Combination Radioimmunotherapy Strategies for Solid Tumors


Abstract

1. Introduction
2. Isotopes for RIT
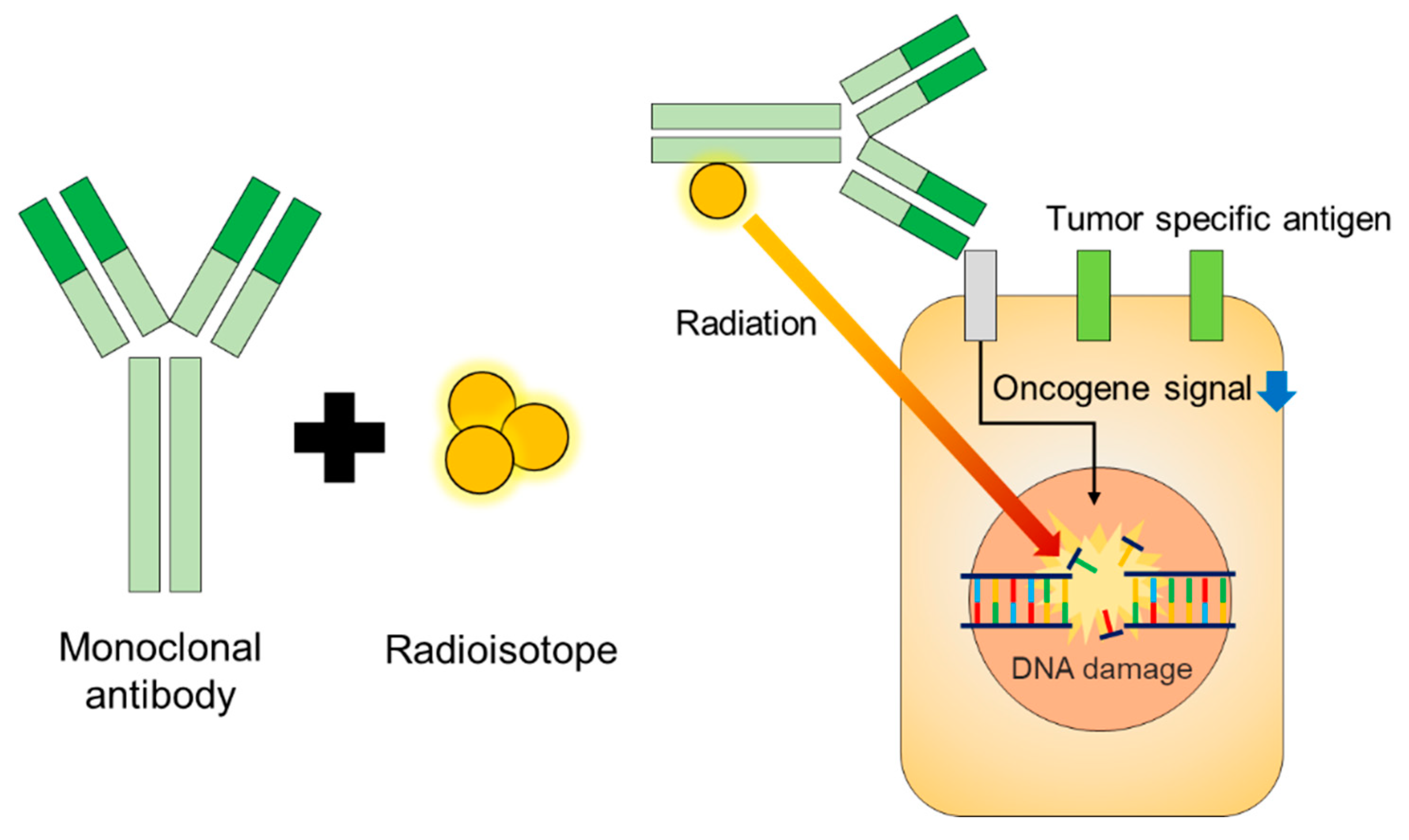
3. Mechanism of Therapeutic Monoclonal Antibodies
3.1. Antibody-Dependent Cellular Cytotoxicity (ADCC)
3.2. Complement-Dependent Cytotoxicity (CDC)
4. Mechanism of Currently Commercialized mAb for Radioimmunotherapy
4.1. Trastuzumab
4.2. Bevacizumab
4.3. Cetuximab
4.4. Rituximab
4.5. Immune Suppression Checkpoint Inhibitor
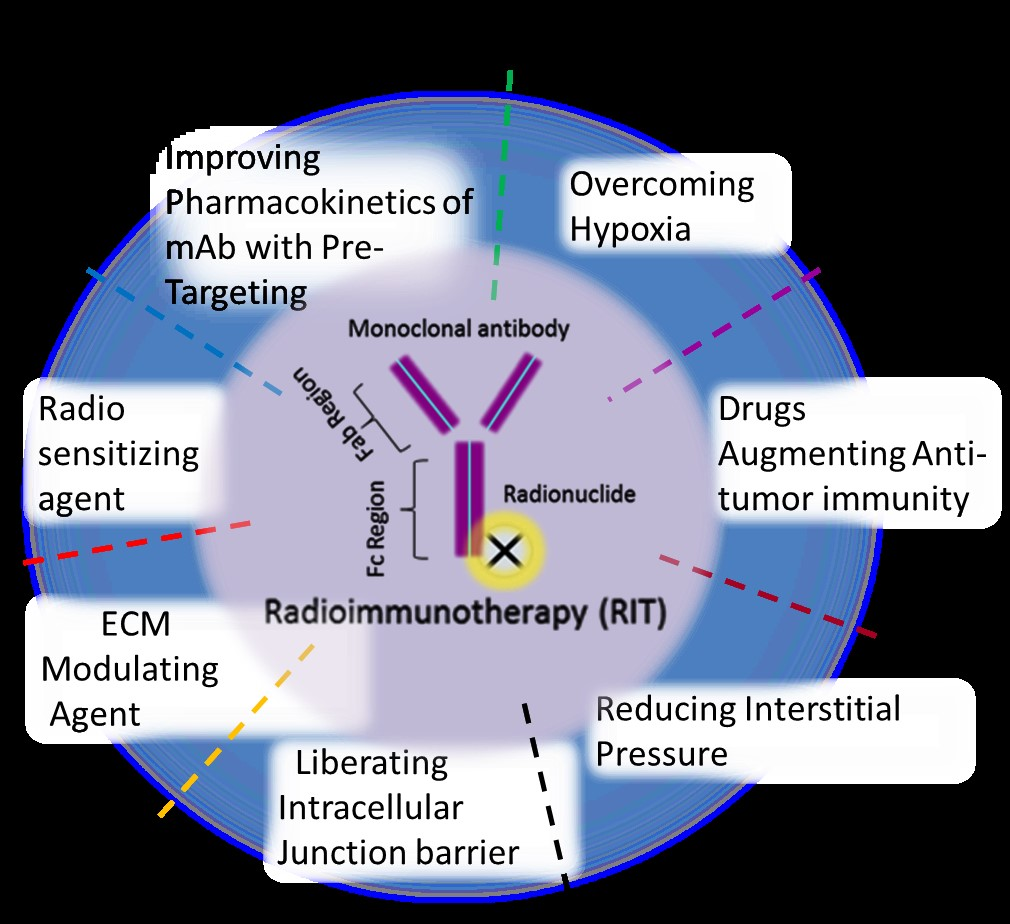
5. Limitation of RIT for Solid Tumors and Combination RIT Strategies
5.1. Extracellular Matrix (ECM)
5.2. Cell-to-Cell Junctions
5.3. High Interstitial Pressure
5.4. Immune Surveillance
5.5. Hypoxia
5.6. Combination of RIT with Other Agents
5.7. Improving Pharmacokinetics of mAb with Pre-Targeting
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RIT | Radioimmunotherapy |
| mAb | Monoclonal Antibody |
| ADCC | Antibody-dependent cellular cytotoxicity |
| ADCP | Antibody-dependent cellular phagocytosis |
| MAC | Membrane attack complex |
| CDC | Complement-dependent cytotoxicity |
| PI3K | Phosphatidylinositol 3-kinase |
| PLCv | Phospholipase v |
| PKC | Protein kinase C |
| MAPK | Mitogen activated protein kinase |
| KRAS | Kristen rat sarcoma |
| CMC | Cell-mediated cytotoxicity |
References
- Boiardi, A.; Bartolomei, M.; Silvani, A.; Eoli, M.; Salmaggi, A.; Lamperti, E.; Milanesi, I.; Botturi, A.; Rocca, P.; Bodei, L.; et al. Intratumoral delivery of mitoxantrone in association with 90-Y radioimmunotherapy (RIT) in recurrent glioblastoma. J. Neuro Oncol. 2005, 72, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Dietlein, M.; Dressler, J.; Farahati, J.; Leisner, B.; Moser, E.; Reiners, C.; Schicha, H.; Schober, O. Guidelines for radioiodine therapy (RIT) in differentiated thyroid cancer. Nuklearmedizin 1999, 38, 221–222. [Google Scholar] [PubMed]
