VEGFR-1 Regulates EGF-R to Promote Proliferation in Colon Cancer Cells

Abstract
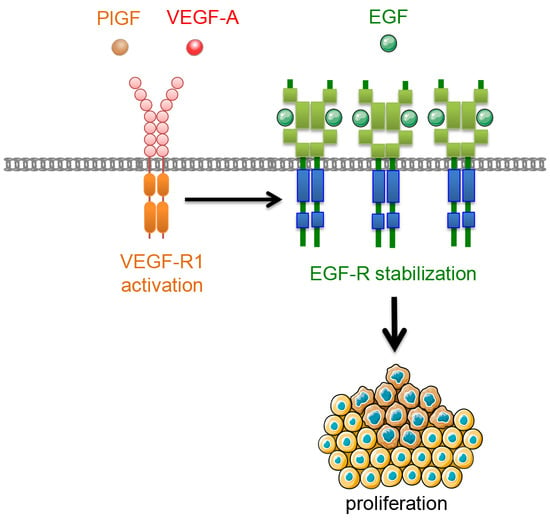
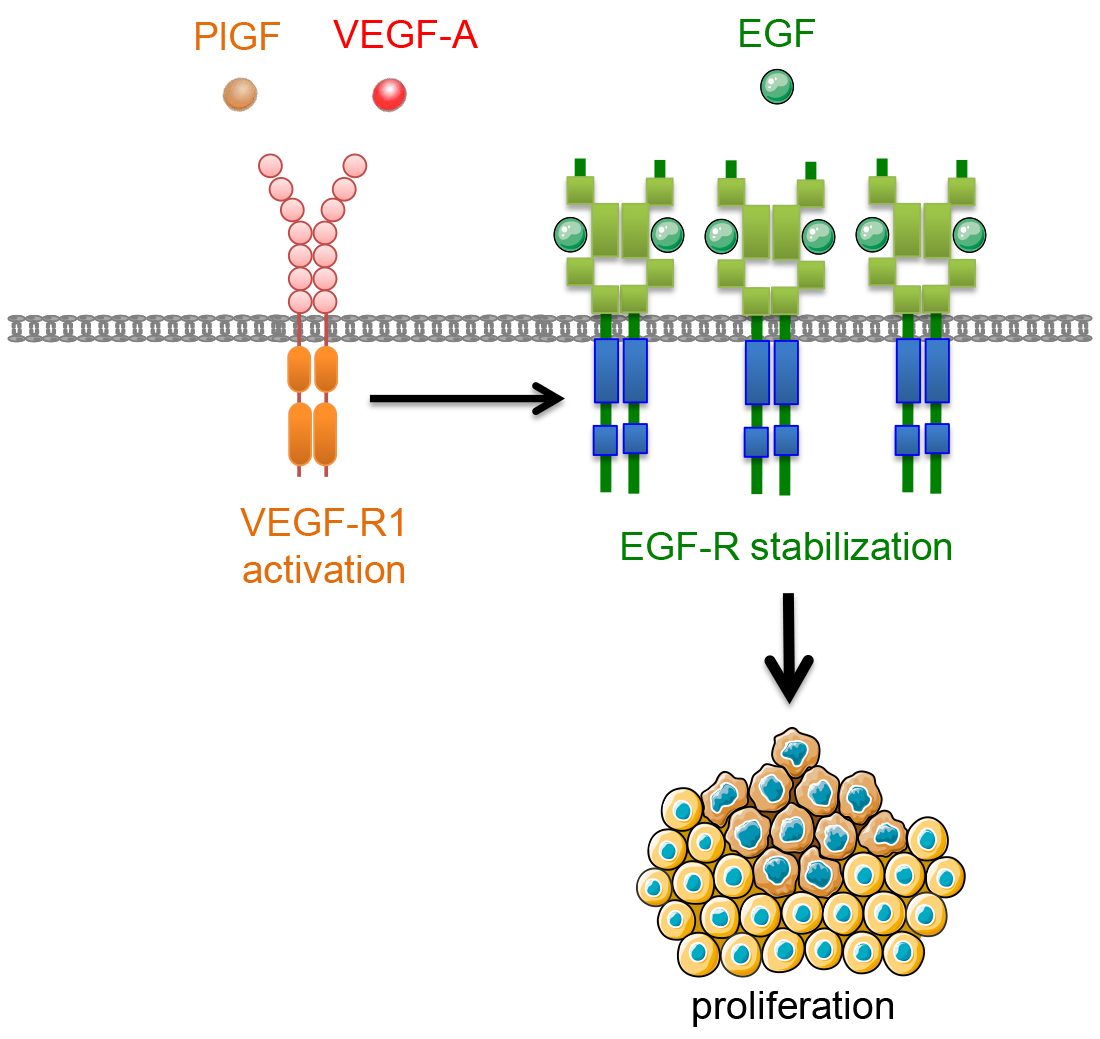{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
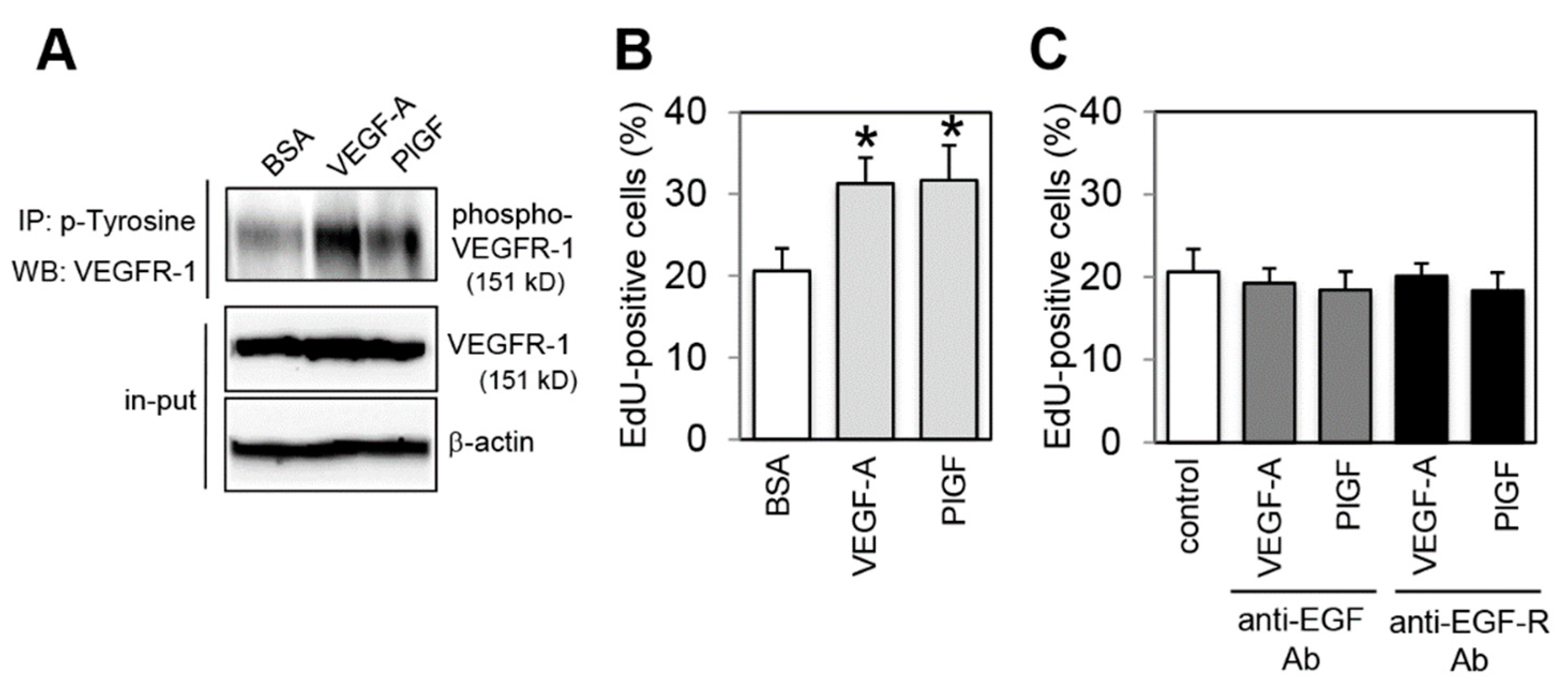
2.1. Effect of VEGFR-1 Activation on Cell Proliferation in Colon Cancer Cells
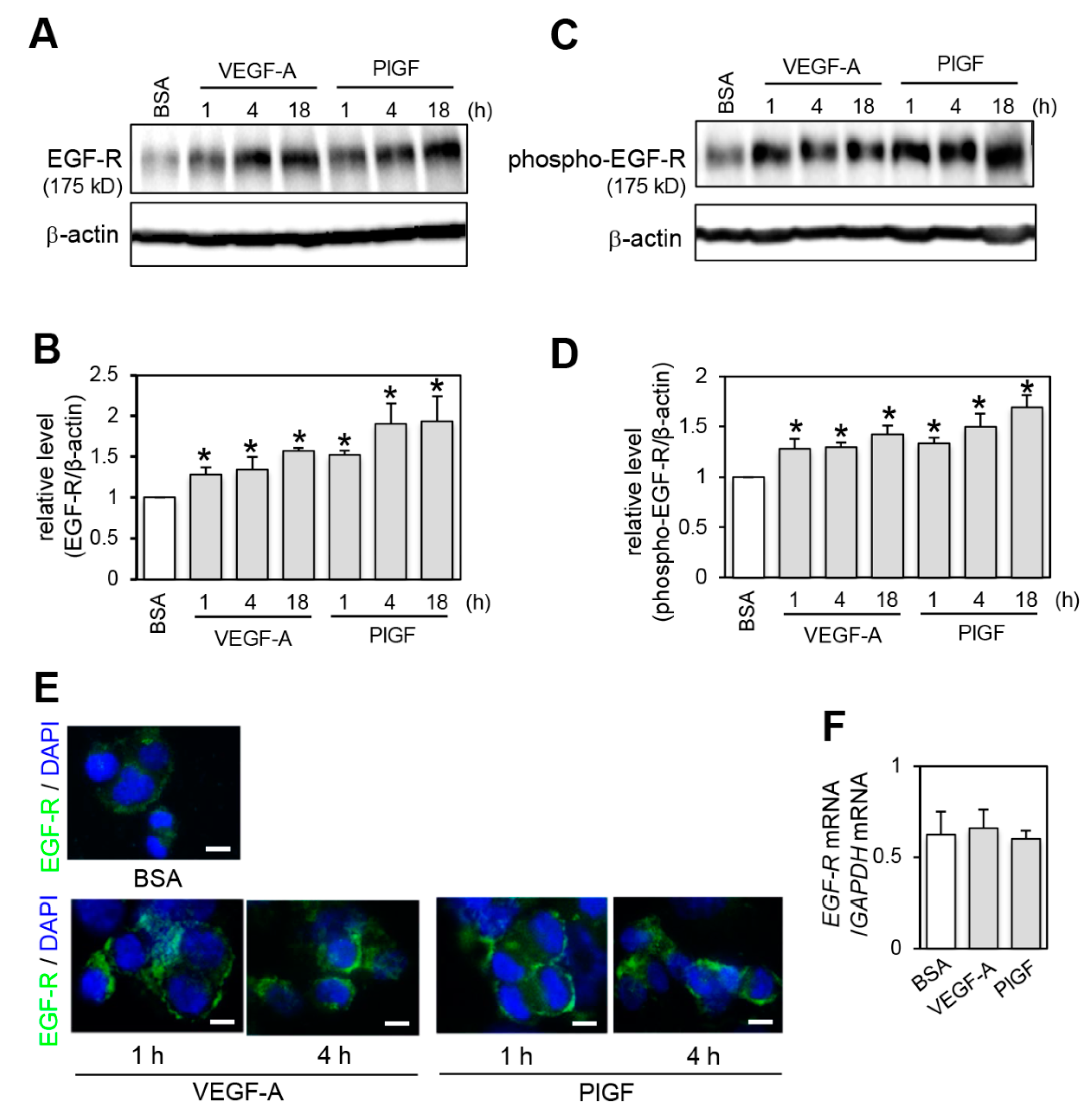
2.2. Effect of VEGFR-1 Activation on EGF-R Expression
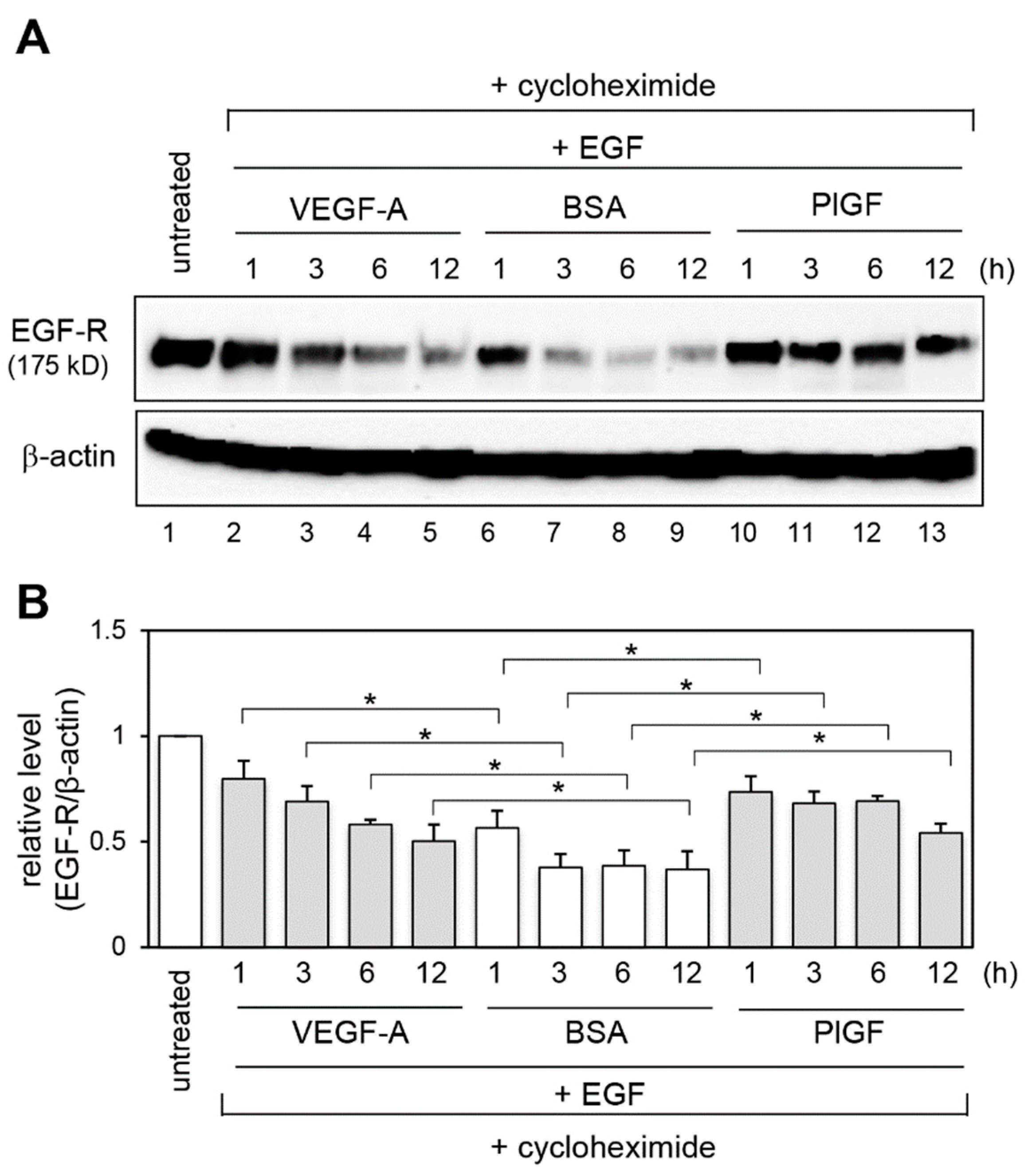
2.3. Effect of VEGFR-1 Activation on EGF-R Stability
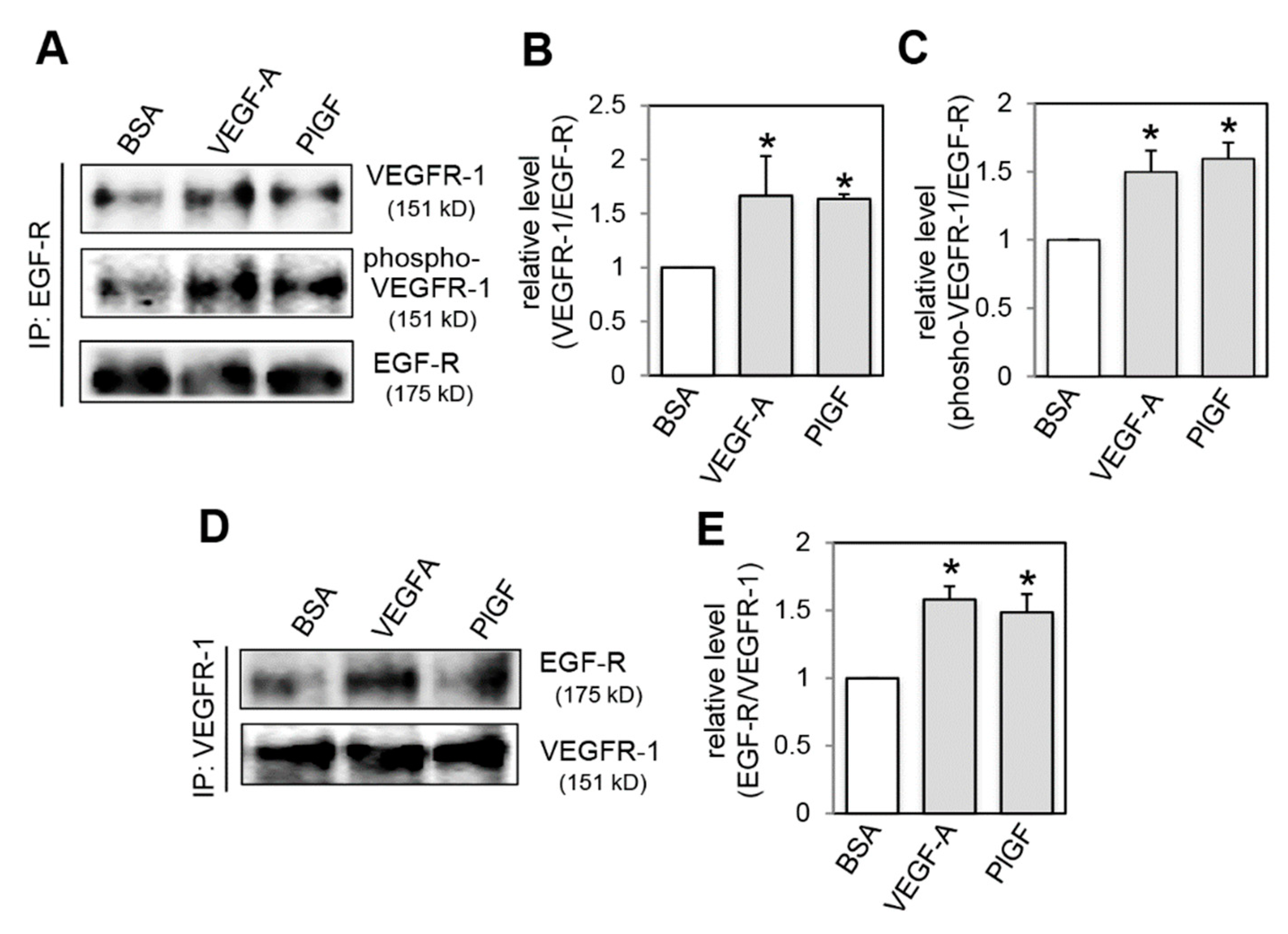
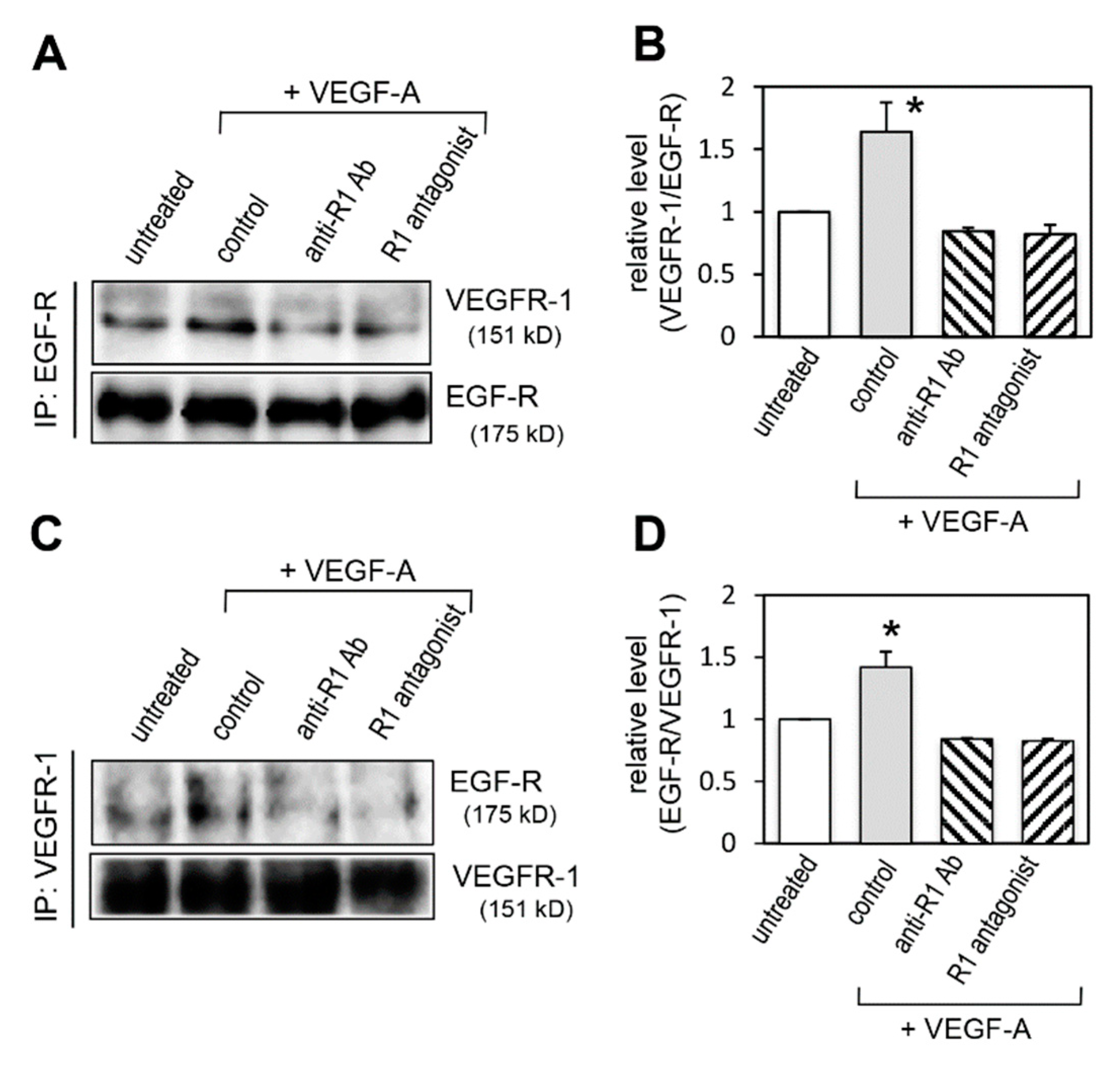
2.4. Effect of VEGFR-1 Activation on Interaction of VEGFR-1 and EGF-R
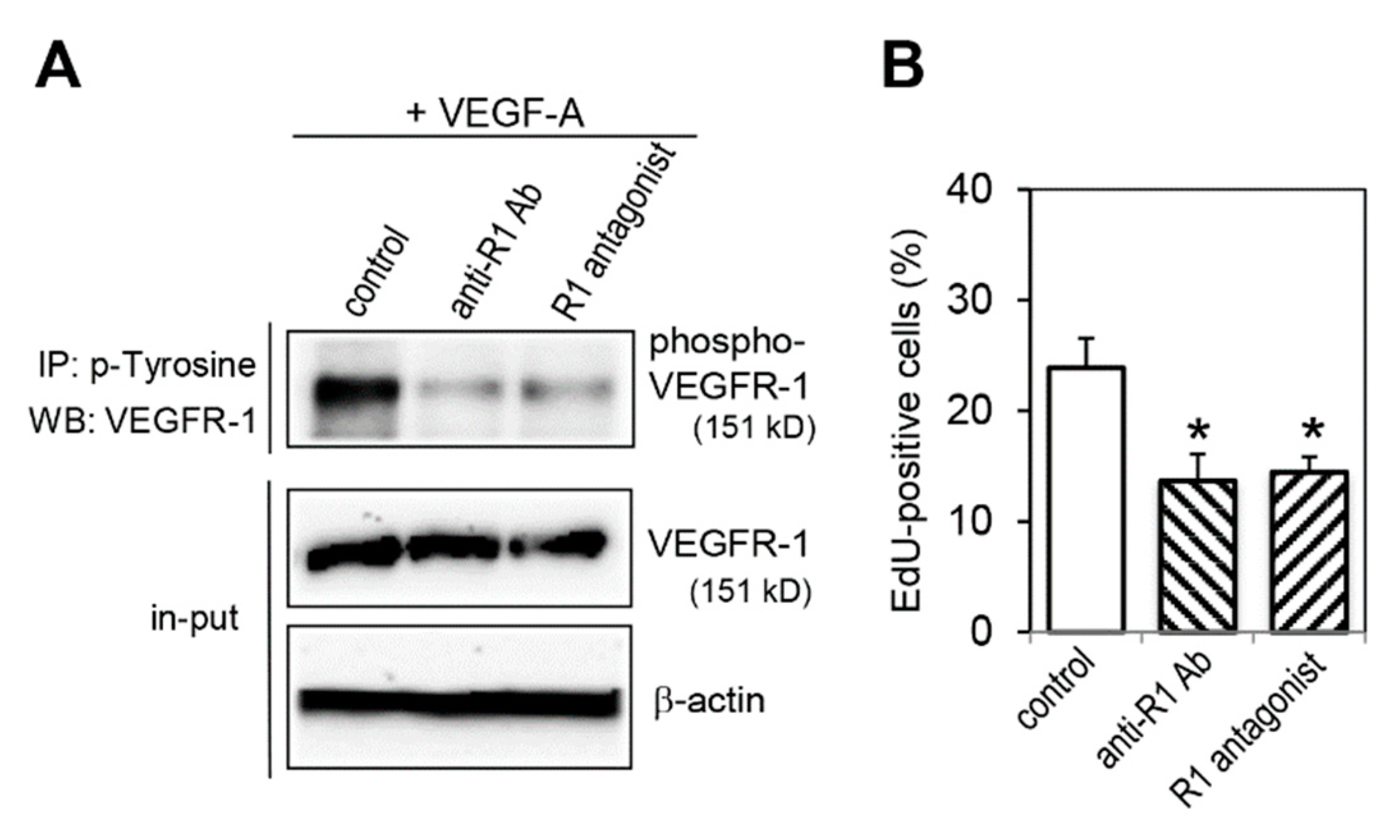
2.5. Effect of VEGFR-1 Blockade on Cell Proliferation Activity
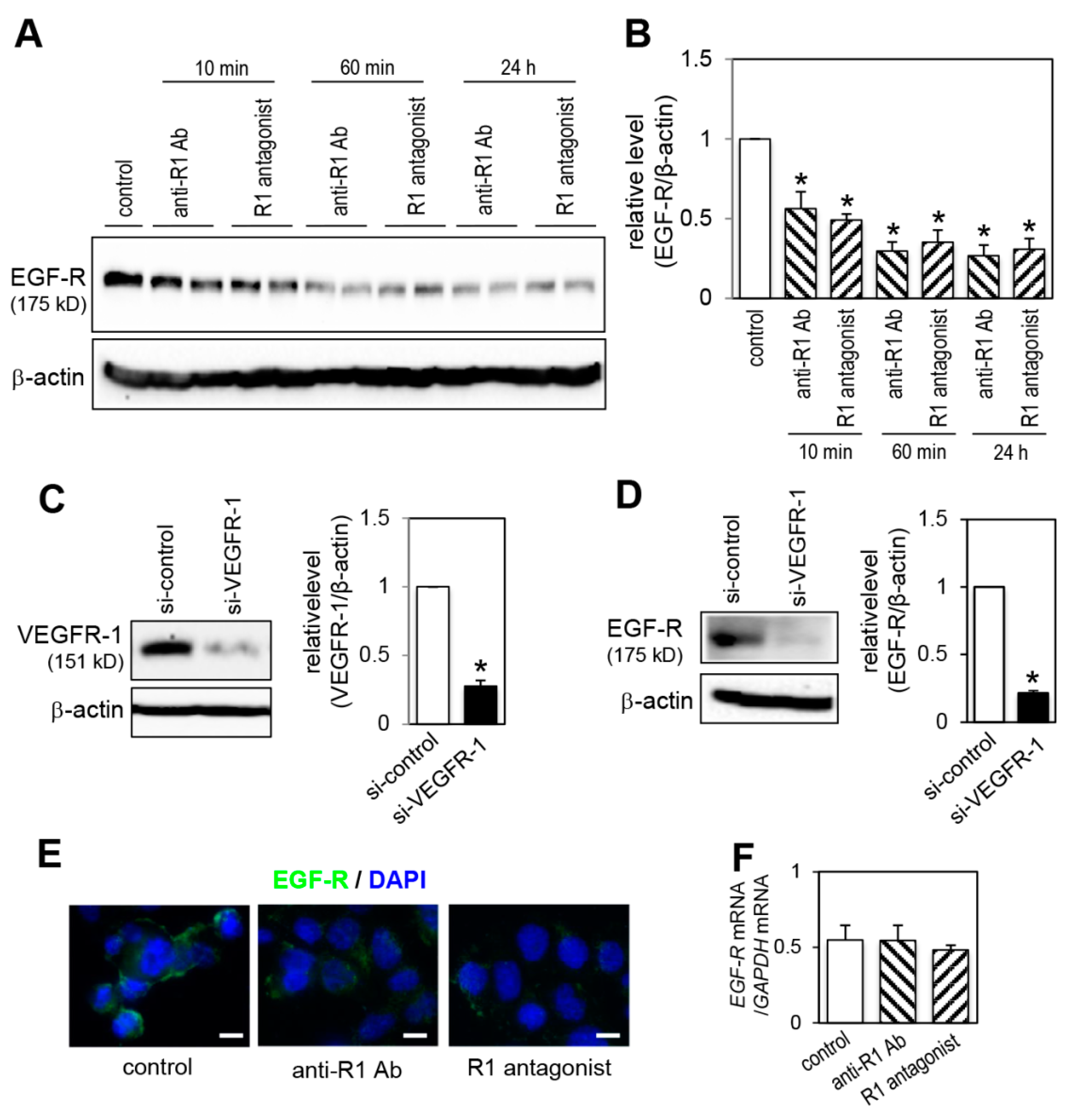
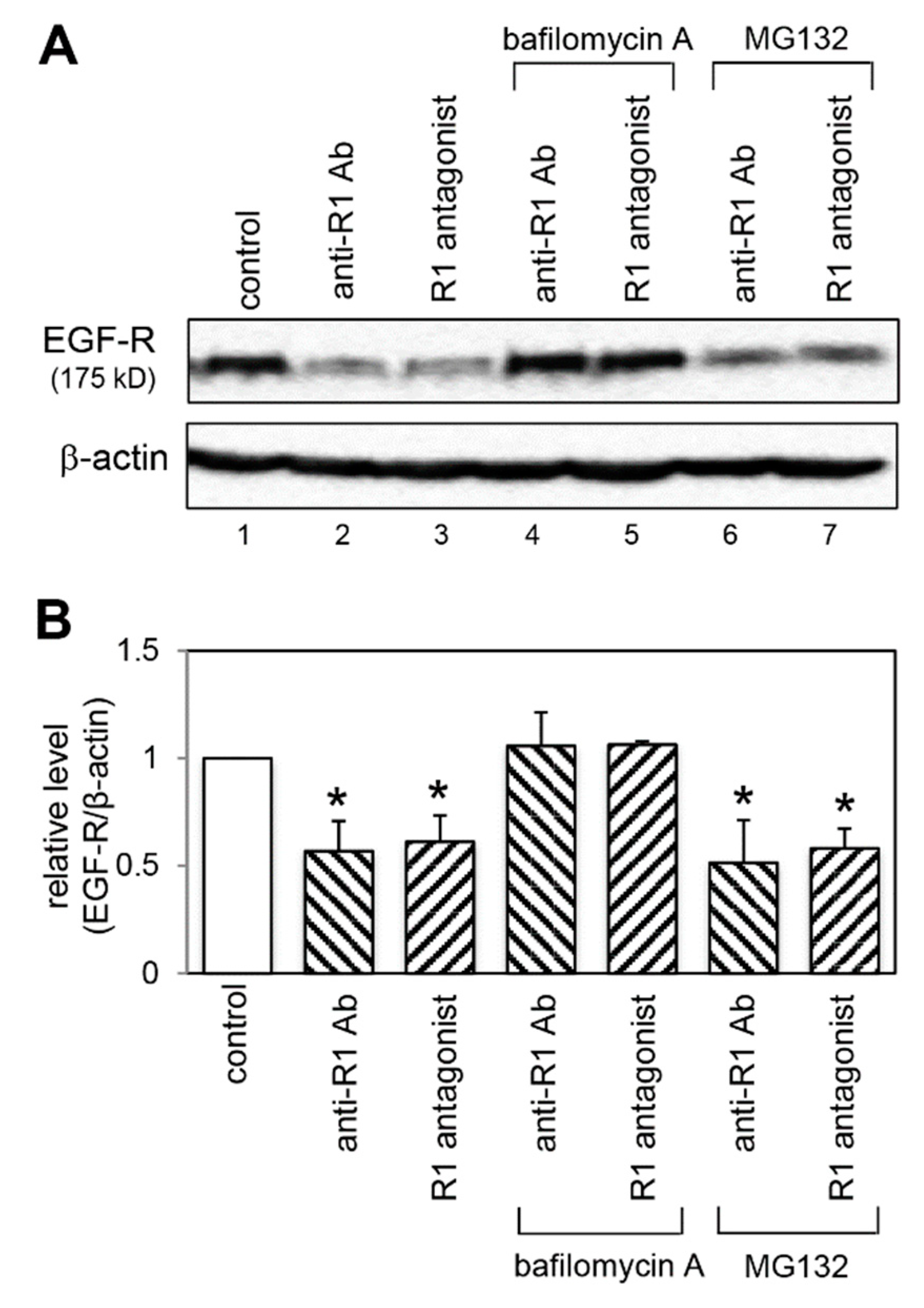
2.6. Effect of VEGFR-1 Blockade on EGF-R Expression
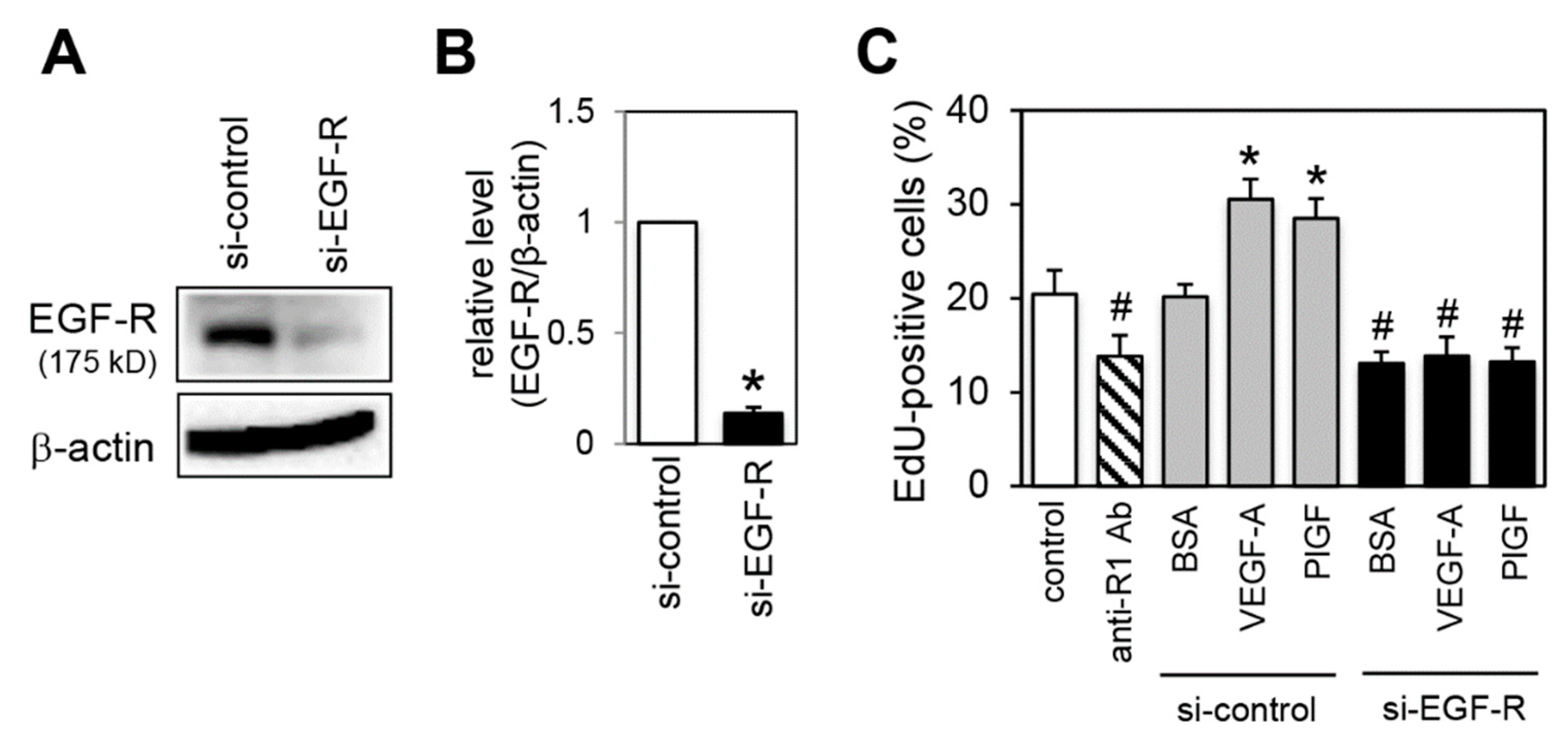
2.7. Effect of EGF-R Knockdown on Proliferation Activity
2.8. Effect of a Lysosomal and Proteasomal Inhibitor on EGF-R Expression
2.9. Effect of VEGFR-1 Blockade on Interaction of VEGFR-1 and EGF-R
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Treatment
4.3. Cell Proliferation Assay
4.4. Cycloheximide Chase Assay
4.5. Western Blot Analysis
4.6. Immunofluorescence Cell Staining
4.7. siRNA and Transfection
4.8. Immunoprecipitation Analysis
4.9. Quantitative RT-PCR
4.10. Flow Cytometric Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| VEGF | vascular endothelial growth factor |
