- Tabibzadeh, S.; Zupi, E.; Babaknia, A.; Liu, R.; Marconi, D.; Romanini, C. Site and menstrual cycle-dependent expression of proteins of the tumour necrosis factor (TNF) receptor family, and BCL-2 oncoprotein and phase-specific production of TNF alpha in human endometrium. Hum. Reprod. Oxf. Engl. 1995, 10, 277–286. [Google Scholar] [CrossRef]
- Taniguchi, F.; Kaponis, A.; Izawa, M.; Kiyama, T.; Deura, I.; Ito, M.; Iwabe, T.; Adonakis, G.; Terakawa, N.; Harada, T. Apoptosis and endometriosis. Front. Biosci. Elite Ed. 2011, 3, 648–662. [Google Scholar] [CrossRef]
- Gebel, H.M.; Braun, D.P.; Tambur, A.; Frame, D.; Rana, N.; Dmowski, W.P. Spontaneous apoptosis of endometrial tissue is impaired in women with endometriosis. Fertil. Steril. 1998, 69, 1042–1047. [Google Scholar] [CrossRef]
- Béliard, A.; Noël, A.; Foidart, J.-M. Reduction of apoptosis and proliferation in endometriosis. Fertil. Steril. 2004, 82, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Lac, V.; Verhoef, L.; Aguirre-Hernandez, R.; Nazeran, T.M.; Tessier-Cloutier, B.; Praetorius, T.; Orr, N.L.; Noga, H.; Lum, A.; Khattra, J.; et al. Iatrogenic endometriosis harbors somatic cancer-driver mutations. Hum. Reprod. Oxf. Engl. 2019, 34, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Vetvicka, V.; Laganà, A.S.; Salmeri, F.M.; Triolo, O.; Palmara, V.I.; Vitale, S.G.; Sofo, V.; Králíčková, M. Regulation of apoptotic pathways during endometriosis: From the molecular basis to the future perspectives. Arch. Gynecol. Obstet. 2016, 294, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Sturlese, E.; Salmeri, F.M.; Retto, G.; Pizzo, A.; De Dominici, R.; Ardita, F.V.; Borrielli, I.; Licata, N.; Laganà, A.S.; Sofo, V. Dysregulation of the Fas/FasL system in mononuclear cells recovered from peritoneal fluid of women with endometriosis. J. Reprod. Immunol. 2011, 92, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, E.J.; Hill, A.D.K.; Hopkins, A.M. Adhesion in Physiological, Benign and Malignant Proliferative States of the Endometrium: Microenvironment and the Clinical Big Picture. Cells 2018, 7, e43. [Google Scholar] [CrossRef] [PubMed]
- Groothuis, P.G.; Koks, C.A.; de Goeij, A.F.; Dunselman, G.A.; Arends, J.W.; Evers, J.L. Adhesion of human endometrial fragments to peritoneum in vitro. Fertil. Steril. 1999, 71, 1119–1124. [Google Scholar] [CrossRef]
- Koks, C.A.; Groothuis, P.G.; Dunselman, G.A.; de Goeij, A.F.; Evers, J.L. Adhesion of shed menstrual tissue in an in-vitro model using amnion and peritoneum: A light and electron microscopic study. Hum. Reprod. Oxf. Engl. 1999, 14, 816–822. [Google Scholar] [CrossRef][Green Version]
- Dunselman, G.A.; Groothuis, P.G.; de Goeij, A.F.; Evers, J.L. The Mesothelium, Teflon or Velcro? Mesothelium in endometriosis pathogenesis. Hum. Reprod. Oxf. Engl. 2001, 16, 605–607. [Google Scholar] [CrossRef]
- Canis, M.; Bourdel, N.; Houlle, C.; Gremeau, A.-S.; Botchorishvili, R.; Matsuzaki, S. Trauma and endometriosis. A review. May we explain surgical phenotypes and natural history of the disease? J. Gynecol. Obstet. Hum. Reprod. 2017, 46, 219–227. [Google Scholar] [CrossRef]
- Koks, C.A.; Demir Weusten, A.Y.; Groothuis, P.G.; Dunselman, G.A.; de Goeij, A.F.; Evers, J.L. Menstruum induces changes in mesothelial cell morphology. Gynecol. Obstet. Investig. 2000, 50, 13–18. [Google Scholar] [CrossRef]
- Demir Weusten, A.Y.; Groothuis, P.G.; Dunselman, G.A.; de Goeij, A.F.; Arends, J.W.; Evers, J.L. Morphological changes in mesothelial cells induced by shed menstrual endometrium in vitro are not primarily due to apoptosis or necrosis. Hum. Reprod. Oxf. Engl. 2000, 15, 1462–1468. [Google Scholar] [CrossRef] [PubMed]
- Demir, A.Y.; Groothuis, P.G.; Nap, A.W.; Punyadeera, C.; de Goeij, A.F.P.M.; Evers, J.L.H.; Dunselman, G.A.J. Menstrual effluent induces epithelial-mesenchymal transitions in mesothelial cells. Hum. Reprod. Oxf. Engl. 2004, 19, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Bilyk, O.; Coatham, M.; Jewer, M.; Postovit, L.-M. Epithelial-to-Mesenchymal Transition in the Female Reproductive Tract: From Normal Functioning to Disease Pathology. Front. Oncol. 2017, 5, 145. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-M.; Yang, W.-X. Epithelial-to-mesenchymal transition in the development of endometriosis. Oncotarget 2017, 8, 41679–41689. [Google Scholar] [CrossRef]
- Lessey, B.A.; Damjanovich, L.; Coutifaris, C.; Castelbaum, A.; Albelda, S.M.; Buck, C.A. Integrin adhesion molecules in the human endometrium. Correlation with the normal and abnormal menstrual cycle. J. Clin. Investig. 1992, 90, 188–195. [Google Scholar] [CrossRef]
- Lessey, B.A. Endometrial integrins and the establishment of uterine receptivity. Hum. Reprod. 1998, 13, 247–258. [Google Scholar] [CrossRef]
- Tabibzadeh, S. Patterns of expression of integrin molecules in human endometrium throughout the menstrual cycle. Hum. Reprod. 1992, 7, 876–882. [Google Scholar] [CrossRef]
- van der Linden, P.J.Q.; de Goeij, A.F.P.M.; Dunselman, G.A.J.; Erkens, H.W.H.; Evers, J.L.H. Expression of cadherins and integrins in human endometrium throughout the menstrual cycle**Supported in part by a research grant from Organon International B.V., Oss, The Netherlands. Fertil. Steril. 1995, 63, 1210–1216. [Google Scholar] [CrossRef]
- Lessey, B.A.; Castelbaum, A.J.; Sawin, S.W.; Buck, C.A.; Schinnar, R.; Bilker, W.; Strom, B.L. Aberrant integrin expression in the endometrium of women with endometriosis. J. Clin. Endocrinol. Metab. 1994, 79, 643–649. [Google Scholar]
- Lessey, B.A.; Kim, J.J. Endometrial receptivity in the eutopic endometrium of women with endometriosis: It is affected, and let me show you why. Fertil. Steril. 2017, 108, 19–27. [Google Scholar] [CrossRef]
- Germeyer, A.; Klinkert, M.S.; Huppertz, A.-G.; Clausmeyer, S.; Popovici, R.M.; Strowitzki, T.; von Wolff, M. Expression of syndecans, cell-cell interaction regulating heparan sulfate proteoglycans, within the human endometrium and their regulation throughout the menstrual cycle. Fertil. Steril. 2007, 87, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Chelariu-Raicu, A.; Wilke, C.; Brand, M.; Starzinski-Powitz, A.; Kiesel, L.; Schüring, A.N.; Götte, M. Syndecan-4 expression is upregulated in endometriosis and contributes to an invasive phenotype. Fertil. Steril. 2016, 106, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Starzinski-Powitz, A.; Handrow-Metzmacher, H.; Kotzian, S. The putative role of cell adhesion molecules in endometriosis: Can we learn from tumour metastasis? Mol. Med. Today 1999, 5, 304–309. [Google Scholar] [CrossRef]
- Gaetje, R.; Kotzian, S.; Herrmann, G.; Baumann, R.; Starzinski-Powitz, A. Nonmalignant epithelial cells, potentially invasive in human endometriosis, lack the tumor suppressor molecule E-cadherin. Am. J. Pathol. 1997, 150, 461–467. [Google Scholar]
