Physiological and Transcriptional Responses of Industrial Rapeseed (Brassica napus) Seedlings to Drought and Salinity Stress

Abstract
1. Introduction
2. Results
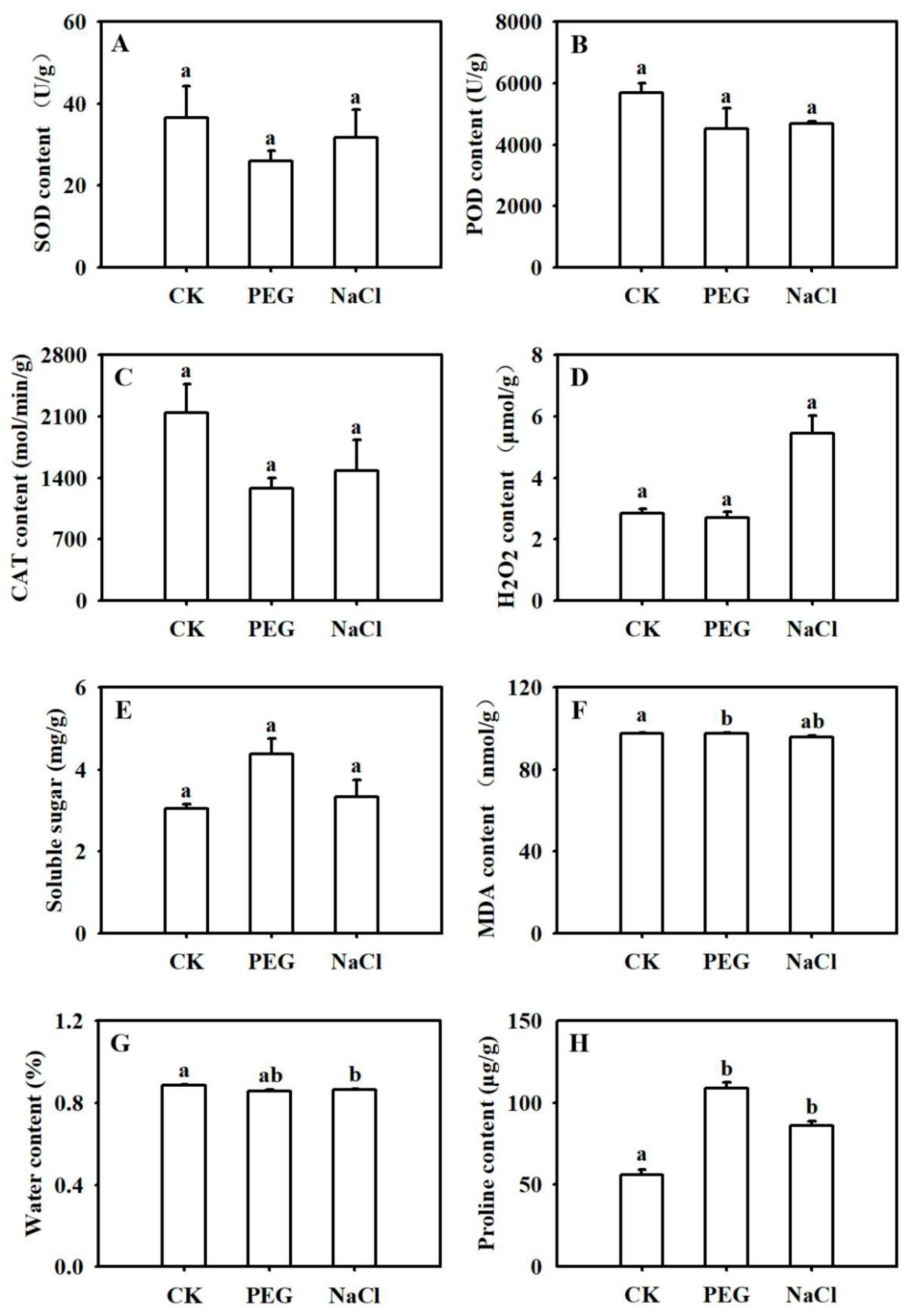
2.1. Physiological Characteristics of B. napus under Abiotic Stresses
2.2. Transcriptomic Sequencing and Unigene Assembly
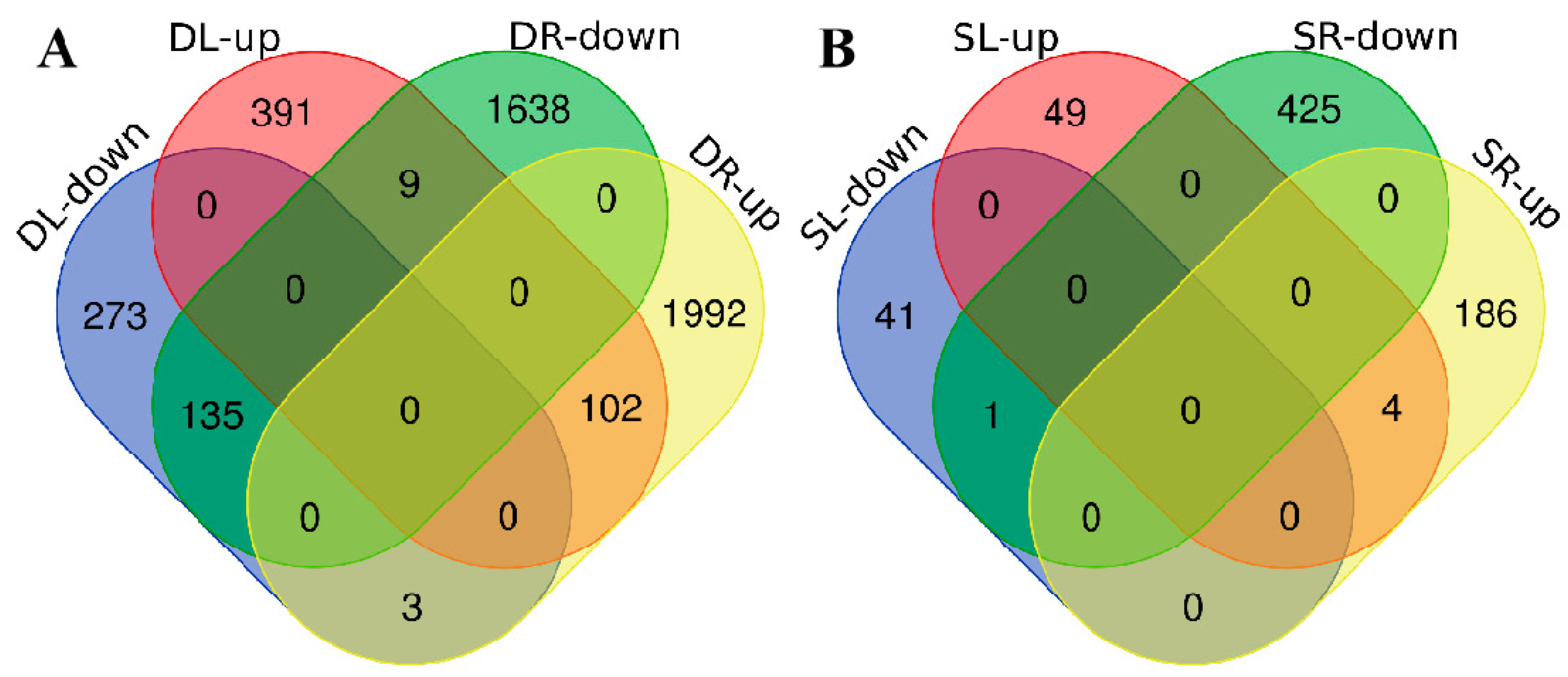
2.3. Analysis of Differentially Expressed Genes
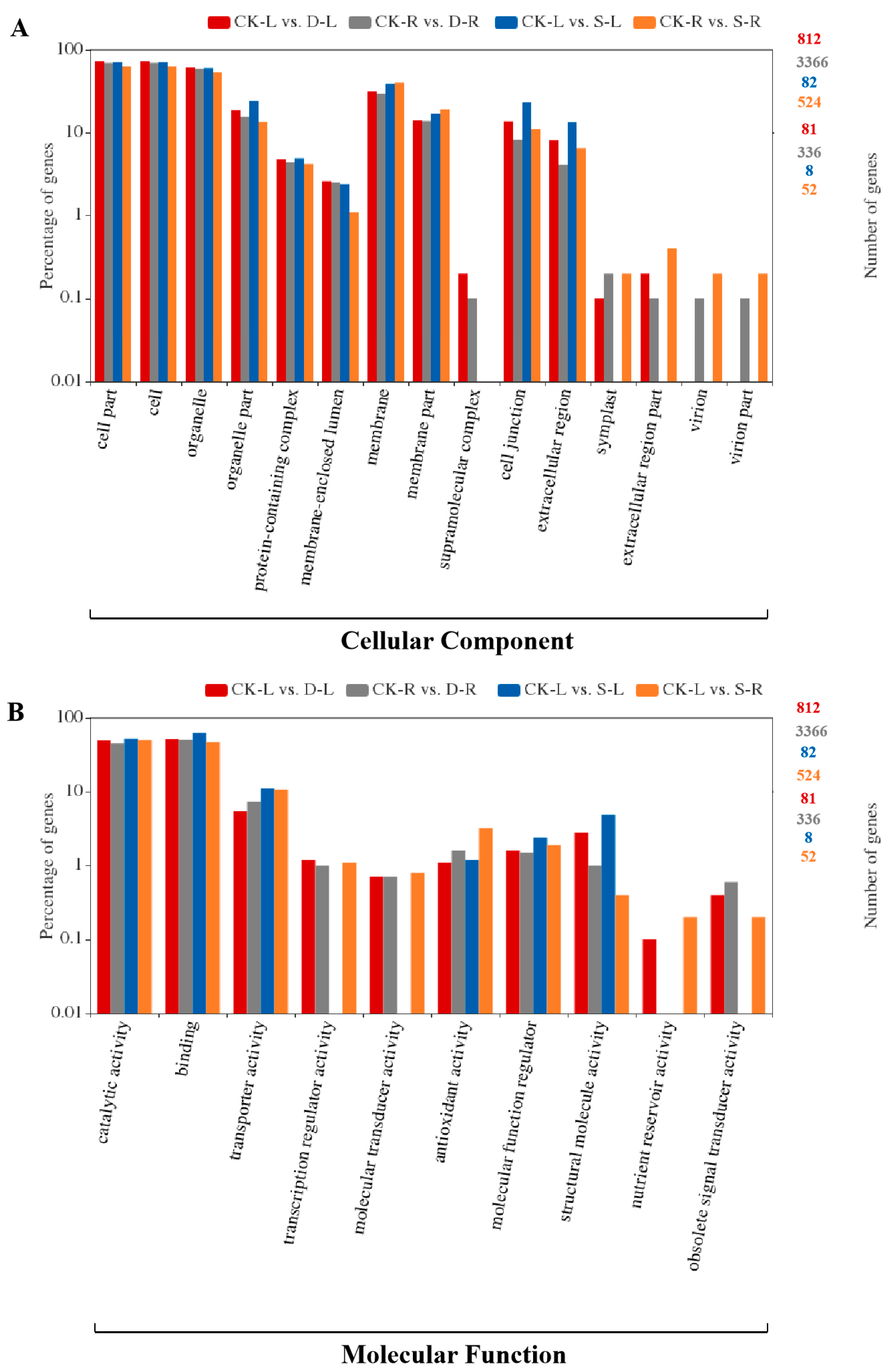
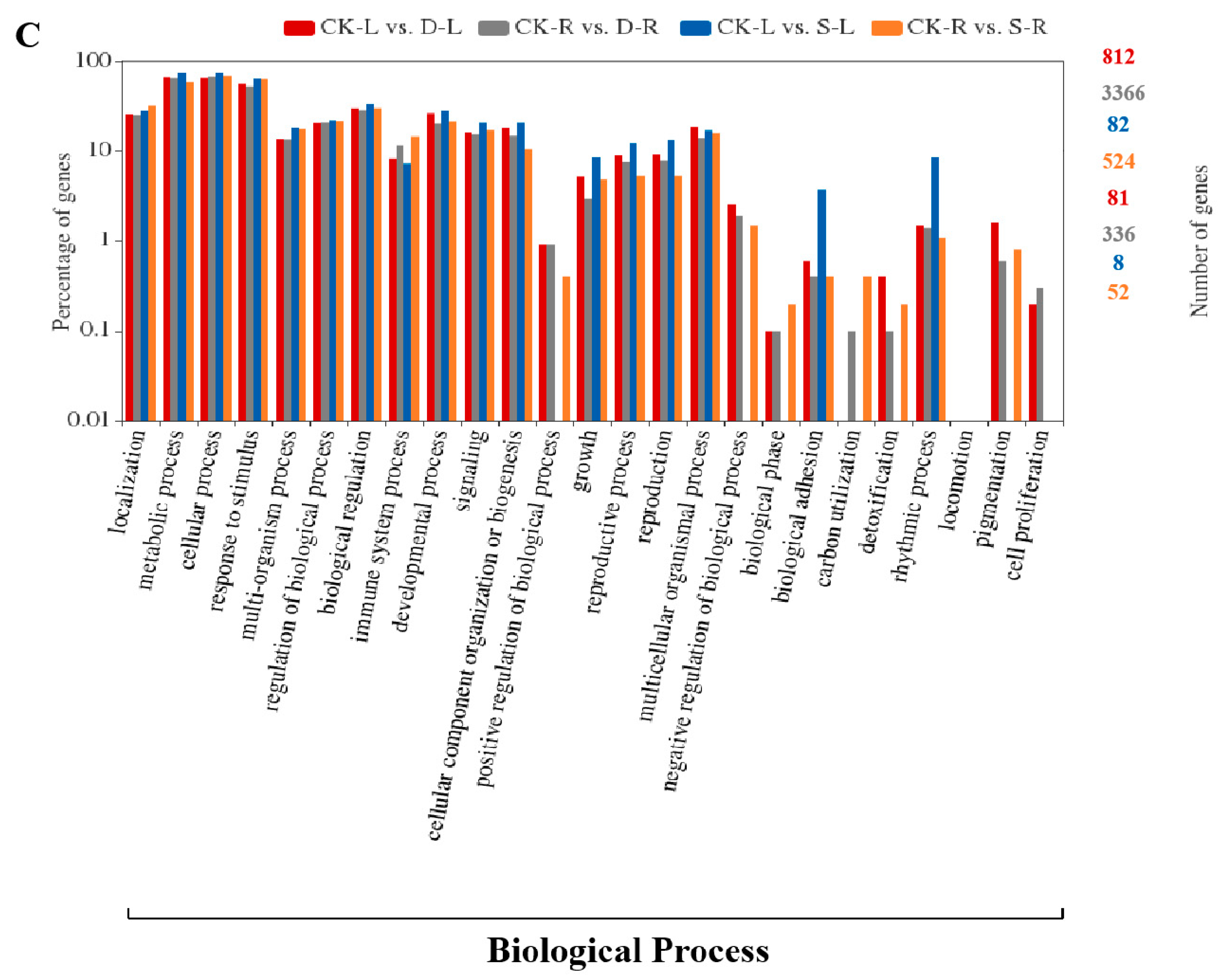
2.4. GO and KEGG Pathway Enrichment Analysis
2.5. Analysis of Differentially Expressed Transcription Factors
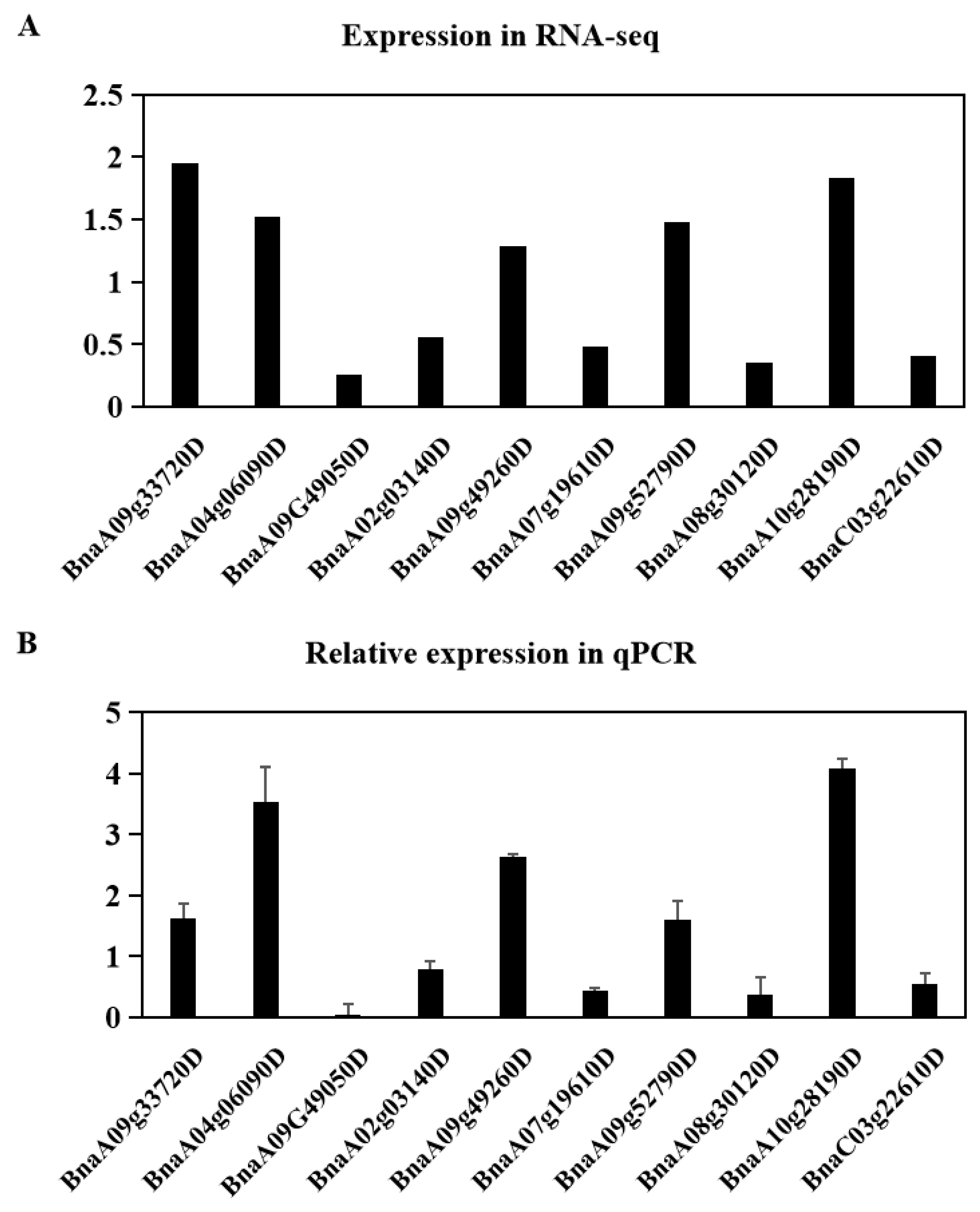
2.6. Validation of Differentially Expressed Genes by Quantitative RT-PCR Analysis
3. Discussion
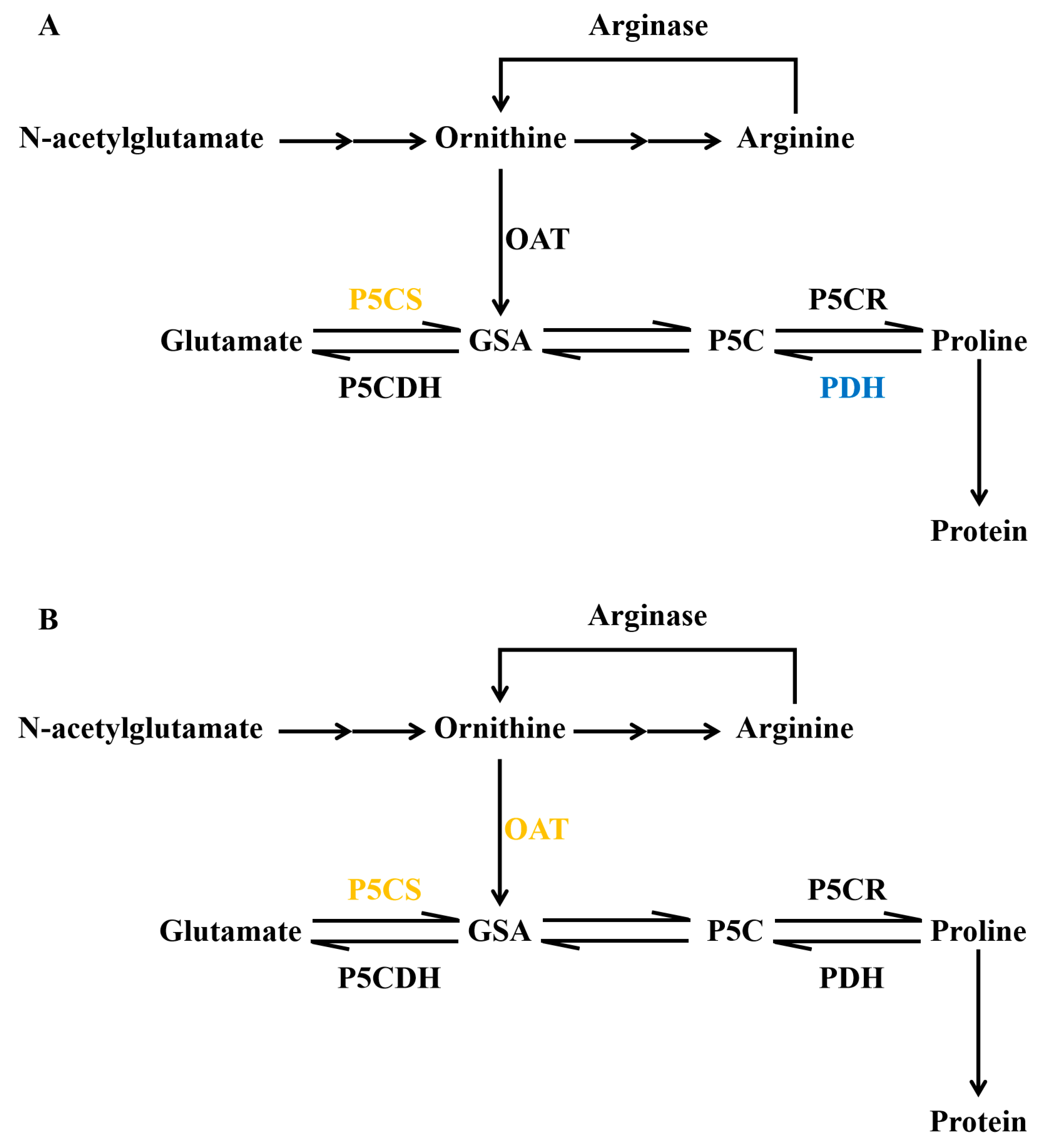
3.1. The Important Role of Proline in Abiotic Stress Response
3.2. The Multiple Transcripts in Response to Salinity and Drought Stresses
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Physiological and Biochemical Analyses
4.3. RNA Extraction, cDNA Library Construction, and Sequencing
4.4. Genome Mapping and Quantification of Gene Expression
4.5. Identification and Functional Enrichment Analysis of DEGs
4.6. Prediction of DEGs Encoding Transcription Factors
4.7. Analysis of Putative Cis-Regulatory Elements Involved in Proline Metabolism
4.8. Verification of Differential Gene Expression by Quantitative Reverse-Transcription PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kreps, J.A.; Wu, Y.; Chang, H.S.; Zhu, T.; Wang, X.; Harper, J.F. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002, 130, 2129–2141. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, G.; Long, W.; Zou, X.; Li, F.; Nishio, T. Recent progress in drought and salt tolerance studies in Brassica crops. Breed Sci. 2014, 64, 60–73. [Google Scholar] [CrossRef]
- Rengasamy, P. Soil processes affecting crop production in salt-affected soils. Funct. Plant Biol. 2010, 37, 613–620. [Google Scholar] [CrossRef]
- Seki, M.; Narusaka, M.; Abe, H.; Kasuga, M.; Yamaguchi-Shinozaki, K.; Carninci, P.; Hayashizaki, Y.; Shinozaki, K. Monitoring the expression pattern of 1300 Arabidopsis genes under drought and cold stresses by using a full-length cDNA microarray. Plant Cell 2001, 13, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Monroe, J.G.; Suhail, Y.; Villiers, F.; Mullen, J.; Pater, D.; Hauser, F.; Jeon, B.W.; Bader, J.S.; Kwak, J.M.; et al. Molecular and systems approaches towards drought-tolerant canola crops. New Phytol. 2016, 210, 1169–1189. [Google Scholar] [CrossRef]
- Wang, D.; Yang, C.; Dong, L.; Zhu, J.; Wang, J.; Zhang, S. Comparative transcriptome analyses of drought-resistant and -susceptible Brassica napus L. and development of EST-SSR markers by RNA-Seq. J. Plant Biol. 2015, 58, 259–269. [Google Scholar] [CrossRef]