| PlGF | placental growth factor |
| VEGFR-1 | vascular endothelial growth factor receptor-1 |
| EGF | epidermal growth factor |
| EGF-R | epidermal growth factor receptor |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Mercurio, A. VEGF/Neuropilin Signaling in Cancer Stem Cells. Int. J. Mol. Sci. 2019, 20, 490. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Hicklin, D.J.; Ellis, L.M. Antiangiogenic therapy—evolving view based on clinical trial results. Nat. Rev. Clin. Oncol. 2012, 9, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. VEGF-VEGFR System as a Target for Suppressing Inflammation and other Diseases. Endocrine, Metab. Immune Disord. Drug Targets 2015, 15, 135–144. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Simon, T.; Gagliano, T.; Giamas, G. Direct Effects of Anti-Angiogenic Therapies on Tumor Cells: VEGF Signaling. Trends Mol. Med. 2017, 23, 282–292. [Google Scholar] [CrossRef]
- Lacal, P.M.; Graziani, G. Therapeutic implication of vascular endothelial growth factor receptor-1 (VEGFR-1) targeting in cancer cells and tumor microenvironment by competitive and non-competitive inhibitors. Pharmacol. Res. 2018, 136, 97–107. [Google Scholar] [CrossRef]
- Fan, F.; Wey, J.S.; McCarty, M.F.; Belcheva, A.; Liu, W.; Bauer, T.W.; Somcio, R.J.; Wu, Y.; Hooper, A.; Hicklin, D.J.; et al. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene 2005, 24, 2647–2653. [Google Scholar] [CrossRef]