- Guilford, P. E-cadherin downregulation in cancer: Fuel on the fire? Mol. Med. Today 1999, 5, 172–177. [Google Scholar] [CrossRef]
- Wijnhoven, B.P.; Dinjens, W.N.; Pignatelli, M. E-cadherin-catenin cell-cell adhesion complex and human cancer. Br. J. Surg. 2000, 87, 992–1005. [Google Scholar] [CrossRef]
- van der Linden, P.J.; de Goeij, A.F.; Dunselman, G.A.; van der Linden, E.P.; Ramaekers, F.C.; Evers, J.L. Expression of integrins and E-cadherin in cells from menstrual effluent, endometrium, peritoneal fluid, peritoneum, and endometriosis. Fertil. Steril. 1994, 61, 85–90. [Google Scholar] [CrossRef]
- Mashayekhi, F.; Aryaee, H.; Mirzajani, E.; Yasin, A.A.; Fathi, A. Soluble CD44 concentration in the serum and peritoneal fluid samples of patients with different stages of endometriosis. Arch. Gynecol. Obstet. 2015, 292, 641–645. [Google Scholar] [CrossRef]
- Karousou, E.; Misra, S.; Ghatak, S.; Dobra, K.; Götte, M.; Vigetti, D.; Passi, A.; Karamanos, N.K.; Skandalis, S.S. Roles and targeting of the HAS/hyaluronan/CD44 molecular system in cancer. Matrix Biol. J. Int. Soc. Matrix Biol. 2017, 59, 3–22. [Google Scholar] [CrossRef]
- Rodgers, A.K.; Nair, A.; Binkley, P.A.; Tekmal, R.; Schenken, R.S. Inhibition of CD44 N- and O-linked glycosylation decreases endometrial cell lines attachment to peritoneal mesothelial cells. Fertil. Steril. 2011, 95, 823–825. [Google Scholar] [CrossRef][Green Version]
- Knudtson, J.F.; Tekmal, R.R.; Santos, M.T.; Binkley, P.A.; Krishnegowda, N.; Valente, P.; Schenken, R.S. Impaired Development of Early Endometriotic Lesions in CD44 Knockout Mice. Reprod. Sci. Thousand Oaks Calif 2016, 23, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Olivares, C.N.; Alaniz, L.D.; Menger, M.D.; Barañao, R.I.; Laschke, M.W.; Meresman, G.F. Inhibition of Hyaluronic Acid Synthesis Suppresses Angiogenesis in Developing Endometriotic Lesions. PloS ONE 2016, 11, e0152302. [Google Scholar] [CrossRef] [PubMed]
- Bałkowiec, M.; Maksym, R.B.; Włodarski, P.K. The bimodal role of matrix metalloproteinases and their inhibitors in etiology and pathogenesis of endometriosis. Mol. Med. Rep. 2018, 18, 3123–3136. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Willenbrock, F.; Crabbe, T.; O’Shea, M.; Ward, R.; Atkinson, S.; O’Connell, J.; Docherty, A. Regulation of matrix metalloproteinase activity. Ann. N. Y. Acad. Sci. 1994, 732, 31–41. [Google Scholar] [CrossRef]
- Nagase, H. Activation mechanisms of matrix metalloproteinases. Biol. Chem. 1997, 378, 151–160. [Google Scholar]
- Smigiel, K.S.; Parks, W.C. Matrix Metalloproteinases and Leukocyte Activation. Prog. Mol. Biol. Transl. Sci. 2017, 147, 167–195. [Google Scholar]
- Marbaix, E.; Kokorine, I.; Henriet, P.; Donnez, J.; Courtoy, P.J.; Eeckhout, Y. The expression of interstitial collagenase in human endometrium is controlled by progesterone and by oestradiol and is related to menstruation. Biochem. J. 1995, 305, 1027–1030. [Google Scholar] [CrossRef]
- Hulboy, D.L.; Rudolph, L.A.; Matrisian, L.M. Matrix metalloproteinases as mediators of reproductive function. Mol. Hum. Reprod. 1997, 3, 27–45. [Google Scholar] [CrossRef]
- Rodgers, W.H.; Matrisian, L.M.; Giudice, L.C.; Dsupin, B.; Cannon, P.; Svitek, C.; Gorstein, F.; Osteen, K.G. Patterns of matrix metalloproteinase expression in cycling endometrium imply differential functions and regulation by steroid hormones. J. Clin. Investig. 1994, 94, 946–953. [Google Scholar] [CrossRef]
- Bruner, K.L.; Eisenberg, E.; Gorstein, F.; Osteen, K.G. Progesterone and transforming growth factor-β coordinately regulate suppression of endometrial matrix metalloproteinases in a model of experimental endometriosis. Steroids 1999, 64, 648–653. [Google Scholar] [CrossRef]
- Spuijbroek, M.D.; Dunselman, G.A.; Menheere, P.P.; Evers, J.L. Early endometriosis invades the extracellular matrix. Fertil. Steril. 1992, 58, 929–933. [Google Scholar] [CrossRef]
- Osteen, K.G.; Bruner-Tran, K.L.; Keller, N.R.; Eisenberg, E. Progesterone-Mediated Endometrial Maturation Limits Matrix Metalloproteinase (MMP) Expression in an Inflammatory-like Environment. Ann. N. Y. Acad. Sci. 2002, 955, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Salamonsen, L.A.; Woolley, D.E. Matrix metalloproteinases in normal menstruation. Hum. Reprod. Oxf. Engl. 1996, 11 (Suppl. 2), 124–133. [Google Scholar] [CrossRef]
- Salamonsen, L.A.; Zhang, J.; Hampton, A.; Lathbury, L. Regulation of matrix metalloproteinases in human endometrium. Hum. Reprod. Oxf. Engl. 2000, 15 (Suppl. 3), 112–119. [Google Scholar] [CrossRef]
- Bruner, K.L.; Matrisian, L.M.; Rodgers, W.H.; Gorstein, F.; Osteen, K.G. Suppression of matrix metalloproteinases inhibits establishment of ectopic lesions by human endometrium in nude mice. J. Clin. Investig. 1997, 99, 2851–2857. [Google Scholar] [CrossRef] [PubMed]
- Nap, A.W.; Dunselman, G.A.J.; de Goeij, A.F.P.M.; Evers, J.L.H.; Groothuis, P.G. Inhibiting MMP activity prevents the development of endometriosis in the chicken chorioallantoic membrane model. Hum. Reprod. 2004, 19, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Osteen, K.G.; Bruner, K.L.; Sharpe-Timms, K.L. Steroid and growth factor regulation of matrix metalloproteinase expression and endometriosis. Semin. Reprod. Endocrinol. 1996, 14, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Sillem, M.; Prifti, S.; Neher, M.; Runnebaum, B. Extracellular matrix remodelling in the endometrium and its possible relevance to the pathogenesis of endometriosis. Hum. Reprod. Update 1998, 4, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Sharpe-Timms, K.L.; Keisler, L.W.; McIntush, E.W.; Keisler, D.H. Tissue inhibitor of metalloproteinase-1 concentrations are attenuated in peritoneal fluid and sera of women with endometriosis and restored in sera by gonadotropin-releasing hormone agonist therapy. Fertil. Steril. 1998, 69, 1128–1134. [Google Scholar] [CrossRef]
- Cox, K.E.; Piva, M.; Sharpe-Timms, K.L. Differential regulation of matrix metalloproteinase-3 gene expression in endometriotic lesions compared with endometrium. Biol. Reprod. 2001, 65, 1297–1303. [Google Scholar] [CrossRef]
