Suppression of Human Platelet Activation via Integrin αIIbβ3 Outside-In Independent Signal and Reduction of the Mortality in Pulmonary Thrombosis by Auraptene

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results
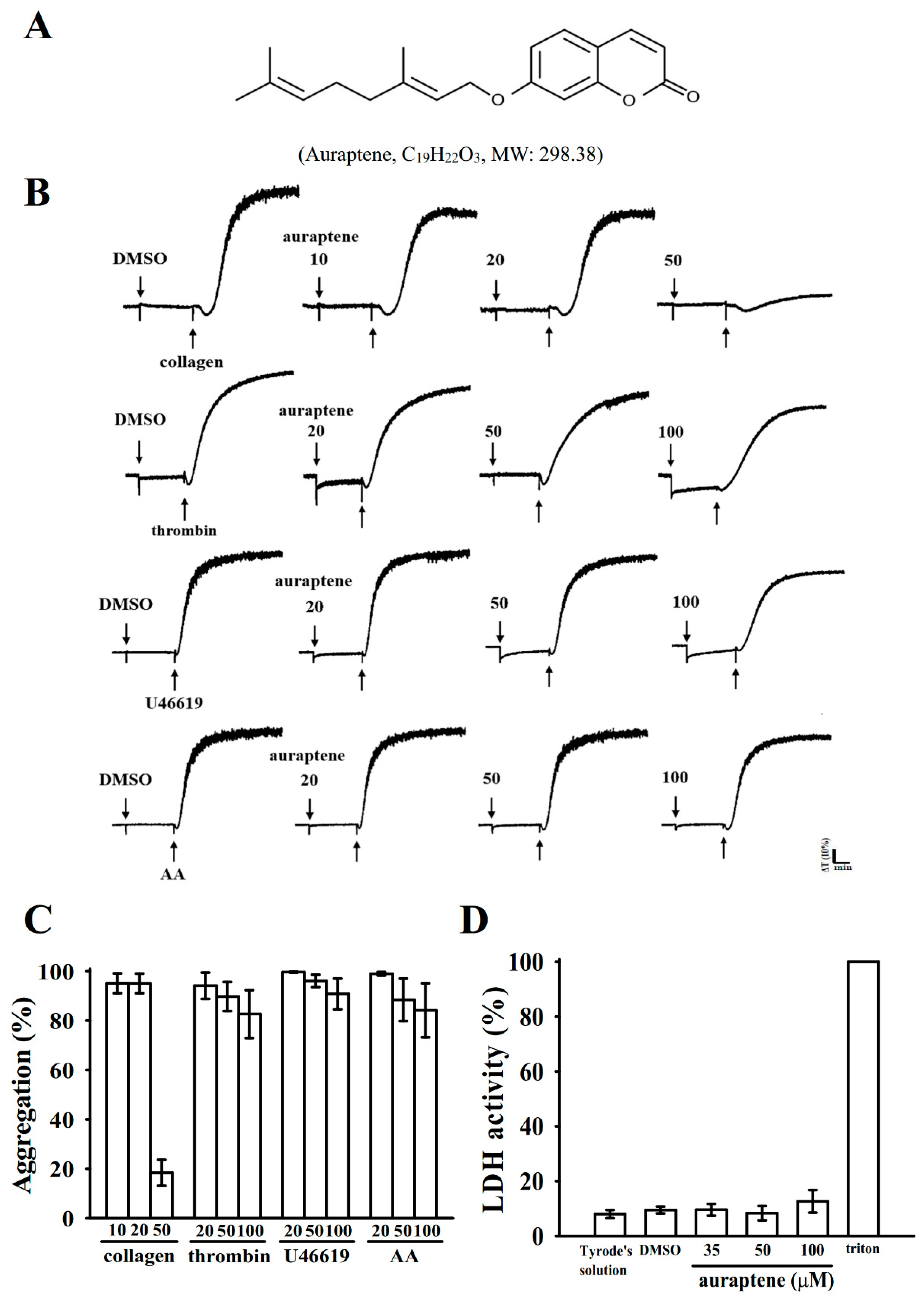
2.1. Inhibitory Profiles of Auraptene in Agonist-Stimulated Washed Human Platelets
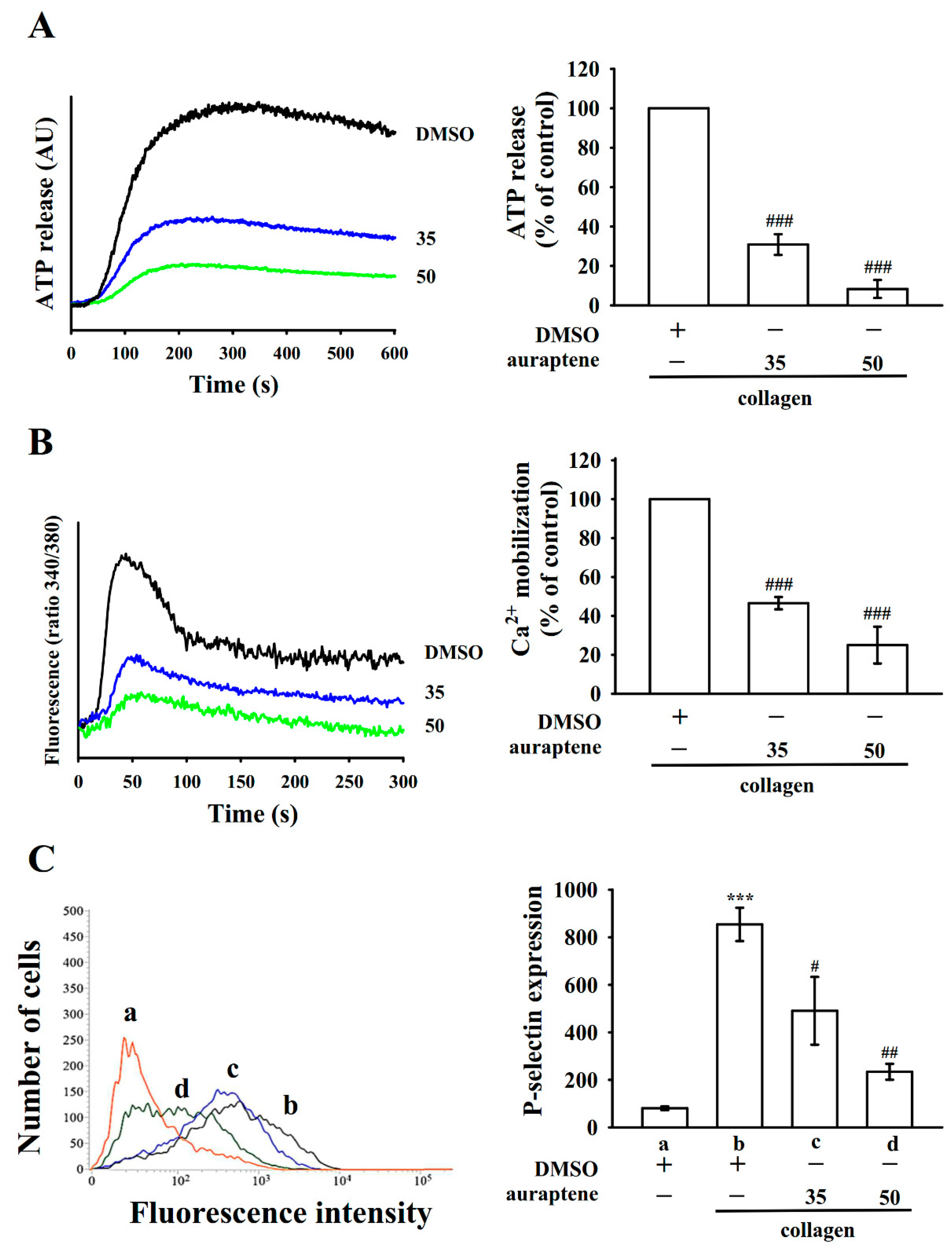
2.2. Regulatory Characteristics of Platelet Activation by Auraptene
2.3. Role of Auraptene in Integrin αIIbβ3 Activation
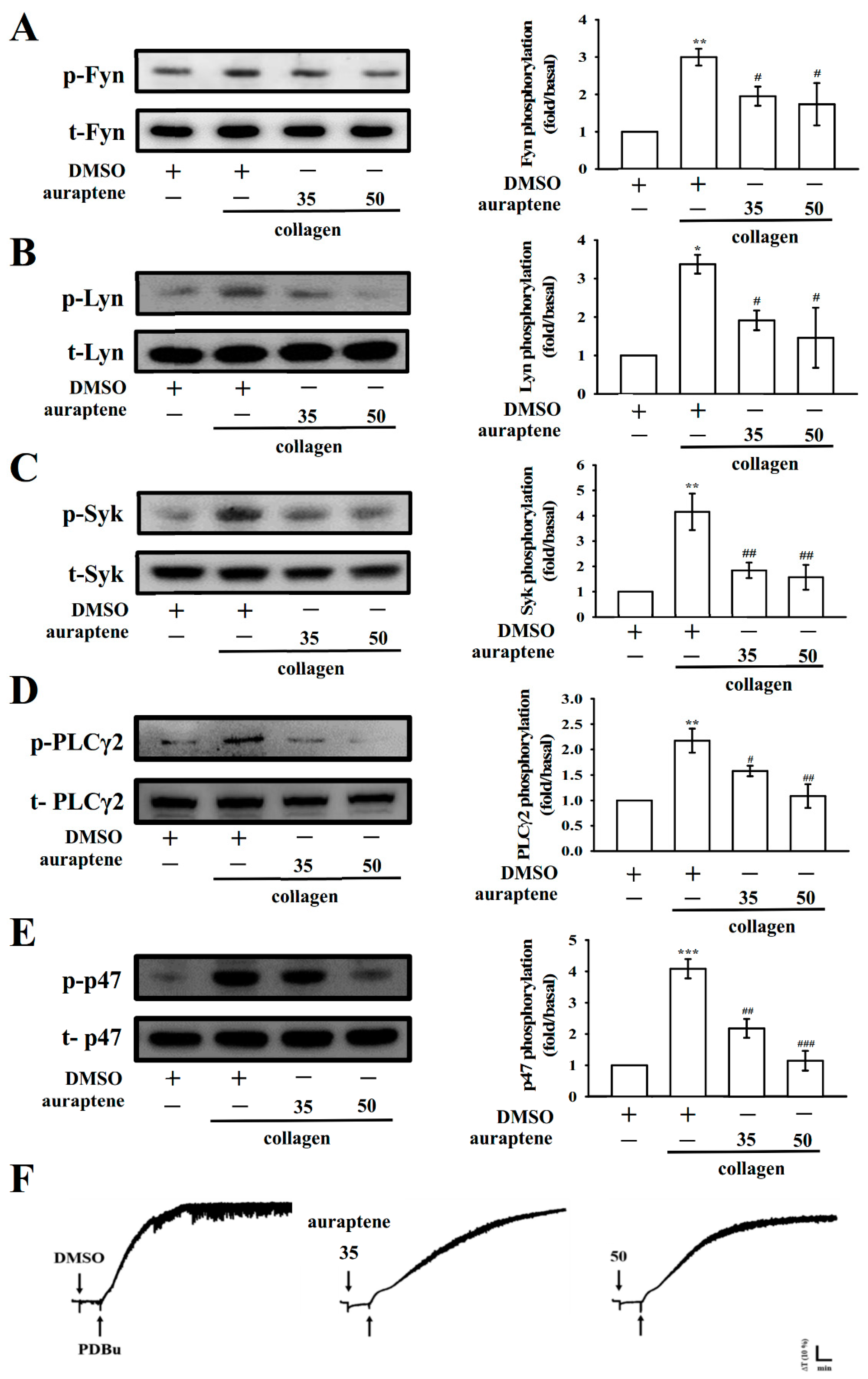
2.4. Effects of Auraptene on Fyn, Lyn, Syk, and PLCγ2/PKC Signaling
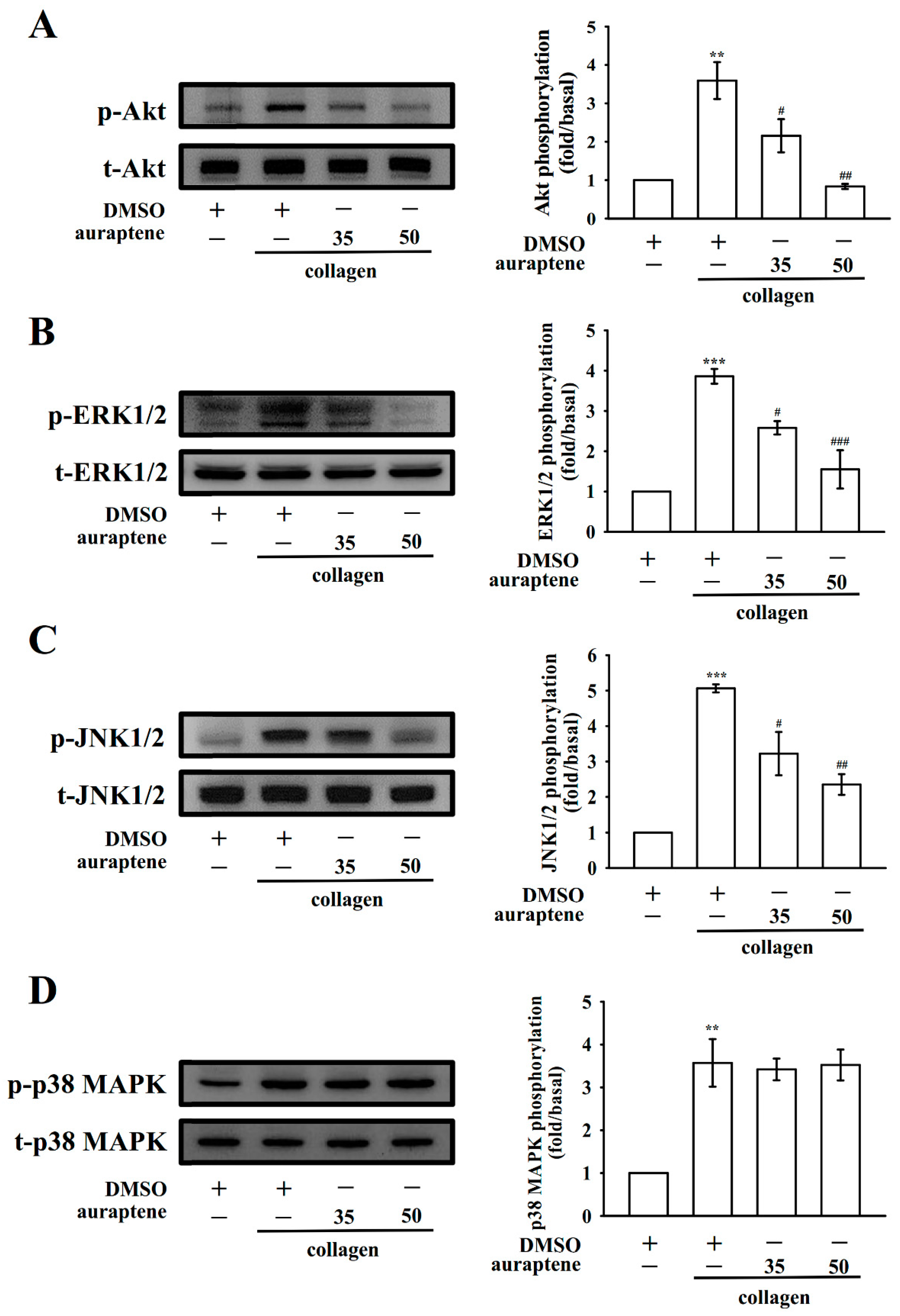
2.5. Auraptene Inhibits Akt, ERK1/2, and JNK1/2 Activation
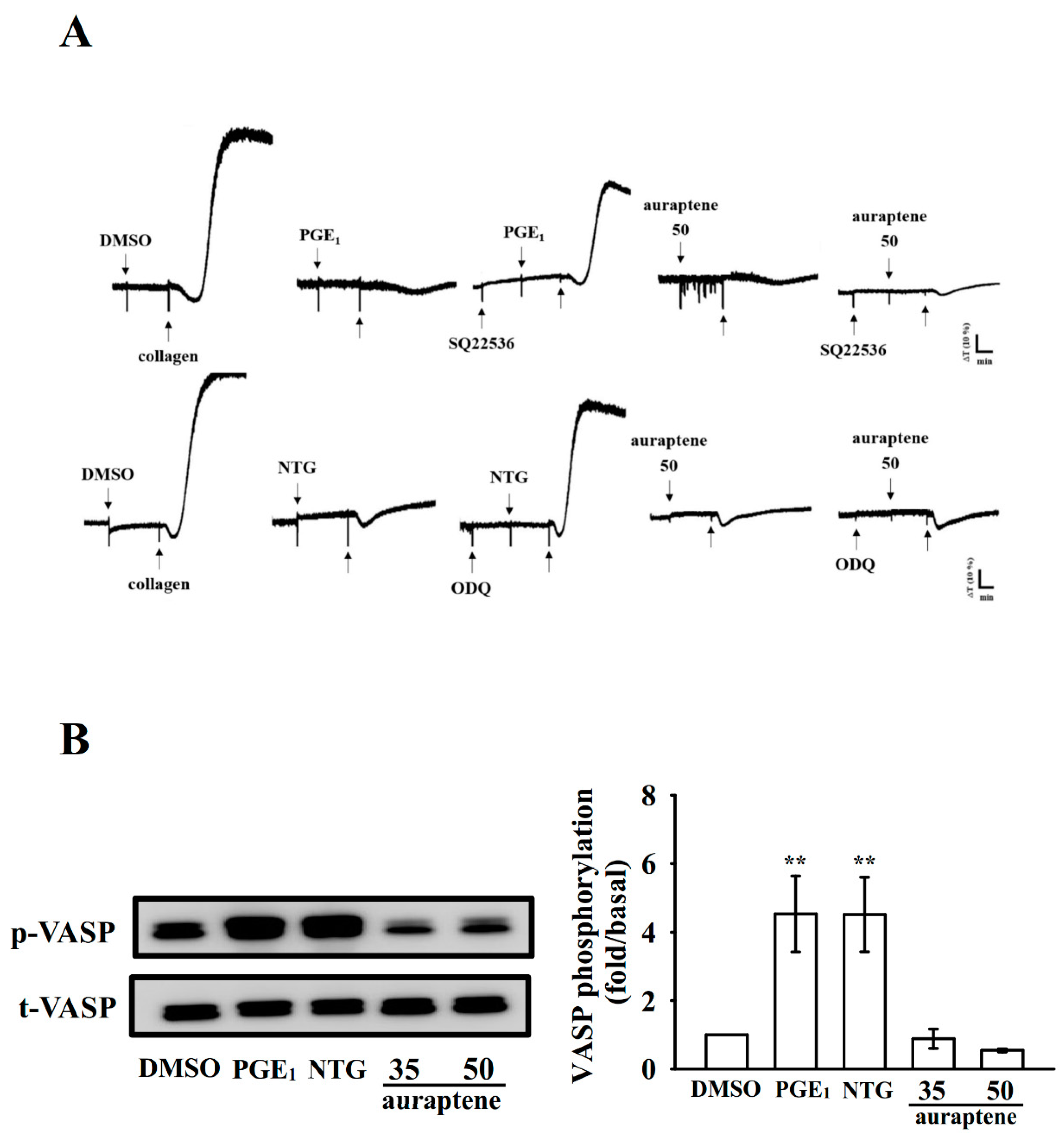
2.6. Effectiveness of Auraptene in Cyclic Nucleotide Formation and Acute Pulmonary Thromboembolism In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Platelet Preparation, Aggregation, and ATP Release
4.3. Intracellular [Ca2+] Mobilization Using Fura 2-AM Fluorescence
4.4. Lactate Dehydrogenase Assay
4.5. Flow Cytometry Analysis for P-Selectin Expression and Integrin αIIbβ3 Activation
4.6. Platelet Adhesion and Spreading Analysis on Immobilized Fibrinogen
4.7. Platelet-Mediated Clot Retraction
4.8. Immunoblotting
4.9. Acute Pulmonary Thromboembolism Stimulated by ADP in Mice