- Long, W.; Zou, X.; Zhang, X. Transcriptome analysis of canola (Brassica napus) under salt stress at the germination stage. PLoS ONE 2015, 10, e0116217. [Google Scholar] [CrossRef] [PubMed]
- Verkest, A.; Byzova, M.; Martens, C.; Willems, P.; Verwulgen, T.; Slabbinck, B.; Rombaut, D.; Van de Velde, J.; Vandepoele, K.; Standaert, E.; et al. Selection for improved energy use efficiency and drought tolerance in canola results in distinct transcriptome and epigenome changes. Plant Physiol. 2015, 168, 1338–1350. [Google Scholar] [CrossRef]
- Trick, M. Bioinformatics Resources for the Brassica Species. In Genetics and Genomics of the Brassicaceae; Schmidt, R., Bancroft, I., Eds.; Springer: New York, NY, USA, 2011; pp. 597–615. [Google Scholar]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.Y.; Liu, L.J.; Wu, J.Y.; Zhao, Y.H.; Bai, J.; Ma, L.; Yue, J.L.; Jin, J.J.; Niu, Z.X.; Fang, Y.; et al. Transcriptome profile analysis of winter rapeseed (Brassica napus L.) in response to freezing stress, reveal potentially connected events to freezing stress. Int. J. Mol. Sci. 2019, 20, 24. [Google Scholar] [CrossRef]
- Haddad, C.; Trouverie, J.; Arkoun, M.; Yvin, J.C.; Caius, J.; Brunaud, V.; Laine, P.; Etienne, P. Silicon supply affects the root transcriptome of Brassica napus L. Planta 2019, 249, 1645–1651. [Google Scholar] [CrossRef]
- Pan, Y.; Zhu, M.; Wang, S.; Ma, G.; Huang, X.; Qiao, C.; Wang, R.; Xu, X.; Liang, Y.; Lu, K.; et al. Genome-wide characterization and analysis of metallothionein family genes that function in metal stress tolerance in Brassica napus L. Int. J. Mol. Sci. 2018, 19, 2181. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, X.; Zhang, K.; An, H.; Hu, K.; Wen, J.; Shen, J.; Ma, C.; Yi, B.; Tu, J. Comparative analysis of the Brassica napus root and leaf transcript profiling in response to drought stress. Int. J. Mol. Sci. 2015, 16, 18752–18777. [Google Scholar] [CrossRef]
- Lu, S.; Fadlalla, T.; Tang, S.; Li, L.; Ali, U.; Li, Q.; Guo, L. Genome-wide analysis of Phospholipase D gene family and profiling of phospholipids under abiotic stresses in Brassica napus. Plant Cell Physiol. 2019, 60, 1556–1566. [Google Scholar] [CrossRef]
- Zhao, G.; Zhao, Y.; Yu, X.; Kiprotich, F.; Han, H.; Guan, R.; Wang, R.; Shen, W. Nitric Oxide is required for melatonin-enhanced tolerance against salinity stress in rapeseed (Brassica napus L.) seedlings. Int. J. Mol. Sci. 2018, 19, 1912. [Google Scholar] [CrossRef]
- Delauney, A.J.; Verma, D.P.S. Proline biosynthesis and osmoreguation in plants. Plant J. 1993, 4, 215–223. [Google Scholar] [CrossRef]