- Sharpe-Timms, K.L.; Cox, K.E. Paracrine regulation of matrix metalloproteinase expression in endometriosis. Ann. N. Y. Acad. Sci. 2002, 955, 147–156; discussion 157–158, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Pitsos, M.; Kanakas, N. The role of matrix metalloproteinases in the pathogenesis of endometriosis. Reprod. Sci. Thousand Oaks Calif 2009, 16, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-E.; Nothnick, W.B. The relevancy of the matrix metalloproteinase system to the pathophysiology of endometriosis. Front. Biosci. J. Virtual Libr. 2005, 10, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Osteen, K.G.; Bruner-Tran, K.L.; Eisenberg, E. Reduced progesterone action during endometrial maturation: A potential risk factor for the development of endometriosis. Fertil. Steril. 2005, 83, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, I. Extracellular matrix remodelling: The role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Maas, J.W.; Le Noble, F.A.; Dunselman, G.A.; de Goeij, A.F.; Struyker Boudier, H.A.; Evers, J.L. The chick embryo chorioallantoic membrane as a model to investigate the angiogenic properties of human endometrium. Gynecol. Obstet. Investig. 1999, 48, 108–112. [Google Scholar] [CrossRef]
- Griffioen, A.W.; Molema, G. Angiogenesis: Potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol. Rev. 2000, 52, 237–268. [Google Scholar]
- McLaren, J. Vascular endothelial growth factor and endometriotic angiogenesis. Hum. Reprod. Update 2000, 6, 45–55. [Google Scholar] [CrossRef]
- Charnock-Jones, D.S.; Sharkey, A.M.; Rajput-Williams, J.; Burch, D.; Schofield, J.P.; Fountain, S.A.; Boocock, C.A.; Smith, S.K. Identification and localization of alternately spliced mRNAs for vascular endothelial growth factor in human uterus and estrogen regulation in endometrial carcinoma cell lines. Biol. Reprod. 1993, 48, 1120–1128. [Google Scholar] [CrossRef]
- Smith, S.K. Regulation of angiogenesis in the endometrium. Trends Endocrinol. Metab. TEM 2001, 12, 147–151. [Google Scholar] [CrossRef]
- Donnez, J.; Smoes, P.; Gillerot, S.; Casanas-Roux, F.; Nisolle, M. Vascular endothelial growth factor (VEGF) in endometriosis. Hum. Reprod. Oxf. Engl. 1998, 13, 1686–1690. [Google Scholar] [CrossRef] [PubMed]
- Wingfield, M.; Macpherson, A.; Healy, D.L.; Rogers, P.A. Cell proliferation is increased in the endometrium of women with endometriosis. Fertil. Steril. 1995, 64, 340–346. [Google Scholar] [CrossRef]
- Bourlev, V.; Volkov, N.; Pavlovitch, S.; Lets, N.; Larsson, A.; Olovsson, M. The relationship between microvessel density, proliferative activity and expression of vascular endothelial growth factor-A and its receptors in eutopic endometrium and endometriotic lesions. Reproduction 2006, 132, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, S.; Canis, M.; Murakami, T.; Dechelotte, P.; Bruhat, M.A.; Okamura, K. Immunohistochemical analysis of the role of angiogenic status in the vasculature of peritoneal endometriosis. Fertil. Steril. 2001, 76, 712–716. [Google Scholar] [CrossRef]
- Hull, M.L.; Charnock-Jones, D.S.; Chan, C.L.K.; Bruner-Tran, K.L.; Osteen, K.G.; Tom, B.D.M.; Fan, T.-P.D.; Smith, S.K. Antiangiogenic agents are effective inhibitors of endometriosis. J. Clin. Endocrinol. Metab. 2003, 88, 2889–2899. [Google Scholar] [CrossRef]
- Nap, A.W.; Dunselman, G.A.J.; Griffioen, A.W.; Mayo, K.H.; Evers, J.L.H.; Groothuis, P.G. Angiostatic agents prevent the development of endometriosis-like lesions in the chicken chorioallantoic membrane. Fertil. Steril. 2005, 83, 793–795. [Google Scholar] [CrossRef]
- Nap, A.W.; Griffioen, A.W.; Dunselman, G.A.J.; Bouma-Ter Steege, J.C.A.; Thijssen, V.L.J.L.; Evers, J.L.H.; Groothuis, P.G. Antiangiogenesis therapy for endometriosis. J. Clin. Endocrinol. Metab. 2004, 89, 1089–1095. [Google Scholar] [CrossRef]
- Van Langendonckt, A.; Donnez, J.; Defrère, S.; Dunselman, G.A.J.; Groothuis, P.G. Antiangiogenic and vascular-disrupting agents in endometriosis: Pitfalls and promises. Mol. Hum. Reprod. 2008, 14, 259–268. [Google Scholar] [CrossRef]
- Becker, C.M.; D’Amato, R.J. Angiogenesis and antiangiogenic therapy in endometriosis. Microvasc. Res. 2007, 74, 121–130. [Google Scholar] [CrossRef]
- May, K.; Becker, C.M. Endometriosis and angiogenesis. Minerva Ginecol. 2008, 60, 245–254. [Google Scholar]
- Taylor, R.N.; Yu, J.; Torres, P.B.; Schickedanz, A.C.; Park, J.K.; Mueller, M.D.; Sidell, N. Mechanistic and Therapeutic Implications of Angiogenesis in Endometriosis. Reprod. Sci. Thousand Oaks Calif 2009, 16, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Körbel, C.; Gerstner, M.D.; Menger, M.D.; Laschke, M.W. Notch signaling controls sprouting angiogenesis of endometriotic lesions. Angiogenesis 2018, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Yoshimura, Y. Stem cell theory for the pathogenesis of endometriosis. Front. Biosci. Elite Ed. 2012, 4, 2754–2763. [Google Scholar] [CrossRef] [PubMed]
- Gargett, C.E.; Masuda, H. Adult stem cells in the endometrium. Mol. Hum. Reprod. 2010, 16, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.S. Endometrial cells derived from donor stem cells in bone marrow transplant recipients. JAMA 2004, 292, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Matsuzaki, Y.; Hiratsu, E.; Ono, M.; Nagashima, T.; Kajitani, T.; Arase, T.; Oda, H.; Uchida, H.; Asada, H.; et al. Stem Cell-Like Properties of the Endometrial Side Population: Implication in Endometrial Regeneration. PLoS ONE 2010, 5, e10387. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Basir, Z.; Kajdacsy-Balla, A.; Strawn, E.; Macias, V.; Montgomery, K.; Guo, S.-W. Resolution of clonal origins for endometriotic lesions using laser capture microdissection and the human androgen receptor (HUMARA) assay. Fertil. Steril. 2003, 79, 710–717. [Google Scholar] [CrossRef]
- Nabeshima, H.; Murakami, T.; Yoshinaga, K.; Sato, K.; Terada, Y.; Okamura, K. Analysis of the clonality of ectopic glands in peritoneal endometriosis using laser microdissection. Fertil. Steril. 2003, 80, 1144–1150. [Google Scholar] [CrossRef]
- Mayr, D.; Amann, G.; Siefert, C.; Diebold, J.; Anderegg, B. Does endometriosis really have premalignant potential? A clonal analysis of laser-microdissected tissue. FASEB J. 2003, 17, 693–695. [Google Scholar] [CrossRef]
- Maruyama, T.; Masuda, H.; Ono, M.; Kajitani, T.; Yoshimura, Y. Human uterine stem/progenitor cells: Their possible role in uterine physiology and pathology. Reprod. Camb. Engl. 2010, 140, 11–22. [Google Scholar] [CrossRef]
- Wang, Y.; Sacchetti, A.; van Dijk, M.R.; van der Zee, M.; van der Horst, P.H.; Joosten, R.; Burger, C.W.; Grootegoed, J.A.; Blok, L.J.; Fodde, R. Identification of Quiescent, Stem-Like Cells in the Distal Female Reproductive Tract. PLOS ONE 2012, 7, e40691. [Google Scholar] [CrossRef] [PubMed]
- Laganà, A.S.; Salmeri, F.M.; Vitale, S.G.; Triolo, O.; Götte, M. Stem Cell Trafficking During Endometriosis: May Epigenetics Play a Pivotal Role? Reprod. Sci. Thousand Oaks Calif 2018, 25, 978–979. [Google Scholar] [CrossRef] [PubMed]