- Wu, Y.; Hooper, A.T.; Zhong, Z.; Witte, L.; Bohlen, P.; Rafii, S.; Hicklin, D.J. The vascular endothelial growth factor receptor (VEGFR-1) supports growth and survival of human breast carcinoma. Int. J. Cancer 2006, 119, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Dallas, N.A.; Fan, F.; Gray, M.J.; Van Buren, G.; Lim, S.J.; Xia, L.; Ellis, L.M. Functional significance of vascular endothelial growth factor receptors on gastrointestinal cancer cells. Cancer Metastasis Rev. 2007, 26, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Ohba, T.; Cates, J.M.M.; Cole, H.A.; Slosky, D.A.; Haro, H.; Ando, T.; Schwartz, H.S.; Schoenecker, J.G. Autocrine VEGF/VEGFR1 Signaling in a Subpopulation of Cells Associates with Aggressive Osteosarcoma. Mol. Cancer Res. 2014, 12, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Wu, X.; Zhuang, G.; Kasman, I.M.; Vogt, T.; Phan, V.; Shibuya, M.; Ferrara, N.; Bais, C. Expression of a functional VEGFR-1 in tumor cells is a major determinant of anti-PlGF antibodies efficacy. Proc. Natl. Acad. Sci. USA 2011, 108, 11590–11595. [Google Scholar] [CrossRef]
- Frank, N.Y.; Schatton, T.; Kim, S.; Zhan, Q.; Wilson, B.J.; Ma, J.; Saab, K.R.; Osherov, V.; Widlund, H.R.; Gasser, M.; et al. VEGFR-1 expressed by malignant melanoma-initiating cells is required for tumor growth. Cancer Res. 2011, 71, 1474–1485. [Google Scholar] [CrossRef]
- Masood, R.; Cai, J.; Zheng, T.; Lynne Smith, D.; Hinton, D.R.; Gill, P.S. Vascular endothelial growth factor (VEGF) is an autocrine growth factor for VEGF receptor-positive human tumors. Blood 2001, 98, 1904–1913. [Google Scholar] [CrossRef]