- Szabados, L.; Savoure, A. Proline: A multifunctional amino acid. Trends Plant Sci. 2010, 15, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Zhao, L.; Zhao, S.M.; Wang, J.; Shi, H.Z. Biochemical and transcriptomic analyses of drought stress responses of LY1306 tobacco strain. Sci. Rep. 2017, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Koca, H.; Bor, M.; Ozdemir, F.; Turkan, I. The effect of salt stress on lipid peroxidation, antioxidative enzymes and proline content of sesame cultivars. Environ. Exp. Bot. 2007, 60, 344–351. [Google Scholar] [CrossRef]
- Planchet, E.; Verdu, I.; Delahaie, J.; Cukier, C.; Girard, C.; Morere-Le Paven, M.C.; Limami, A.M. Abscisic acid-induced nitric oxide and proline accumulation in independent pathways under water-deficit stress during seedling establishment in Medicago truncatula. J. Exp. Bot. 2014, 65, 2161–2170. [Google Scholar] [CrossRef]
- Lutts, S.; Majerus, V.; Kinet, J.M. NaCl effects on proline metabolism in rice (Oryza sativa) seedlings. Physiol. Plant. 1999, 105, 450–458. [Google Scholar] [CrossRef]
- Li, J.J.; Ma, J.J.; Guo, H.L.; Zong, J.Q.; Chen, J.B.; Wang, Y.; Li, D.D.; Li, L.; Wang, J.J.; Liu, J.X. Growth and physiological responses of two phenotypically distinct accessions of centipedegrass (Eremochloa ophiuroides (Munro) Hack.) to salt stress. Plant Physiol. Biochem. 2018, 126, 1–10. [Google Scholar] [CrossRef]
- Pavlovic, I.; Pencik, A.; Novak, O.; Vujcic, V.; Brkanac, S.R.; Lepedus, H.; Strnad, M.; Salopek-Sondi, B. Short-term salt stress in Brassica rapa seedlings causes alterations in auxin metabolism. Plant Physiol. Biochem. 2018, 125, 74–84. [Google Scholar] [CrossRef]
- Yin, S.Y.; Han, Y.; Huang, L.; Hong, Y.; Zhang, G.P. Overexpression of HvCBF7 and HvCBF9 changes salt and drought tolerance in Arabidopsis. Plant Growth Regul. 2018, 85, 281–292. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Nahar, K.; Anee, T.I.; Khan, M.I.R.; Fujita, M. Silicon-mediated regulation of antioxidant defense and glyoxalase systems confers drought stress tolerance in Brassica napus L. S. Afr. J. Bot. 2018, 115, 50–57. [Google Scholar] [CrossRef]
- Toorchi, M.; Naderi, R.; Kanbar, A.; Shakiba, M.R. Response of spring canola cultivars to sodium chloride stress. Ann. Biol. Res. 2011, 2, 312–322. [Google Scholar]
- Delauney, A.J.; Hu, C.A.A.; Kishor, P.B.K.; Verma, D.P.S. Cloning of ornithine Δ-aminotransferase cDNA from Vigna aconitifolia by trans-complementation in Escherichchia coli and regulation of proline biosynthesis. J. Biol. Chem. 1993, 268, 18673–18678. [Google Scholar]