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Peluso, M.R. Flavonoids attenuate cardiovascular disease, inhibit phosphodiesterase, and modulate lipid homeostasis in adipose tissue and liver. Exp. Biol. Med. 2006, 231, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Estevez, B.; Du, X. New concepts and mechanisms of platelet activation signaling. Physiology 2017, 32, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Payrastre, B.; Missy, K.; Trumel, C.; Bodin, S.; Plantavid, M.; Chap, H. The integrin αIIb/β3 in human platelet signal transduction. Biochem. Pharmacol. 2000, 60, 1069–1074. [Google Scholar] [CrossRef]
- Genovese, S.; Epifano, F. Auraptene: A natural biologically active compound with multiple targets. Curr. Drug Targets 2011, 12, 381–386. [Google Scholar] [CrossRef]
- Marumoto, S.; Miyazawa, M. Structure-activity relationships for naturally occurring coumarins as—secretase inhibitor. Bioorg. Med. Chem. 2012, 20, 784–788. [Google Scholar] [CrossRef]
- Okuyama, S.; Minami, S.; Shimada, N.; Makihata, N.; Nakajima, M.; Furukawa, Y. Anti-inflammatory and neuroprotective effects of auraptene, acitrus coumarin, following cerebral global ischemia in mice. Eur. J. Pharmacol. 2013, 699, 118–123. [Google Scholar] [CrossRef]
- Derosa, G.; Maffioli, P.; Sahebkar, A. Auraptene and its role in chronic diseases. Adv. Exp. Med. Biol. 2016, 929, 399–407. [Google Scholar]