- Dietlein, M.; Dressler, J.; Joseph, K.; Leisner, B.; Moser, E.; Reiners, C.; Schicha, H.; Schneider, P.; Schober, O. Guidelines for radioiodine therapy (RIT) in benign thyroid diseases. Nuklearmedizin 1999, 38, 219–220. [Google Scholar] [PubMed]
- Kim, J.S. Combination Radioimmunotherapy Approaches and Quantification of Immuno-PET. Nucl. Med. Mol. Imaging 2016, 50, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Kraeber-Bodéré, F.; Barbet, J.; Chatal, J.F. Radioimmunotherapy: From Current Clinical Success to Future Industrial Breakthrough? J. Nucl. Med. 2018, 115, 329–331. [Google Scholar] [CrossRef]
- Larson, S.M.; Carrasquillo, J.A.; Cheung, N.V.; Press, O.W. Radioimmunotherapy of human tumours. Nat. Rev. Cancer 2015, 15, 347–360. [Google Scholar] [CrossRef]
- Jacene, H.; Crandall, J.; Kasamon, Y.L.; Ambinder, R.F.; Piantadosi, S.; Serena, D.; Kasecamp, W.; Wahl, R.L. Initial Experience with Tositumomab and I-131-Labeled Tositumomab for Treatment of Relapsed/Refractory Hodgkin Lymphoma. Mol. Imaging Biol. 2017, 19. [Google Scholar] [CrossRef]
- Lim, I.; Park, J.Y.; Kang, H.J.; Hwang, J.P.; Lee, S.S.; Kim, K.M.; Choi, T.H.; Yang, S.H.; Kim, B.I.; Choi, C.W.; et al. Prognostic significance of pretreatment F-18-FDG PET/CT in patients with relapsed/refractory B-cell non-Hodgkin’s lymphoma treated by radioimmunotherapy using I-131-rituximab. Acta Haematol. 2013, 130, 74–82. [Google Scholar] [CrossRef]
- Provencio, M.F.F.; Gómez-Codina, J.; Quero Blanco, C.; Llanos, M.; Garcia-Arroyo, F.; de la Cruz, L.; Gumá, J.; Delgado, J.R.; Álvarez, R.; Chacón, J.I.; et al. Consolidation treatment with yttrium-90 ibritumomab tiuxetan after new induction regimen in advanced stage follicular lymphoma: Update results from the Spanish Lymphoma Oncology Group trial after a median follow-up of 8.5-years. Leuk. Lymphoma 2018, 20, 1–4. [Google Scholar] [CrossRef]