- Schüring, A.N.; Dahlhues, B.; Korte, A.; Kiesel, L.; Titze, U.; Heitkötter, B.; Ruckert, C.; Götte, M. The endometrial stem cell markers notch-1 and numb are associated with endometriosis. Reprod. Biomed. Online 2018, 36, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Wolf, M.; Staebler, A.; Buchweitz, O.; Kiesel, L.; Schüring, A.N. Aberrant expression of the pluripotency marker SOX-2 in endometriosis. Fertil. Steril. 2011, 95, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Adammek, M.; Greve, B.; Kässens, N.; Schneider, C.; Brüggemann, K.; Schüring, A.N.; Starzinski-Powitz, A.; Kiesel, L.; Götte, M. MicroRNA miR-145 inhibits proliferation, invasiveness, and stem cell phenotype of an in vitro endometriosis model by targeting multiple cytoskeletal elements and pluripotency factors. Fertil. Steril. 2013, 99, 1346–1355. [Google Scholar] [CrossRef]
- Eggers, J.C.; Martino, V.; Reinbold, R.; Schäfer, S.D.; Kiesel, L.; Starzinski-Powitz, A.; Schüring, A.N.; Kemper, B.; Greve, B.; Götte, M. microRNA miR-200b affects proliferation, invasiveness and stemness of endometriotic cells by targeting ZEB1, ZEB2 and KLF4. Reprod. Biomed. Online 2016, 32, 434–445. [Google Scholar] [CrossRef]
- Sapkota, Y.; Steinthorsdottir, V.; Morris, A.P.; Fassbender, A.; Rahmioglu, N.; De Vivo, I.; Buring, J.E.; Zhang, F.; Edwards, T.L.; Jones, S.; et al. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat. Commun. 2017, 8, 15539. [Google Scholar] [CrossRef]
- Bouaziz, J.; Mashiach, R.; Cohen, S.; Kedem, A.; Baron, A.; Zajicek, M.; Feldman, I.; Seidman, D.; Soriano, D. How Artificial Intelligence Can Improve Our Understanding of the Genes Associated with Endometriosis: Natural Language Processing of the PubMed Database. BioMed Res. Int. 2018, 2018, 6217812. [Google Scholar] [CrossRef]
- Gajbhiye, R.; Fung, J.N.; Montgomery, G.W. Complex genetics of female fertility. NPJ Genomic Med. 2018, 3, 29. [Google Scholar] [CrossRef]
- Kennedy, S. The genetics of endometriosis. J. Reprod. Med. 1998, 43, 263–268. [Google Scholar]
- Simpson, J.L.; Bischoff, F.Z. Heritability and molecular genetic studies of endometriosis. Ann. N. Y. Acad. Sci. 2002, 955, 239–251; discussion 293–295, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, H.; Geirsson, R.T.; Steinthorsdottir, V.; Jonsson, H.; Manolescu, A.; Kong, A.; Ingadottir, G.; Gulcher, J.; Stefansson, K. Genetic factors contribute to the risk of developing endometriosis. Hum. Reprod. Oxf. Engl. 2002, 17, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Treloar, S.; Hadfield, R.; Montgomery, G.; Lambert, A.; Wicks, J.; Barlow, D.H.; O’Connor, D.T.; Kennedy, S.; International Endogene Study Group. The International Endogene Study: A collection of families for genetic research in endometriosis. Fertil. Steril. 2002, 78, 679–685. [Google Scholar] [CrossRef]
- Zondervan, K.T.; Cardon, L.R.; Kennedy, S.H. The genetic basis of endometriosis. Curr. Opin. Obstet. Gynecol. 2001, 13, 309–314. [Google Scholar] [CrossRef]
- Zondervan, K.T.; Weeks, D.E.; Colman, R.; Cardon, L.R.; Hadfield, R.; Schleffler, J.; Trainor, A.G.; Coe, C.L.; Kemnitz, J.W.; Kennedy, S.H. Familial aggregation of endometriosis in a large pedigree of rhesus macaques. Hum. Reprod. Oxf. Engl. 2004, 19, 448–455. [Google Scholar] [CrossRef]
- Kennedy, S.; Mardon, H.; Barlow, D. Familial endometriosis. J. Assist. Reprod. Genet. 1995, 12, 32–34. [Google Scholar] [CrossRef]
- Hadfield, R.M.; Mardon, H.J.; Barlow, D.H.; Kennedy, S.H. Endometriosis in monozygotic twins. Fertil. Steril. 1997, 68, 941–942. [Google Scholar] [CrossRef]
- Moen, M.H. Endometriosis in monozygotic twins. Acta Obstet. Gynecol. Scand. 1994, 73, 59–62. [Google Scholar] [CrossRef]
- Treloar, S.A.; O’Connor, D.T.; O’Connor, V.M.; Martin, N.G. Genetic influences on endometriosis in an Australian twin sample. Fertil. Steril. 1999, 71, 701–710. [Google Scholar] [CrossRef]
- Di, W.; Guo, S.-W. The search for genetic variants predisposing women to endometriosis. Curr. Opin. Obstet. Gynecol. 2007, 19, 395–401. [Google Scholar] [CrossRef]
- Falconer, H.; D’Hooghe, T.; Fried, G. Endometriosis and genetic polymorphisms. Obstet. Gynecol. Surv. 2007, 62, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-W. Glutathione S-transferases M1/T1 gene polymorphisms and endometriosis: A meta-analysis of genetic association studies. Mol. Hum. Reprod. 2005, 11, 729–743. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-W. The association of endometriosis risk and genetic polymorphisms involving dioxin detoxification enzymes: A systematic review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2006, 124, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-W. Association of endometriosis risk and genetic polymorphisms involving sex steroid biosynthesis and their receptors: A meta-analysis. Gynecol. Obstet. Investig. 2006, 61, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, G.W.; Nyholt, D.R.; Zhao, Z.Z.; Treloar, S.A.; Painter, J.N.; Missmer, S.A.; Kennedy, S.H.; Zondervan, K.T. The search for genes contributing to endometriosis risk. Hum. Reprod. Update 2008, 14, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Çayan, F.; Ayaz, L.; Aban, M.; Dilek, S.; Gümüş, L.T. Role of CYP2C19 polymorphisms in patients with endometriosis. Gynecol. Endocrinol. 2009, 25, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Bozdag, G.; Alp, A.; Saribas, Z.; Tuncer, S.; Aksu, T.; Gurgan, T. CYP17 and CYP2C19 gene polymorphisms in patients with endometriosis. Reprod. Biomed. Online 2010, 20, 286–290. [Google Scholar] [CrossRef]
- Guo, S.-W.; Simsa, P.; Kyama, C.M.; Mihályi, A.; Fülöp, V.; Othman, E.-E.R.; D’Hooghe, T.M. Reassessing the evidence for the link between dioxin and endometriosis: From molecular biology to clinical epidemiology. MHR Basic Sci. Reprod. Med. 2009, 15, 609–624. [Google Scholar] [CrossRef]
- Ioannidis, J.P.A.; Ntzani, E.E.; Trikalinos, T.A.; Contopoulos-Ioannidis, D.G. Replication validity of genetic association studies. Nat. Genet. 2001, 29, 306–309. [Google Scholar] [CrossRef]
- Lohmueller, K.E.; Pearce, C.L.; Pike, M.; Lander, E.S.; Hirschhorn, J.N. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat. Genet. 2003, 33, 177–182. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.I.; Abecasis, G.R.; Cardon, L.R.; Goldstein, D.B.; Little, J.; Ioannidis, J.P.A.; Hirschhorn, J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008, 9, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Montgomery, G.W. Genome-wide association studies and human disease: From trickle to flood. JAMA 2009, 302, 2028–2029. [Google Scholar] [CrossRef] [PubMed]
- Zondervan, K.; Cardon, L.; Desrosiers, R.; Hyde, D.; Kemnitz, J.; Mansfield, K.; Roberts, J.; Scheffler, J.; Weeks, D.E.; Kennedy, S. The Genetic Epidemiology of Spontaneous Endometriosis in the Rhesus Monkey. Ann. N. Y. Acad. Sci. 2002, 955, 233–238. [Google Scholar] [CrossRef]