- Lichtenberger, B.M.; Tan, P.K.; Niederleithner, H.; Ferrara, N.; Petzelbauer, P.; Sibilia, M. Autocrine VEGF Signaling Synergizes with EGFR in Tumor Cells to Promote Epithelial Cancer Development. Cell 2010, 140, 268–279. [Google Scholar] [CrossRef]
- Tomida, C.; Yamagishi, N.; Nagano, H.; Uchida, T.; Ohno, A.; Hirasaka, K.; Nikawa, T.; Da Teshima-Kondo, S. (corresponding author) VEGF pathway-Targeting drugs induce evasive adaptation by activation of neuropilin-1/cMet in colon cancer cells. Int. J. Oncol. 2018, 52, 1350–1362. [Google Scholar]
- Endo, A.; Fukuhara, S.; Masuda, M.; Ohmori, T.; Mochizuki, N. Selective inhibition of vascular endothelial growth factor receptor-2 (VEGFR-2) identifies a central role for VEGFR-2 in human aortic endothelial cell responses to VEGF. J. Recept. Signal. Transduct. Res. 2003, 23, 239–254. [Google Scholar] [CrossRef]
- Ciardiello, F.; Troiani, T.; Bianco, R.; Orditura, M.; Morgillo, F.; Martinelli, E.; Morelli, M.P.; Cascone, T.; Tortora, G. Interaction between the epidermal growth factor receptor (EGFR) and the vascular endothelial growth factor (VEGF) pathways: A rational approach for multi-target anticancer therapy. Ann. Oncol. 2006, 17, vii109–vii114. [Google Scholar] [CrossRef]