- Chen, I.S.; Lin, Y.C.; Tsai, I.L.; Teng, C.M.; Ko, F.N.; Ishikawa, T.; Ishii, H. Coumarins and anti-platelet aggregation constituents from Zanthoxylum schinifolium. Phytochemistry 1995, 39, 1091–1097. [Google Scholar] [CrossRef]
- Teng, C.M.; Li, H.L.; Wu, T.S.; Huang, S.C.; Huang, T.F. Antiplatelet actions of some coumarin compounds isolated from plant sources. Thromb. Res. 1992, 66, 549–557. [Google Scholar] [CrossRef]
- Majerus, P.W.; Broze, G.J.; Miletich, J.P.; Tollefsen, D.M. Anticoagulant, thrombolytic, and antiplatelet drugs. In Goodman & Gilman’s the Pharmacological Basis of Therapeutics, 8th ed.; Gilman, A.G., Goodman, L.S., Gilman, A., Eds.; Pergamon Press: New York, NY, USA, 1991; pp. 1311–1331. [Google Scholar]
- Cosemans, J.M.; Iserbyt, B.F.; Deckmyn, H.; Heemskerk, J.W. Multiple ways to switch platelet integrins on and off. J. Thromb. Haemost. 2008, 6, 1253–1261. [Google Scholar] [CrossRef]
- Shattil, S.J. The beta3 integrin cytoplasmic tail: protein scaffold and control freak. J. Thromb. Haemost. 2009, 7, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Quek, L.S.; Pasquet, J.M.; Hers, I.; Cornall, R.; Knight, G.; Barnes, M.; Hibbs, M.L.; Dunn, A.R.; Lowell, C.A.; Watson, S.P. Fyn and Lyn phosphorylate the Fc receptor gamma chain downstream of glycoprotein VI in murine platelets, and Lyn regulates