- Zondervan, K.T.; Cardon, L.R. The complex interplay among factors that influence allelic association. Nat. Rev. Genet. 2004, 5, 89–100. [Google Scholar] [CrossRef]
- Hirschhorn, J.N.; Lohmueller, K.; Byrne, E.; Hirschhorn, K. A comprehensive review of genetic association studies. Genet. Med. 2002, 4, 45–61. [Google Scholar] [CrossRef]
- Kruglyak, L.; Lander, E.S. Complete multipoint sib-pair analysis of qualitative and quantitative traits. Am. J. Hum. Genet. 1995, 57, 439–454. [Google Scholar]
- Lander, E.; Kruglyak, L. Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nat. Genet. 1995, 11, 241. [Google Scholar] [CrossRef]
- Risch, N. Linkage strategies for genetically complex traits. II. The power of affected relative pairs. Am. J. Hum. Genet. 1990, 46, 229–241. [Google Scholar]
- Treloar, S.A.; Wicks, J.; Nyholt, D.R.; Montgomery, G.W.; Bahlo, M.; Smith, V.; Dawson, G.; Mackay, I.J.; Weeks, D.E.; Bennett, S.T.; et al. Genomewide Linkage Study in 1,176 Affected Sister Pair Families Identifies a Significant Susceptibility Locus for Endometriosis on Chromosome 10q26. Am. J. Hum. Genet. 2005, 77, 365–376. [Google Scholar] [CrossRef]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789. [Google Scholar] [CrossRef] [PubMed]
- Zondervan, K.T.; Treloar, S.A.; Lin, J.; Weeks, D.E.; Nyholt, D.R.; Mangion, J.; MacKay, I.J.; Cardon, L.R.; Martin, N.G.; Kennedy, S.H.; et al. Significant evidence of one or more susceptibility loci for endometriosis with near-Mendelian inheritance on chromosome 7p13–15. Hum. Reprod. 2007, 22, 717–728. [Google Scholar] [CrossRef] [PubMed]
- The International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature 2007, 449, 851–861. [CrossRef]
- Treloar, S.A.; Zhao, Z.Z.; Le, L.; Zondervan, K.T.; Martin, N.G.; Kennedy, S.; Nyholt, D.R.; Montgomery, G.W. Variants in EMX2 and PTEN do not contribute to risk of endometriosis. MHR Basic Sci. Reprod. Med. 2007, 13, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Daftary, G.S.; Taylor, H.S. EMX2 Gene Expression in the Female Reproductive Tract and Aberrant Expression in the Endometrium of Patients with Endometriosis. J. Clin. Endocrinol. Metab. 2004, 89, 2390–2396. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Troy, P.J.; Daftary, G.S.; Bagot, C.N.; Taylor, H.S. Transcriptional Repression of Peri-Implantation EMX2 Expression in Mammalian Reproduction by HOXA10. Mol. Cell. Biol. 2003, 23, 1–13. [Google Scholar] [CrossRef]
- Maxwell, G.L.; Risinger, J.I.; Gumbs, C.; Shaw, H.; Bentley, R.C.; Barrett, J.C.; Berchuck, A.; Futreal, P.A. Mutation of the PTEN Tumor Suppressor Gene in Endometrial Hyperplasias. Cancer Res. 1998, 58, 2500–2503. [Google Scholar]
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.P.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007, 447, 1087–1093. [Google Scholar] [CrossRef]
- Pollock, P.M.; Gartside, M.G.; Dejeza, L.C.; Powell, M.A.; Mallon, M.A.; Cancer Genome Project; Davies, H.; Mohammadi, M.; Futreal, P.A.; Stratton, M.R.; et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 2007, 26, 7158–7162. [Google Scholar] [CrossRef]
- Zhao, Z.Z.; Pollock, P.M.; Thomas, S.; Treloar, S.A.; Nyholt, D.R.; Montgomery, G.W. Common variation in the fibroblast growth factor receptor 2 gene is not associated with endometriosis risk. Hum. Reprod. 2008, 23, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Zondervan, K.T.; Cardon, L.R. Designing candidate gene and genome-wide case–control association studies. Nat. Protoc. 2007, 2, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Zöllner, S.; Pritchard, J.K. Overcoming the Winner’s Curse: Estimating Penetrance Parameters from Case-Control Data. Am. J. Hum. Genet. 2007, 80, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef]
- Uno, S.; Zembutsu, H.; Hirasawa, A.; Takahashi, A.; Kubo, M.; Akahane, T.; Aoki, D.; Kamatani, N.; Hirata, K.; Nakamura, Y. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat. Genet. 2010, 42, 707–710. [Google Scholar] [CrossRef]
- Kondera-Anasz, Z.; Sikora, J.; Mielczarek-Palacz, A.; Jońca, M. Concentrations of interleukin (IL)-1α, IL-1 soluble receptor type II (IL-1 sRII) and IL-1 receptor antagonist (IL-1 Ra) in the peritoneal fluid and serum of infertile women with endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2005, 123, 198–203. [Google Scholar] [CrossRef]
- Adachi, S.; Tajima, A.; Quan, J.; Haino, K.; Yoshihara, K.; Masuzaki, H.; Katabuchi, H.; Ikuma, K.; Suginami, H.; Nishida, N.; et al. Meta-analysis of genome-wide association scans for genetic susceptibility to endometriosis in Japanese population. J. Hum. Genet. 2010, 55, 816–821. [Google Scholar] [CrossRef]
- Zanatta, A.; Rocha, A.M.; Carvalho, F.M.; Pereira, R.M.A.; Taylor, H.S.; Motta, E.L.A.; Baracat, E.C.; Serafini, P.C. The role of the Hoxa10/HOXA10 gene in the etiology of endometriosis and its related infertility: A review. J. Assist. Reprod. Genet. 2010, 27, 701–710. [Google Scholar] [CrossRef]
- Naillat, F.; Prunskaite-Hyyryläinen, R.; Pietilä, I.; Sormunen, R.; Jokela, T.; Shan, J.; Vainio, S.J. Wnt4/5a signalling coordinates cell adhesion and entry into meiosis during presumptive ovarian follicle development. Hum. Mol. Genet. 2010, 19, 1539–1550. [Google Scholar] [CrossRef]
- Boyer, A.; Lapointe, É.; Zheng, X.; Cowan, R.G.; Li, H.; Quirk, S.M.; DeMayo, F.J.; Richards, J.S.; Boerboom, D. WNT4 is required for normal ovarian follicle development and female fertility. FASEB J. 2010, 24, 3010–3025. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Painter, J.N.; Anderson, C.A.; Nyholt, D.R.; Macgregor, S.; Lin, J.; Lee, S.H.; Lambert, A.; Zhao, Z.Z.; Roseman, F.; Guo, Q.; et al. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat. Genet. 2011, 43, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, W.; Bonilla, C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008, 40, 695–701. [Google Scholar] [CrossRef]
- Gorlov, I.P.; Gorlova, O.Y.; Sunyaev, S.R.; Spitz, M.R.; Amos, C.I. Shifting Paradigm of Association Studies: Value of Rare Single-Nucleotide Polymorphisms. Am. J. Hum. Genet. 2008, 82, 100–112. [Google Scholar] [CrossRef]
- Curtin, K.; Iles, M.M.; Camp, N.J. Identifying Rarer Genetic Variants for Common Complex Diseases: Diseased Versus Neutral Discovery Panels. Ann. Hum. Genet. 2009, 73, 54–60. [Google Scholar] [CrossRef]
- Iles, M.M. What Can Genome-Wide Association Studies Tell Us about the Genetics of Common Disease? PLOS Genet. 2008, 4, e33. [Google Scholar] [CrossRef]
- Mettler, L.; Salmassi, A.; Schollmeyer, T.; Schmutzler, A.G.; Püngel, F.; Jonat, W. Comparison of c-DNA microarray analysis of gene expression between eutopic endometrium and ectopic endometrium (endometriosis). J. Assist. Reprod. Genet. 2007, 24, 249–258. [Google Scholar] [CrossRef][Green Version]