- Ciardiello, F.; Bianco, R.; Damiano, V.; Fontanini, G.; Caputo, R.; Pomatico, G.; Sabino De Placido; Bianco, A.R.; Mendelsohn, J.; Tortora, G. Antiangiogenic and Antitumor Activity of Anti-Epidermal Growth Factor Receptor C225 Monoclonal Antibody in Combination with Vascular Endothelial Growth Factor Antisense Oligonucleotide in Human GEO Colon Cancer Cells. Clin. Cancer Res. 2000, 6, 3739–3747. [Google Scholar] [PubMed]
- Akagi, M.; Kawaguchi, M.; Liu, W.; McCarty, M.F.; Takeda, A.; Fan, F.; Stoeltzing, O.; Parikh, A.A.; Jung, Y.D.; Bucana, C.D.; et al. Induction of neuropilin-I and vascular endothelial growth factor by epidermal growth factor in human gastric cancer cells. Br. J. Cancer 2003, 88, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Sanada, F.; Taniyama, Y.; Iekushi, K.; Azuma, J.; Okayama, K.; Kusunoki, H.; Koibuchi, N.; Doi, T.; Aizawa, Y.; Morishita, R. Negative action of hepatocyte growth factor/c-met system on angiotensin ii signaling via ligand-dependent epithelial growth factor receptor degradation mechanism in vascular smooth muscle cells. Circ. Res. 2009, 105, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Taniyama, Y.; Sanada, F.; Azuma, J.; Iwabayashi, M.; Iekushi, K.; Rakugi, H.; Morishita, R. Hepatocyte growth factor inhibits lipopolysaccharide-induced oxidative stress via epithelial growth factor receptor degradation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2687–2693. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Haendeler, J.; Hojo, Y.; Yamamoto, K.; Berk, B.C. Receptor Heterodimerization: Essential Mechanism for Platelet-Derived Growth Factor-Induced Epidermal Growth Factor Receptor Transactivation. Mol. Cell. Biol. 2001, 21, 6387–6394. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.; Spits, M.; Neefjes, J.; Berlin, I. The EGFR odyssey – from activation to destruction in space and time. J. Cell Sci. 2017, 130, 4087–4096. [Google Scholar] [CrossRef]
- Caldieri, G.; Malabarba, M.G.; Di Fiore, P.P.; Sigismund, S. EGFR Trafficking in Physiology and Cancer. Prog. Mol. Subcell. Biol. 2018, 57, 235–272. [Google Scholar]
- Bae, D.G.; Kim, T.D.; Li, G.; Yoon, W.H.; Chae, C.B. Anti-Flt1 peptide, a vascular endothelial growth factor receptor 1-specific hexapeptide, inhibits tumor growth and metastasis. Clin. Cancer Res. 2005, 11, 2651–2661. [Google Scholar] [CrossRef]
- Petit, A.M.; Rak, J.; Hung, M.C.; Rockwell, P.; Goldstein, N.; Fendly, B.; Kerbel, R.S. Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: Angiogenic implications for signal transduction therapy of solid tumors. Am. J. Pathol. 1997, 151, 1523–1530. [Google Scholar]
- Domigan, C.K.; Ziyad, S.; Iruela-Arispe, M.L. Canonical and noncanonical vascular endothelial growth factor pathways: New developments in biology and signal transduction. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 30–39. [Google Scholar] [CrossRef]
- Modest, D.P.; Pant, S.; Sartore-Bianchi, A. Treatment sequencing in metastatic colorectal cancer. Eur. J. Cancer 2019, 109, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Bianco, R.; Rosa, R.; Damiano, V.; Daniele, G.; Gelardi, T.; Garofalo, S.; Tarallo, V.; De