a novel feedback pathway. Blood 2000, 96, 4246–4253. [Google Scholar] [CrossRef] [PubMed]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, T.; Ding, K.; Liu, Z.; Li, Y.; He, T.; Zhang, W.; Fan, Y.; Ma, W.; Cui, L.; et al. Phospholipase Cγ2 signaling cascade contribute to the antiplatelet effect of notoginsenoside Fc. Front. Pharmacol. 2018, 9, 1293. [Google Scholar] [CrossRef]
- Chen, W.; Thielmann, I.; Gupta, S.; Subramanian, H.; Stegner, D.; van Kruchten, R.; Dietrich, A.; Gambaryan, S.; Heemskerk, J.W.; Hermanns, H.M.; et al. Orai1-induced store-operated Ca2+ entry enhances phospholipase activity and modulates canonical transient receptor potential channel 6 function in murine platelets. J. Thromb. Haemost. 2014, 12, 528–539. [Google Scholar] [CrossRef]
- Ragab, A.; Severin, S.; Gratacap, M.P.; Aguado, E.; Malissen, M.; Jandrot-Perrus, M.; Malissen, B.; Ragab-Thomas, J.; Payrastre, B. Roles of the C-terminal tyrosine residues of LAT in GPVI-induced platelet activation: insights into the mechanism of PLC gamma 2 activation. Blood 2007, 110, 2466–2474. [Google Scholar] [CrossRef]
- Kim, S.; Jin, J.; Kunapuli, S.P. Relative contribution of G-protein-coupled pathways to protease-activated receptor-mediated Akt phosphorylation in platelets. Blood 2006, 107, 947–954. [Google Scholar] [CrossRef]