- Hu, C.A.A.; Delauney, A.J.; Verma, D.P.S. A bifunctional enzyme (Δ1-pyrroline-5-carboxylate synthetase) catalyzes the first two steps in proline biosynthesis in plants. Proc. Natl. Acad. Sci. USA 1992, 89, 9354–9358. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Rigo, G.; Szekely, G.; Nagy, R.; Koncz, C.; Szabados, L. Light-dependent induction of proline biosynthesis by abscisic acid and salt stress is inhibited by brassinosteroid in Arabidopsis. Plant Mol. Biol. 2003, 51, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.L.; Lan, S.S.; Gong, M. Hydrogen peroxide-induced proline and metabolic pathway of its accumulation in maize seedlings. J. Plant Physiol. 2009, 166, 1694–1699. [Google Scholar] [CrossRef]
- Fichman, Y.; Gerdes, S.Y.; Kovacs, H.; Szabados, L.; Zilberstein, A.; Csonka, L.N. Evolution of proline biosynthesis: Enzymology, bioinformatics, genetics, and transcriptional regulation. Biol. Rev. 2015, 90, 1065–1099. [Google Scholar] [CrossRef] [PubMed]
- Kubala, S.; Wojtyla, L.; Quinet, M.; Lechowska, K.; Lutts, S.; Garnczarska, M. Enhanced expression of the proline synthesis gene P5CSA in relation to seed osmopriming improvement of Brassica napus germination under salinity stress. J. Plant Physiol. 2015, 183, 1–12. [Google Scholar] [CrossRef]
- Zarattini, M.; Forlani, G. Toward unveiling the mechanisms for transcriptional regulation of proline biosynthesis in the plant cell response to biotic and abiotic stress conditions. Front. Plant Sci. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Song, Y.G.; Zhao, Z.; Qiu, N.W.; Liu, X.J.; Guo, W.H. The Medicago truncatula R2R3-MYB transcription factor gene MtMYBS1 enhances salinity tolerance when constitutively expressed in Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 2017, 490, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.Y.; Wang, Y.C.; Wang, L.Q.; Hu, P.; Wang, Y.M.; Jia, Y.Y.; Zhang, C.R.; Zhang, Y.; Zhang, Y.M.; Wang, C.; et al. Expression of the MYB transcription factor gene BplMYB46 affects abiotic stress tolerance and secondary cell wall deposition in Betula platyphylla. Plant Biotechnol. J. 2017, 15, 107–121. [Google Scholar] [CrossRef]
- Sharma, S.; Verslues, P.E. Mechanisms independent of abscisic acid (ABA) or proline feedback have a predominant role in transcriptional regulation of proline metabolism during low water potential and stress recovery. Plant Cell Environ. 2010, 33, 1838–1851. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Li, F.; Li, D.J.; Zhang, H.W.; Huang, R.F. Expression of ethylene response factor JERF1 in rice improves tolerance to drought. Planta 2010, 232, 765–774. [Google Scholar] [CrossRef]
- Li, S.F.; Fan, C.M.; Li, Y.; Zhang, J.H.; Sun, J.S.; Chen, Y.H.; Tian, C.Y.; Su, X.H.; Lu, M.Z.; Liang, C.Z.; et al. Effects of drought and salt-stresses on gene expression in Caragana korshinskii seedlings revealed by RNA-seq. BMC Genom. 2016, 17, 200. [Google Scholar] [CrossRef]
- Rastogi, S.; Shah, S.; Kumar, R.; Vashisth, D.; Akhtar, M.Q.; Kumar, A.; Dwivedi, U.N.; Shasany, A.K. Ocimum metabolomics in response to abiotic stresses: Cold, flood, drought and salinity. PLoS ONE 2019, 14, e0210903. [Google Scholar] [CrossRef]