Falco, S.; Melisi, D.; Benelli, R.; et al. Vascular endothelial growth factor receptor-1 contributes to resistance to anti-epidermal growth factor receptor drugs in human cancer cells. Clin. Cancer Res. 2008, 14, 5069–5080. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Samuel, S.; Gaur, P.; Lu, J.; Dallas, N.A.; Xia, L.; Bose, D.; Ramachandran, V.; Ellis, L.M. Chronic exposure of colorectal cancer cells to bevacizumab promotes compensatory pathways that mediate tumour cell migration. Br. J. Cancer 2011, 104, 1270–1277. [Google Scholar] [CrossRef] [PubMed]
- Lieu, C.H.; Jiang, Z.-Q.; Mao, M.; Overman, M.J.; Eng, C.; Kopetz, S.; Tran, H.; Heymach, J.V.; Morris, J.; et al. The Association of Alternate VEGF Ligands with Resistance to Anti-VEGF Therapy in Metastatic Colorectal Cancer. PLoS ONE 2013, 8, e77117. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; Troiani, T.; Morgillo, F.; Orditura, M.; De Vita, F.; Belli, G.; Ciardiello, F. Emerging VEGF-receptor inhibitors for colorectal cancer. Expert Opin. Emerg. Drugs 2013, 18, 25–37. [Google Scholar] [CrossRef]
- Syed, Y.Y.; McKeage, K. Aflibercept: A Review in Metastatic Colorectal Cancer. Drugs 2015, 75, 1435–1445. [Google Scholar] [CrossRef]
- Mitchell, E.P. Targeted Therapy for Metastatic Colorectal Cancer: Role of Aflibercept. Clin. Colorectal Cancer 2013, 12, 73–85. [Google Scholar] [CrossRef]
- Macarulla, T.; Sauri, T.; Tabernero, J. Evaluation of aflibercept in the treatment of metastatic colorectal cancer. Expert Opin. Biol. Ther. 2014, 14, 1493–1505. [Google Scholar] [CrossRef]