- Garcia, A.; Shankar, H.; Murugappan, S.; Kim, S.; Kunapuli, S.P. Regulation and functional consequences of ADP receptor-mediated ERK2 activation in platelets. Biochem. J. 2007, 404, 299–308. [Google Scholar] [CrossRef]
- Fan, X.; Wang, C.; Shi, P.; Gao, W.; Gu, J.; Geng, Y.; Yang, W.; Wu, N.; Wang, Y.; Xu, Y.; et al. Platelet MEKK3 regulates arterial thrombosis and myocardial infarct expansion in mice. Blood Adv. 2018, 2, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.E.; Renshaw, M.W.; Pfaff, M.; Forsyth, J.; Keivens, V.M.; Schwartz, M.A.; Ginsberg, M.H. Suppression of integrin activation: a novel function of a Ras/Raf-initiated MAP kinase pathway. Cell 1997, 88, 521–530. [Google Scholar] [CrossRef]
- Adam, F.; Kauskot, A.; Rosa, J.P.; Bryckaert, M. Mitogen-activated protein kinases in hemostasis and thrombosis. J. Thromb. Haemost. 2008, 6, 2007–2016. [Google Scholar] [CrossRef] [PubMed]
- Coulon, L.; Calzada, C.; Moulin, P.; Véricel, E.; Lagarde, M. Activation of p38 mitogen-activated protein kinase/cytosolic phospholipase A2 cascade in hydroperoxide-stressed platelets. Free Radic. Biol. Med. 2003, 35, 616–625. [Google Scholar] [CrossRef]
- Woulfe, D.S. Akt signaling in platelets and thrombosis. Expert Rev. Hematol. 2010, 3, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Jayakumara, T.; Chen, W.F.; Lu, W.J.; Chou, D.S.; Hsiao, G.; Hsu, C.Y.; Sheu, J.R.; Hsieh, C.Y. A novel antithrombotic effect of sulforaphane via activation of platelet adenylate cyclase: ex vivo and in vivo studies. J. Nutr. Biochem. 2013, 24, 1086–1095. [Google Scholar] [CrossRef]