- Eyster, K.M.; Klinkova, O.; Kennedy, V.; Hansen, K.A. Whole genome deoxyribonucleic acid microarray analysis of gene expression in ectopic versus eutopic endometrium. Fertil. Steril. 2007, 88, 1505–1533. [Google Scholar] [CrossRef]
- Flores, I.; Rivera, E.; Ruiz, L.A.; Santiago, O.I.; Vernon, M.W.; Appleyard, C.B. Molecular profiling of experimental endometriosis identified gene expression patterns in common with human disease. Fertil. Steril. 2007, 87, 1180–1199. [Google Scholar] [CrossRef]
- Hull, M.L.; Escareno, C.R.; Godsland, J.M.; Doig, J.R.; Johnson, C.M.; Phillips, S.C.; Smith, S.K.; Tavaré, S.; Print, C.G.; Charnock-Jones, D.S. Endometrial-Peritoneal Interactions during Endometriotic Lesion Establishment. Am. J. Pathol. 2008, 173, 700–715. [Google Scholar] [CrossRef] [PubMed]
- Zafrakas, M.; Tarlatzis, B.C.; Streichert, T.; Pournaropoulos, F.; Wölfle, U.; Smeets, S.J.; Wittek, B.; Grimbizis, G.; Brakenhoff, R.H.; Pantel, K.; et al. Genome-wide microarray gene expression, array-CGH analysis, and telomerase activity in advanced ovarian endometriosis: A high degree of differentiation rather than malignant potential. Int. J. Mol. Med. 2008, 21, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Pelch, K.E.; Schroder, A.L.; Kimball, P.A.; Sharpe-Timms, K.L.; Davis, J.W.; Nagel, S.C. Aberrant gene expression profile in a mouse model of endometriosis mirrors that observed in women. Fertil. Steril. 2010, 93, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, M.; Tanaka, N.; Tainaka, H.; Takeda, K.; Ihara, T.; Sugamata, M. Microarray analysis provides insight into the early steps of pathophysiology of mouse endometriosis model induced by autotransplantation of endometrium. Life Sci. 2009, 84, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Bruner-Tran, K.L.; Gnecco, J.; Ding, T.; Glore, D.R.; Pensabene, V.; Osteen, K.G. Exposure to the environmental endocrine disruptor TCDD and human reproductive dysfunction: Translating lessons from murine models. Reprod. Toxicol. Elmsford N 2017, 68, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef]
- Gabory, A.; Attig, L.; Junien, C. Epigenetic mechanisms involved in developmental nutritional programming. World J. Diabetes 2011, 2, 164–175. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Robertson, K.D.; Wolffe, A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef]
- Rodenhiser, D.; Mann, M. Epigenetics and human disease: Translating basic biology into clinical applications. CMAJ Can. Med. Assoc. J. J. Assoc. Medicale Can. 2006, 174, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Li, E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 2002, 3, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, W. microRNA Target Prediction. Methods Mol. Biol. Clifton NJ 2017, 1513, 193–200. [Google Scholar]
- Sun, K.; Lai, E.C. Adult-specific functions of animal microRNAs. Nat. Rev. Genet. 2013, 14, 535–548. [Google Scholar] [CrossRef]
- Kurokawa, R.; Rosenfeld, M.G.; Glass, C.K. Transcriptional regulation through noncoding RNAs and epigenetic modifications. RNA Biol. 2009, 6, 233–236. [Google Scholar] [CrossRef][Green Version]
- Saare, M.; Rekker, K.; Laisk-Podar, T.; Rahmioglu, N.; Zondervan, K.; Salumets, A.; Götte, M.; Peters, M. Challenges in endometriosis miRNA studies - From tissue heterogeneity to disease specific miRNAs. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2282–2292. [Google Scholar] [CrossRef]
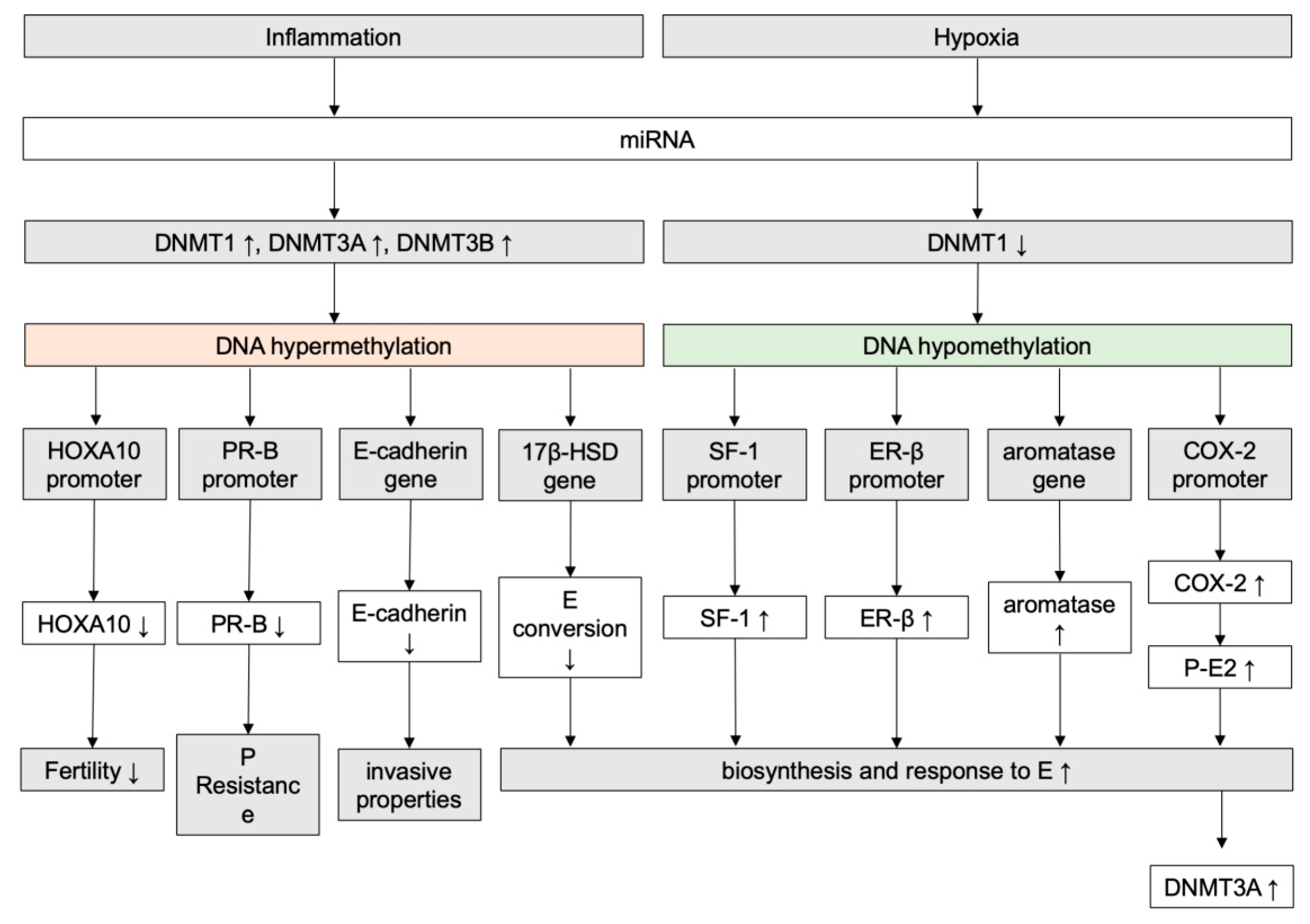
- Munro, S.K.; Farquhar, C.M.; Mitchell, M.D.; Ponnampalam, A.P. Epigenetic regulation of endometrium during the menstrual cycle. Mol. Hum. Reprod. 2010, 16, 297–310. [Google Scholar] [CrossRef]
- Yamagata, Y.; Asada, H.; Tamura, I.; Lee, L.; Maekawa, R.; Taniguchi, K.; Taketani, T.; Matsuoka, A.; Tamura, H.; Sugino, N. DNA methyltransferase expression in the human endometrium: Down-regulation by progesterone and estrogen. Hum. Reprod. Oxf. Engl. 2009, 24, 1126–1132. [Google Scholar] [CrossRef]
- Krusche, C.A.; Vloet, A.J.; Classen-Linke, I.; von Rango, U.; Beier, H.M.; Alfer, J. Class I histone deacetylase expression in the human cyclic endometrium and endometrial adenocarcinomas. Hum. Reprod. Oxf. Engl. 2007, 22, 2956–2966. [Google Scholar] [CrossRef]
- Sakai, N.; Maruyama, T.; Sakurai, R.; Masuda, H.; Yamamoto, Y.; Shimizu, A.; Kishi, I.; Asada, H.; Yamagoe, S.; Yoshimura, Y. Involvement of histone acetylation in ovarian steroid-induced decidualization of human endometrial stromal cells. J. Biol. Chem. 2003, 278, 16675–16682. [Google Scholar] [CrossRef]
- Uchida, H.; Maruyama, T.; Ohta, K.; Ono, M.; Arase, T.; Kagami, M.; Oda, H.; Kajitani, T.; Asada, H.; Yoshimura, Y. Histone deacetylase inhibitor-induced glycodelin enhances the initial step of implantation. Hum. Reprod. Oxf. Engl. 2007, 22, 2615–2622. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Maruyama, T.; Nagashima, T.; Ono, M.; Masuda, H.; Arase, T.; Sugiura, I.; Onouchi, M.; Kajitani, T.; Asada, H.; et al. Human endometrial cytodifferentiation by histone deacetylase inhibitors. Hum. Cell 2006, 19, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Galliano, D.; Pellicer, A. MicroRNA and implantation. Fertil. Steril. 2014, 101, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Hull, M.L.; Nisenblat, V. Tissue and circulating microRNA influence reproductive function in endometrial disease. Reprod. Biomed. Online 2013, 27, 515–529. [Google Scholar] [CrossRef]
- Pan, Q.; Chegini, N. MicroRNA signature and regulatory functions in the endometrium during normal and disease states. Semin. Reprod. Med. 2008, 26, 479–493. [Google Scholar] [CrossRef]
- Kuokkanen, S.; Chen, B.; Ojalvo, L.; Benard, L.; Santoro, N.; Pollard, J.W. Genomic profiling of microRNAs and messenger RNAs reveals hormonal regulation in microRNA expression in human endometrium. Biol. Reprod. 2010, 82, 791–801. [Google Scholar] [CrossRef]
- Lessey, B.A. Fine tuning of endometrial function by estrogen and progesterone through microRNAs. Biol. Reprod. 2010, 82, 653–655. [Google Scholar] [CrossRef]
- Pastorelli, L.M.; Wells, S.; Fray, M.; Smith, A.; Hough, T.; Harfe, B.D.; McManus, M.T.; Smith, L.; Woolf, A.S.; Cheeseman, M.; et al. Genetic analyses reveal a requirement for Dicer1 in the mouse urogenital tract. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2009, 20, 140–151. [Google Scholar] [CrossRef]
- Hong, X.; Luense, L.J.; McGinnis, L.K.; Nothnick, W.B.; Christenson, L.K. Dicer1 is essential for female fertility and normal development of the female reproductive system. Endocrinology 2008, 149, 6207–6212. [Google Scholar] [CrossRef]
- Koninckx, P.R.; Ussia, A.; Adamyan, L.; Wattiez, A.; Gomel, V.; Martin, D.C. Pathogenesis of endometriosis: The genetic/epigenetic theory. Fertil. Steril. 2019, 111, 327–340. [Google Scholar] [CrossRef]