- Selmar, D.; Kleinwachter, M. Stress enhances the synthesis of secondary plant products: The impact of stress-related over-reduction on the accumulation of natural products. Plant Cell Physiol. 2013, 54, 817–826. [Google Scholar] [CrossRef]
- Do, T.H.T.; Martinoia, E.; Lee, Y. Functions of ABC transporters in plant growth and development. Curr. Opin. Plant Biol. 2018, 41, 32–38. [Google Scholar] [CrossRef]
- Zhao, G.C.; Shi, J.X.; Liang, W.Q.; Xue, F.Y.; Luo, Q.; Zhu, L.; Qu, G.R.; Chen, M.J.; Schreiber, L.; Zhang, D.B. Two ATP binding cassette G transporters, rice ATP binding cassette G26 and ATP binding cassette G15, collaboratively regulate rice male reproduction. Plant Physiol. 2015, 169, 2064–2079. [Google Scholar]
- Liu, J.; Osbourn, A.; Ma, P. MYB transcription factors as regulators of phenylpropanoid metabolism in plants. Mol. Plant 2015, 8, 689–708. [Google Scholar] [CrossRef] [PubMed]
- Baldoni, E.; Genga, A.; Cominelli, E. Plant MYB transcription factors: Their role in drought response mechanisms. Int. J. Mol. Sci. 2015, 16, 15811–15851. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Pireyre, M.; Burow, M. Regulation of MYB and bHLH transcription factors: A glance at the protein level. Mol. Plant 2015, 8, 378–388. [Google Scholar] [CrossRef]
- Olsen, A.N.; Ernst Haleggio, L.L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10, 79–87. [Google Scholar] [CrossRef]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S.J.F.M. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4, 248. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Meyerowitz, E.M. The AP2/EREBP family of plant transcription factors. Biol. Chem. 1998, 379, 633–646. [Google Scholar] [PubMed]
- Kizis, D.; Lumbreras, V.; Pagès, M. Role of AP2/EREBP transcription factors in gene regulation during abiotic stress. FEBS Lett. 2001, 498, 187–189. [Google Scholar] [CrossRef]
- Agarwal, P.K.; Gupta, K.; Lopato, S.; Agarwal, P. Dehydration responsive element binding transcription factors and their applications for the engineering of stress tolerance. J. Exp. Bot. 2017, 68, 2135–2148. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Yin, X.; Lin, Z.; Zheng, Q.; Liu, G.; Zhao, G. Identification and characterization of NF-Y transcription factor families in Canola (Brassica napus L.). Planta 2014, 239, 107–126. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Clean Reads (Mb) | Total Clean Bases (Gb) | Clean Reads Q30 (%) | Total Mapping Ratio |
|---|---|---|---|---|
| CK-L-1 | 44.93 | 6.74 | 95.03 | 60.59% |
| CK-L-2 | 44.07 | 6.61 | 96.06 | 64.09% |
| CK-L-3 | 45 | 6.75 | 95.94 | 64.47% |
| CK-R-1 | 44.27 | 6.64 | 95.34 | 62.21% |
| CK-R-2 | 44.42 | 6.66 | 95.81 | 59.86% |
| CK-R-3 | 44.06 | 6.61 | 95.94 | 60.16% |