- Benz, P.M.; Laban, H.; Günther, L.; Walter, U.; Gambaryan, S.; Dib, K. Vasodilator-Stimulated Phosphoprotein (VASP)-dependent and-independent pathways regulate thrombin-induced activation of Rap1b in platelets. Cell Commun. Signal. 2016, 14, 21. [Google Scholar] [CrossRef]
- Przygodzki, T.; Talar, M.; Blazejczyk, A.; Kalchenko, V.; Watala, C. Quantification of the blood platelet reactivity in the ADP-induced model of non-lethal pulmonary thromboembolism in mice with the use of laser doppler flowmeter. PLoS ONE 2016, 11, e0146346. [Google Scholar] [CrossRef]
- Chen, W.F.; Lee, J.J.; Chang, C.C.; Lin, K.H.; Wang, S.H.; Sheu, J.R. Platelet protease-activated receptor (PAR)4, but not PAR1, associated with neutral sphingomyelinase responsible for thrombin-stimulated ceramide-NF-κB signaling in human platelets. Haematologica 2013, 98, 793–801. [Google Scholar] [CrossRef]
- Sheu, J.R.; Lee, C.R.; Lin, C.H.; Hsiao, G.; Ko, W.C.; Chen, Y.C.; Yen, M.H. Mechanisms involved in the antiplatelet activity of Staphylococcus aureus lipoteichoic acid in human platelets. Thromb. Haemost. 2000, 83, 777–784. [Google Scholar]