- Yamagata, Y.; Nishino, K.; Takaki, E.; Sato, S.; Maekawa, R.; Nakai, A.; Sugino, N. Genome-wide DNA methylation profiling in cultured eutopic and ectopic endometrial stromal cells. PloS ONE 2014, 9, e83612. [Google Scholar] [CrossRef] [PubMed]
- Signorile, P.G.; Severino, A.; Santoro, M.; Spyrou, M.; Viceconte, R.; Baldi, A. Methylation analysis of HOXA10 regulatory elements in patients with endometriosis. BMC Res. Notes 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Halverson, G.; Basir, Z.; Strawn, E.; Yan, P.; Guo, S.-W. Aberrant methylation at HOXA10 may be responsible for its aberrant expression in the endometrium of patients with endometriosis. Am. J. Obstet. Gynecol. 2005, 193, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Du, H.; Taylor, H.S. Experimental Murine Endometriosis Induces DNA Methylation and Altered Gene Expression in Eutopic Endometrium. Biol. Reprod. 2009, 80, 79–85. [Google Scholar] [CrossRef]
- Bromer, J.G.; Wu, J.; Zhou, Y.; Taylor, H.S. Hypermethylation of Homeobox A10 by in Utero Diethylstilbestrol Exposure: An Epigenetic Mechanism for Altered Developmental Programming. Endocrinology 2009, 150, 3376–3382. [Google Scholar] [CrossRef][Green Version]
- Bromer, J.G.; Zhou, Y.; Taylor, M.B.; Doherty, L.; Taylor, H.S. Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response. FASEB J. 2010, 24, 2273–2280. [Google Scholar] [CrossRef]
- Kulp, J.L.; Mamillapalli, R.; Taylor, H.S. Aberrant HOXA10 Methylation in Patients With Common Gynecologic Disorders. Reprod. Sci. 2016, 23, 455–463. [Google Scholar] [CrossRef]
- Wu, Y.; Strawn, E.; Basir, Z.; Halverson, G.; Guo, S.-W. Promoter hypermethylation of progesterone receptor isoform B (PR-B) in endometriosis. Epigenetics 2006, 1, 106–111. [Google Scholar] [CrossRef]
- Attia, G.R.; Zeitoun, K.; Edwards, D.; Johns, A.; Carr, B.R.; Bulun, S.E. Progesterone receptor isoform A but not B is expressed in endometriosis. J. Clin. Endocrinol. Metab. 2000, 85, 2897–2902. [Google Scholar] [CrossRef]
- Nie, J.; Liu, X.; Guo, S.W. Promoter hypermethylation of progesterone receptor isoform B (PR-B) in adenomyosis and its rectification by a histone deacetylase inhibitor and a demethylation agent. Reprod. Sci. Thousand Oaks Calif 2010, 17, 995–1005. [Google Scholar]
- Wu, Y.; Strawn, E.; Basir, Z.; Halverson, G.; Guo, S.-W. Aberrant expression of deoxyribonucleic acid methyltransferases DNMT1, DNMT3A, and DNMT3B in women with endometriosis. Fertil. Steril. 2007, 87, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Dyson, M.T.; Kakinuma, T.; Pavone, M.E.; Monsivais, D.; Navarro, A.; Malpani, S.S.; Ono, M.; Bulun, S.E. Aberrant expression and localization of deoxyribonucleic acid methyltransferase 3B in endometriotic stromal cells. Fertil. Steril. 2015, 104, 953–963. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aznaurova, Y.B.; Zhumataev, M.B.; Roberts, T.K.; Aliper, A.M.; Zhavoronkov, A.A. Molecular aspects of development and regulation of endometriosis. Reprod. Biol. Endocrinol. RBE 2014, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Starzinski-Powitz, A.; Guo, S.-W. Trichostatin A, a histone deacetylase inhibitor, attenuates invasiveness and reactivates E-cadherin expression in immortalized endometriotic cells. Reprod. Sci. Thousand Oaks Calif 2007, 14, 374–382. [Google Scholar] [CrossRef]
- KOUKOURA, O.; SIFAKIS, S.; SPANDIDOS, D.A. DNA methylation in endometriosis (Review). Mol. Med. Rep. 2016, 13, 2939–2948. [Google Scholar] [CrossRef]
- Arosh, J.A.; Lee, J.; Starzinski-Powitz, A.; Banu, S.K. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 modulates DNA methylation and histone modification machinery proteins in human endometriotic cells. Mol. Cell. Endocrinol. 2015, 409, 51–58. [Google Scholar] [CrossRef]
- Xue, Q.; Lin, Z.; Yin, P.; Milad, M.P.; Cheng, Y.-H.; Confino, E.; Reierstad, S.; Bulun, S.E. Transcriptional activation of steroidogenic factor-1 by hypomethylation of the 5’ CpG island in endometriosis. J. Clin. Endocrinol. Metab. 2007, 92, 3261–3267. [Google Scholar] [CrossRef]
- Xue, Q.; Lin, Z.; Cheng, Y.-H.; Huang, C.-C.; Marsh, E.; Yin, P.; Milad, M.P.; Confino, E.; Reierstad, S.; Innes, J.; et al. Promoter methylation regulates estrogen receptor 2 in human endometrium and endometriosis. Biol. Reprod. 2007, 77, 681–687. [Google Scholar] [CrossRef]
- Izawa, M.; Taniguchi, F.; Uegaki, T.; Takai, E.; Iwabe, T.; Terakawa, N.; Harada, T. Demethylation of a nonpromoter cytosine-phosphate-guanine island in the aromatase gene may cause the aberrant up-regulation in endometriotic tissues. Fertil. Steril. 2011, 95, 33–39. [Google Scholar] [CrossRef]
- Wang, D.; Chen, Q.; Zhang, C.; Ren, F.; Li, T. DNA hypomethylation of the COX-2 gene promoter is associated with up-regulation of its mRNA expression in eutopic endometrium of endometriosis. Eur. J. Med. Res. 2012, 17, 12. [Google Scholar] [CrossRef]
- Nasu, K.; Kawano, Y.; Kai, K.; Aoyagi, Y.; Abe, W.; Okamoto, M.; Narahara, H. Aberrant histone modification in endometriosis. Front. Biosci. Landmark Ed. 2014, 19, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, J.B.; Colón-Díaz, M.; García, M.; Gutierrez, S.; Colón, M.; Seto, E.; Laboy, J.; Flores, I. Endometriosis Is Characterized by a Distinct Pattern of Histone 3 and Histone 4 Lysine Modifications. Reprod. Sci. 2014, 21, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Xiaomeng, X.; Ming, Z.; Jiezhi, M.; Xiaoling, F. Aberrant histone acetylation and methylation levels in woman with endometriosis. Arch. Gynecol. Obstet. 2013, 287, 487–494. [Google Scholar] [CrossRef]
- Samartzis, E.P.; Noske, A.; Samartzis, N.; Fink, D.; Imesch, P. The Expression of Histone Deacetylase 1, But Not Other Class I Histone Deacetylases, Is Significantly Increased in Endometriosis. Reprod. Sci. 2013, 20, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Nasu, K.; Li, H.; Tsuno, A.; Abe, W.; Takai, N.; Narahara, H. Application of the histone deacetylase inhibitors for the treatment of endometriosis: Histone modifications as pathogenesis and novel therapeutic target. Hum. Reprod. Oxf. Engl. 2011, 26, 2486–2498. [Google Scholar] [CrossRef]
- Imesch, P.; Samartzis, E.P.; Schneider, M.; Fink, D.; Fedier, A. Inhibition of transcription, expression, and secretion of the vascular epithelial growth factor in human epithelial endometriotic cells by romidepsin. Fertil. Steril. 2011, 95, 1579–1583. [Google Scholar] [CrossRef] [PubMed]
- Ayhan, A.; Mao, T.-L.; Seckin, T.; Wu, C.-H.; Guan, B.; Ogawa, H.; Futagami, M.; Mizukami, H.; Yokoyama, Y.; Kurman, R.J.; et al. Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2012, 22, 1310–1315. [Google Scholar] [CrossRef]