| D-L-1 | 44.73 | 6.71 | 95.02 | 63.08% |
| D-L-2 | 45.13 | 6.77 | 95.1 | 63.50% |
| D-L-3 | 44.8 | 6.72 | 95.1 | 63.80% |
| D-R-1 | 44.97 | 6.74 | 95.4 | 60.51% |
| D-R-2 | 44.71 | 6.71 | 95.06 | 61.52% |
| D-R-3 | 41.5 | 6.23 | 95 | 62.03% |
| S-L-1 | 42.78 | 6.42 | 95.18 | 63.53% |
| S-L-2 | 44.42 | 6.66 | 95.18 | 62.31% |
| S-L-3 | 44.99 | 6.75 | 95.2 | 61.65% |
| S-R-1 | 44.9 | 6.73 | 95.25 | 61.69% |
| S-R-2 | 44.91 | 6.74 | 95.63 | 61.21% |
| S-R-3 | 42.11 | 6.32 | 95.09 | 60.84% |
| Total number of novel transcripts | 42,251 | |||
| Coding transcripts | 35,124 | |||
| Noncoding transcripts | 7127 | |||
| Novel genes | 4251 | |||
| 2A. KEGG pathways of DEGs in CK-L vs. D-L | ||||
| # | Pathway | Annotation (713) | p-Value | Pathway ID |
| 1 | Metabolic pathways | 210 (29.45%) | 1.487446e-06 | ko01100 |
| 2 | Biosynthesis of secondary metabolites | 133 (18.65%) | 2.375184e-06 | ko01110 |
| 3 | Pyruvate metabolism | 20 (2.81%) | 0.0003487765 | ko00620 |
| 4 | Arginine and proline metabolism | 13 (1.82%) | 0.001129843 | ko00330 |
| 5 | Glycine, serine and threonine metabolism | 16 (2.24%) | 0.001321938 | ko00260 |
| 6 | Glutathione metabolism | 13 (1.82%) | 0.001345293 | ko00480 |
| 7 | Fatty acid degradation | 10 (1.4%) | 0.001462133 | ko00071 |
| 8 | Glycerolipid metabolism | 20 (2.81%) | 0.001519717 | ko00561 |
| 9 | Indole alkaloid biosynthesis | 6 (0.84%) | 0.001620922 | ko00901 |
| 10 | Regulation of autophagy | 13 (1.82%) | 0.001845656 | ko04140 |
| 11 | Ribosome | 37 (5.19%) | 0.002576793 | ko03010 |
| 12 | Tryptophan metabolism | 12 (1.68%) | 0.003130347 | ko00380 |
| 13 | Caffeine metabolism | 2 (0.28%) | 0.003729469 | ko00232 |
| 14 | Ascorbate and aldarate metabolism | 12 (1.68%) | 0.004263276 | ko00053 |
| 15 | Histidine metabolism | 7 (0.98%) | 0.004263308 | ko00340 |
| 16 | mRNA surveillance pathway | 27 (3.79%) | 0.005933953 | ko03015 |
| 17 | alpha-Linolenic acid metabolism | 9 (1.26%) | 0.007378807 | ko00592 |
| 18 | Ubiquinone and other terpenoid-quinone biosynthesis | 9 (1.26%) | 0.008113964 | ko00130 |
| 19 | Cysteine and methionine metabolism | 15 (2.1%) | 0.01011539 | ko00270 |
| 20 | Steroid biosynthesis | 7 (0.98%) | 0.01100777 | ko00100 |
| 21 | Pentose and glucuronate interconversions | 20 (2.81%) | 0.01620864 | ko00040 |
| 22 | Lysine degradation | 8 (1.12%) | 0.01643623 | ko00310 |
| 23 | Galactose metabolism | 13 (1.82%) | 0.019456 | ko00052 |
| 24 | Carotenoid biosynthesis | 8 (1.12%) | 0.0198106 | ko00906 |
| 2B. KEGG pathways of DEGs in CK-R vs. D-R | ||||
| # | Pathway | Annotation (3034) | p-Value | Pathway ID |
| 1 | Metabolic pathways | 926 (30.52%) | 1.348677e-29 | ko01100 |
| 2 | Biosynthesis of secondary metabolites | 570 (18.79%) | 2.264539e-23 | ko01110 |