- Giordano, G.; Febbraro, A.; Venditti, M.; Campidoglio, S.; Olivieri, N.; Raieta, K.; Parcesepe, P.; Imbriani, G.C.; Remo, A.; Pancione, M. Targeting Angiogenesis and Tumor Microenvironment in Metastatic Colorectal Cancer: Role of Aflibercept. Gastroenterol. Res. Pract. 2014, 2014, 1–13. [Google Scholar] [CrossRef]
- W Stewart, M. Aflibercept (VEGF-TRAP): The Next Anti-VEGF Drug. Inflamm. Allergy-Drug Targets 2012, 10, 497–508. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagano, H.; Tomida, C.; Yamagishi, N.; Teshima-Kondo, S. VEGFR-1 Regulates EGF-R to Promote Proliferation in Colon Cancer Cells. Int. J. Mol. Sci. 2019, 20, 5608. https://doi.org/10.3390/ijms20225608
Nagano H, Tomida C, Yamagishi N, Teshima-Kondo S. VEGFR-1 Regulates EGF-R to Promote Proliferation in Colon Cancer Cells. International Journal of Molecular Sciences. 2019; 20(22):5608. https://doi.org/10.3390/ijms20225608
Chicago/Turabian StyleNagano, Hikaru, Chisato Tomida, Naoko Yamagishi, and Shigetada Teshima-Kondo. 2019. "VEGFR-1 Regulates EGF-R to Promote Proliferation in Colon Cancer Cells" International Journal of Molecular Sciences 20, no. 22: 5608. https://doi.org/10.3390/ijms20225608
APA StyleNagano, H., Tomida, C., Yamagishi, N., & Teshima-Kondo, S. (2019). VEGFR-1 Regulates EGF-R to Promote Proliferation in Colon Cancer Cells. International Journal of Molecular Sciences, 20(22), 5608. https://doi.org/10.3390/ijms20225608