- Hsia, C.H.; Lu, W.J.; Lin, K.H.; Chou, D.S.; Geraldine, P.; Jayakuma, T.; Chang, N.C.; Sheu, J.R. NCTD, a clinical used chemotherapeutic agent, acts as a powerful inhibitor by interfering with fibrinogen-integrin α II bβ3 binding in human platelets. J. Cell. Mol. Med. 2018, 22, 2142–2152. [Google Scholar] [CrossRef] [PubMed]
- Osdoit, S.; Rosa, J.P. Fibrin clot retraction by human platelets correlates with alpha (IIb) beta (3) integrin-dependent protein tyrosine dephosphorylation. J. Biol. Chem. 2001, 276, 6703–6710. [Google Scholar] [CrossRef] [PubMed]
- Hsia, C.W.; Lin, K.C.; Lee, T.Y.; Hsia, C.H.; Chou, D.S.; Jayakumar, T.; Velusamy, M.; Chang, C.C.; Sheu, J.R. Esculetin, a coumarin derivative, prevents thrombosis: inhibitory signaling on PLCγ2–PKC–AKT activation in human platelets. Int. J. Mol. Sci. 2019, 20, 2731. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Number | Number of Deaths | Mortality (%) | Platelet Count (K/μL) | |
|---|---|---|---|---|
| DMSO | 8 | 0 | 0 | 915 ± 24 |
| ADP (0.7 mg/g) | ||||
| + DMSO | 8 | 7 | 87.5 | 770 ± 22 ** |
| + aspirin (mg/kg) | ||||
| 20 | 8 | 2 | 25.0 | 888 ± 29 # |
| + auraptene (mg/kg) | ||||
| 7.5 | 8 | 5 | 62.5 | 819 ± 21 |