| 3 | Photosynthesis–antenna proteins | 26 (0.86%) | 1.749018e-15 | ko00196 |
| 4 | Phenylpropanoid biosynthesis | 132 (4.35%) | 6.312239e-15 | ko00940 |
| 5 | Glycolysis/Gluconeogenesis | 105 (3.46%) | 9.470061e-12 | ko00010 |
| 6 | Carbon metabolism | 151 (4.98%) | 1.339665e-11 | ko01200 |
| 7 | Pyruvate metabolism | 70 (2.31%) | 7.136255e-08 | ko00620 |
| 8 | Carbon fixation in photosynthetic organisms | 60 (1.98%) | 1.608047e-07 | ko00710 |
| 9 | Photosynthesis | 37 (1.22%) | 4.541068e-07 | ko00195 |
| 10 | Plant hormone signal transduction | 172 (5.67%) | 6.098387e-07 | ko04075 |
| 11 | Arginine and proline metabolism | 43 (1.42%) | 3.276459e-06 | ko00330 |
| 12 | Circadian rhythm–plant | 66 (2.18%) | 4.310124e-06 | ko04712 |
| 13 | Nitrogen metabolism | 35 (1.15%) | 2.766522e-05 | ko00910 |
| 14 | Alanine, aspartate and glutamate metabolism | 43 (1.42%) | 3.614134e-05 | ko00250 |
| 15 | Sulfur metabolism | 25 (0.82%) | 0.0001004235 | ko00920 |
| 16 | Valine, leucine and isoleucine degradation | 32 (1.05%) | 0.0001541904 | ko00280 |
| 17 | Fructose and mannose metabolism | 44 (1.45%) | 0.0002872131 | ko00051 |
| 18 | Glycine, serine and threonine metabolism | 46 (1.52%) | 0.001046218 | ko00260 |
| 19 | Inositol phosphate metabolism | 34 (1.12%) | 0.001077126 | ko00562 |
| 20 | Stilbenoid, diarylheptanoid and gingerol biosynthesis | 45 (1.48%) | 0.001329768 | ko00945 |
| 21 | Biosynthesis of unsaturated fatty acids | 19 (0.63%) | 0.001333774 | ko01040 |
| 22 | Galactose metabolism | 45 (1.48%) | 0.002157363 | ko00052 |
| 23 | Starch and sucrose metabolism | 131 (4.32%) | 0.002175542 | ko00500 |
| 24 | Flavonoid biosynthesis | 31 (1.02%) | 0.002204898 | ko00941 |
| 25 | Taurine and hypotaurine metabolism | 14 (0.46%) | 0.002282244 | ko00430 |
| 26 | Zeatin biosynthesis | 18 (0.59%) | 0.003346513 | ko00908 |
| 27 | Citrate cycle (TCA cycle) | 33 (1.09%) | 0.00335235 | ko00020 |
| 28 | Diterpenoid biosynthesis | 25 (0.82%) | 0.003665961 | ko00904 |
| 29 | Pentose phosphate pathway | 44 (1.45%) | 0.004369615 | ko00030 |
| 30 | Biosynthesis of amino acids | 113 (3.72%) | 0.004736771 | ko01230 |
| 31 | beta-Alanine metabolism | 27 (0.89%) | 0.005233566 | ko00410 |
| 32 | Flavone and flavonol biosynthesis | 16 (0.53%) | 0.006075288 | ko00944 |
| 33 | Limonene and pinene degradation | 40 (1.32%) | 0.009214323 | ko00903 |
| 34 | Tyrosine metabolism | 25 (0.82%) | 0.01085029 | ko00350 |
| 35 | Glyoxylate and dicarboxylate metabolism | 44 (1.45%) | 0.01298711 | ko00630 |
| 36 | Amino sugar and nucleotide sugar metabolism | 60 (1.98%) | 0.0143173 | ko00520 |
| 37 | Cysteine and methionine metabolism | 45 (1.48%) | 0.0158524 | ko00270 |
| 38 | Ether lipid metabolism | 17 (0.56%) | 0.01740316 | ko00565 |
| 39 | Glucosinolate biosynthesis | 15 (0.49%) | 0.01936801 | ko00966 |