- Lowery, W.J.; Schildkraut, J.M.; Akushevich, L.; Bentley, R.; Marks, J.R.; Huntsman, D.; Berchuck, A. Loss of ARID1A-associated Protein Expression Is a Frequent Event in Clear Cell and Endometrioid Ovarian Cancers. Int. J. Gynecol. Cancer 2012, 22, 9–14. [Google Scholar] [CrossRef]
- Chene, G.; Ouellet, V.; Rahimi, K.; Barres, V.; Provencher, D.; Mes-Masson, A.M. The ARID1A pathway in ovarian clear cell and endometrioid carcinoma, contiguous endometriosis, and benign endometriosis. Int. J. Gynaecol. Obstet. Off. Organ Int. Fed. Gynaecol. Obstet. 2015, 130, 27–30. [Google Scholar] [CrossRef]
- Er, T.-K.; Su, Y.-F.; Wu, C.-C.; Chen, C.-C.; Wang, J.; Hsieh, T.-H.; Herreros-Villanueva, M.; Chen, W.-T.; Chen, Y.-T.; Liu, T.-C.; et al. Targeted next-generation sequencing for molecular diagnosis of endometriosis-associated ovarian cancer. J. Mol. Med. Berl. Ger. 2016, 94, 835–847. [Google Scholar] [CrossRef]
- Nothnick, W.B. MicroRNAs and Endometriosis: Distinguishing Drivers from Passengers in Disease Pathogenesis. Semin. Reprod. Med. 2017, 35, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Filigheddu, N.; Gregnanin, I.; Porporato, P.E.; Surico, D.; Perego, B.; Galli, L.; Patrignani, C.; Graziani, A.; Surico, N. Differential Expression of MicroRNAs between Eutopic and Ectopic Endometrium in Ovarian Endometriosis. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Burney, R.O.; Hamilton, A.E.; Aghajanova, L.; Vo, K.C.; Nezhat, C.N.; Lessey, B.A.; Giudice, L.C. MicroRNA expression profiling of eutopic secretory endometrium in women with versus without endometriosis. Mol. Hum. Reprod. 2009, 15, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: MicroRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Han, A.R.; Lee, T.H.; Kim, S.; Lee, H.Y. Risk factors and biomarkers for the recurrence of ovarian endometrioma: About the immunoreactivity of progesterone receptor isoform B and nuclear factor kappa B. Gynecol. Endocrinol. Off. J. Int. Soc. Gynecol. Endocrinol. 2017, 33, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Guo, S.-W. Inhibition of Proliferation of Endometrial Stromal Cells by Trichostatin A, RU486, CDB-2914, N-Acetylcysteine, and ICI 182780. Gynecol. Obstet. Investig. 2006, 62, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Guo, S.-W. Histone deacetylase inhibitors trichostatin A and valproic acid induce cell cycle arrest and p21 expression in immortalized human endometrial stromal cells. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 137, 198–203. [Google Scholar] [CrossRef]
- Wu, Y.; Guo, S.-W. Suppression of IL-1β-induced COX-2 expression by trichostatin A (TSA) in human endometrial stromal cells. Eur. J. Obstet. Gynecol. Reprod. Biol. 2007, 135, 88–93. [Google Scholar] [CrossRef]
- Wu, Y.; Guo, S.-W. Peroxisome proliferator-activated receptor-gamma and retinoid X receptor agonists synergistically suppress proliferation of immortalized endometrial stromal cells. Fertil. Steril. 2009, 91, 2142–2147. [Google Scholar] [CrossRef]
- Peeters, L.L.H.; Vigne, J.-L.; Tee, M.K.; Zhao, D.; Waite, L.L.; Taylor, R.N. PPARgamma represses VEGF expression in human endometrial cells: Implications for uterine angiogenesis. Angiogenesis 2006, 8, 373–379. [Google Scholar] [CrossRef]
- Ohama, Y.; Harada, T.; Iwabe, T.; Taniguchi, F.; Takenaka, Y.; Terakawa, N. Peroxisome proliferator-activated receptor-γ ligand reduced tumor necrosis factor-α-induced interleukin-8 production and growth in endometriotic stromal cells. Fertil. Steril. 2008, 89, 311–317. [Google Scholar] [CrossRef]
- Lu, Y.; Nie, J.; Liu, X.; Zheng, Y.; Guo, S.-W. Trichostatin A, a histone deacetylase inhibitor, reduces lesion growth and hyperalgesia in experimentally induced endometriosis in mice. Hum. Reprod. Oxf. Engl. 2010, 25, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Imesch, P.; Fink, D.; Fedier, A. Romidepsin reduces histone deacetylase activity, induces acetylation of histones, inhibits proliferation, and activates apoptosis in immortalized epithelial endometriotic cells. Fertil. Steril. 2010, 94, 2838–2842. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mahmud, S.A.; Bitterman, P.B.; Huo, Y.; Slungaard, A. Histone Deacetylase Inhibitors Suppress TF-κB-dependent Agonist-driven Tissue Factor Expression in Endothelial Cells and Monocytes. J. Biol. Chem. 2007, 282, 28408–28418. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-F.; Song, Q.; Li, L.-Z.; Zhao, C.-L.; Wang, L.-Q. Histone deacetylase inhibitor valproic acid inhibits proliferation and induces apoptosis in KM3 cells via downregulating VEGF receptor. Neuro Endocrinol. Lett. 2007, 28, 775–780. [Google Scholar]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.-M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef]
- Lockwood, C.J.; Krikun, G.; Hickey, M.; Huang, S.J.; Schatz, F. Decidualized Human Endometrial Stromal Cells Mediate Hemostasis, Angiogenesis, and Abnormal Uterine Bleeding. Reprod. Sci. Thousand Oaks Calif 2009, 16, 162–170. [Google Scholar] [CrossRef][Green Version]
- Liu, X.; Guo, S.-W. A pilot study on the off-label use of valproic acid to treat adenomyosis. Fertil. Steril. 2008, 89, 246–250. [Google Scholar] [CrossRef]
- Liu, X.; Yuan, L.; Guo, S.W. Valproic acid as a therapy for adenomyosis: A comparative case series. Reprod. Sci. Thousand Oaks Calif 2010, 17, 904–912. [Google Scholar]
- Liu, M.; Liu, X.; Zhang, Y.; Guo, S.-W. Valproic acid and progestin inhibit lesion growth and reduce hyperalgesia in experimentally induced endometriosis in rats. Reprod. Sci. Thousand Oaks Calif 2012, 19, 360–373. [Google Scholar]
- Barra, F.; Ferrero, S. Epigenetic Drugs in the Treatment of Endometriosis. Reprod. Sci. Thousand Oaks Calif 2018, 1933719118765987. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laganà, A.S.; Garzon, S.; Götte, M.; Viganò, P.; Franchi, M.; Ghezzi, F.; Martin, D.C. The Pathogenesis of Endometriosis: Molecular and Cell Biology Insights. Int. J. Mol. Sci. 2019, 20, 5615. https://doi.org/10.3390/ijms20225615
Laganà AS, Garzon S, Götte M, Viganò P, Franchi M, Ghezzi F, Martin DC. The Pathogenesis of Endometriosis: Molecular and Cell Biology Insights. International Journal of Molecular Sciences. 2019; 20(22):5615. https://doi.org/10.3390/ijms20225615
Chicago/Turabian StyleLaganà, Antonio Simone, Simone Garzon, Martin Götte, Paola Viganò, Massimo Franchi, Fabio Ghezzi, and Dan C. Martin. 2019. "The Pathogenesis of Endometriosis: Molecular and Cell Biology Insights" International Journal of Molecular Sciences 20, no. 22: 5615. https://doi.org/10.3390/ijms20225615
APA StyleLaganà, A. S., Garzon, S., Götte, M., Viganò, P., Franchi, M., Ghezzi, F., & Martin, D. C. (2019). The Pathogenesis of Endometriosis: Molecular and Cell Biology Insights. International Journal of Molecular Sciences, 20(22), 5615. https://doi.org/10.3390/ijms20225615