| 15 | 8 | 2 | 25.0 | 879 ± 26 # |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsia, C.-W.; Tsai, C.-L.; Sheu, J.-R.; Lu, W.-J.; Hsia, C.-H.; Velusamy, M.; Jayakumar, T.; Li, J.-Y. Suppression of Human Platelet Activation via Integrin αIIbβ3 Outside-In Independent Signal and Reduction of the Mortality in Pulmonary Thrombosis by Auraptene. Int. J. Mol. Sci. 2019, 20, 5585. https://doi.org/10.3390/ijms20225585
Hsia C-W, Tsai C-L, Sheu J-R, Lu W-J, Hsia C-H, Velusamy M, Jayakumar T, Li J-Y. Suppression of Human Platelet Activation via Integrin αIIbβ3 Outside-In Independent Signal and Reduction of the Mortality in Pulmonary Thrombosis by Auraptene. International Journal of Molecular Sciences. 2019; 20(22):5585. https://doi.org/10.3390/ijms20225585
Chicago/Turabian StyleHsia, Chih-Wei, Cheng-Lin Tsai, Joen-Rong Sheu, Wan-Jung Lu, Chih-Hsuan Hsia, Marappan Velusamy, Thanasekaran Jayakumar, and Jiun-Yi Li. 2019. "Suppression of Human Platelet Activation via Integrin αIIbβ3 Outside-In Independent Signal and Reduction of the Mortality in Pulmonary Thrombosis by Auraptene" International Journal of Molecular Sciences 20, no. 22: 5585. https://doi.org/10.3390/ijms20225585
APA StyleHsia, C.-W., Tsai, C.-L., Sheu, J.-R., Lu, W.-J., Hsia, C.-H., Velusamy, M., Jayakumar, T., & Li, J.-Y. (2019). Suppression of Human Platelet Activation via Integrin αIIbβ3 Outside-In Independent Signal and Reduction of the Mortality in Pulmonary Thrombosis by Auraptene. International Journal of Molecular Sciences, 20(22), 5585. https://doi.org/10.3390/ijms20225585