| 2C. KEGG pathways of DEGs in CK-L vs. S-L | ||||
| # | Pathway | Annotation (79) | p-Value | Pathway ID |
| 1 | ABC transporters | 5 (6.33%) | 0.0003552804 | ko02010 |
| 2 | Cutin, suberin, and wax biosynthesis | 3 (3.8%) | 0.01149507 | ko00073 |
| 3 | Regulation of autophagy | 3 (3.8%) | 0.01838196 | ko04140 |
| 2D. KEGG pathways of DEGs in CK-R vs. S-R | ||||
| # | Pathway | Annotation (502) | p-Value | Pathway ID |
| 1 | Nitrogen metabolism | 20 (3.98%) | 8.034565e-12 | ko00910 |
| 2 | Endocytosis | 38 (7.57%) | 9.001716e-06 | ko04144 |
| 3 | Phenylpropanoid biosynthesis | 26 (5.18%) | 3.331352e-05 | ko00940 |
| 4 | Ether lipid metabolism | 8 (1.59%) | 0.0002257619 | ko00565 |
| 5 | Glucosinolate biosynthesis | 7 (1.39%) | 0.0004888917 | ko00966 |
| 6 | Biosynthesis of secondary metabolites | 89 (17.73%) | 0.0005704914 | ko01110 |
| 7 | Plant-pathogen interaction | 40 (7.97%) | 0.004413053 | ko04626 |
| 8 | Metabolic pathways | 135 (26.89%) | 0.004841477 | ko01100 |
| 9 | ABC transporters | 10 (1.99%) | 0.005615396 | ko02010 |
| 10 | Arginine and proline metabolism | 9 (1.79%) | 0.006949714 | ko00330 |
| 11 | Flavonoid biosynthesis | 8 (1.59%) | 0.010255 | ko00941 |
| 12 | Indole alkaloid biosynthesis | 4 (0.8%) | 0.01178287 | ko00901 |
| 13 | Protein processing in endoplasmic reticulum | 24 (4.78%) | 0.01324247 | ko04141 |
| 14 | Plant hormone signal transduction | 30 (5.98%) | 0.01431411 | ko04075 |
| 15 | Pyruvate metabolism | 12 (2.39%) | 0.01621669 | ko00620 |
| 16 | Tryptophan metabolism | 8 (1.59%) | 0.01940078 | ko00380 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Jiao, J.; Zhou, M.; Jin, Z.; Yu, Y.; Liang, M. Physiological and Transcriptional Responses of Industrial Rapeseed (Brassica napus) Seedlings to Drought and Salinity Stress. Int. J. Mol. Sci. 2019, 20, 5604. https://doi.org/10.3390/ijms20225604
Wang J, Jiao J, Zhou M, Jin Z, Yu Y, Liang M. Physiological and Transcriptional Responses of Industrial Rapeseed (Brassica napus) Seedlings to Drought and Salinity Stress. International Journal of Molecular Sciences. 2019; 20(22):5604. https://doi.org/10.3390/ijms20225604
Chicago/Turabian StyleWang, Ji, Jiao Jiao, Mengjia Zhou, Zeyang Jin, Yongjian Yu, and Mingxiang Liang. 2019. "Physiological and Transcriptional Responses of Industrial Rapeseed (Brassica napus) Seedlings to Drought and Salinity Stress" International Journal of Molecular Sciences 20, no. 22: 5604. https://doi.org/10.3390/ijms20225604
APA StyleWang, J., Jiao, J., Zhou, M., Jin, Z., Yu, Y., & Liang, M. (2019). Physiological and Transcriptional Responses of Industrial Rapeseed (Brassica napus) Seedlings to Drought and Salinity Stress. International Journal of Molecular Sciences, 20(22), 5604. https://doi.org/10.3390/ijms20225604