- Puronen, C.E.; Cassaday, R.D.; Stevenson, P.A.; Sandmaier, B.M.; Flowers, M.E.; Green, D.J.; Maloney, D.G.; Storb, R.F.; Press, O.W.; Gopal, A.K. Long-Term Follow-Up of Y-90-Ibritumomab Tiuxetan, Fludarabine, and Total Body Irradiation-Based Nonmyeloablative Allogeneic Transplant Conditioning for Persistent High-Risk B Cell Lymphoma. Biol. Blood Marrow Transplant. 2018, 24, 2211–2215. [Google Scholar] [CrossRef]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Factors determining antibody distribution in tumors. Trends Pharmacol. Sci. 2008, 29, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Ocean, A.J.; Pennington, K.L.; Guarino, M.J.; Sheikh, A.; Bekaii-Saab, T.; Serafini, A.N.; Lee, D.; Sung, M.W.; Gulec, S.A.; Goldsmith, S.J.; et al. Fractionated radioimmunotherapy with Y-90- clivatuzumab tetraxetan and low-dose gemcitabine is active in advanced pancreatic cancer: A phase 1 trial. Cancer 2012, 118, 5497–5506. [Google Scholar] [CrossRef] [PubMed]
- Forero-Torres, A.; Shen, S.; Breitz, H.; Sims, R.B.; Axworthy, D.B.; Khazaeli, M.B.; Chen, K.H.; Percent, I.; Besh, S.; LoBuglio, A.F.; et al. Pretargeted radioimmunotherapy (RIT) with a novel anti-TAG-72 fusion protein. Cancer Biother. Radiopharm. 2005, 20, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Buchsbaum, D.J. CD38 pretargeted RIT of B-cell tumors. Blood 2018, 131, 589–590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xing, J.; Zhang, Q.; Song, F.; Li, Y.; Yang, X.; Chen, Z. Optimal design of Ig 5’ primers for construction of diverse phage antibody library established to select anti-HAb18GEF and anti-DOTA-Y Fabs for hepatoma pretargeting RIT. Front. Biosci. 2006, 11, 1733–1749. [Google Scholar] [CrossRef] [PubMed][Green Version]
- DeNardo, S.J.; DeNardo, G.L.; Brush, J.; Carter, P. Phage library-derived human anti-TETA and anti-DOTA ScFv for pretargeting RIT. Hybridoma 1999, 18, 13–21. [Google Scholar] [CrossRef]
- Orlova, A.; Jonsson, A.; Rosik, D.; Lundqvist, H.; Lindborg, M.; Abrahmsen, L.; Ekblad, C.; Frejd, F.Y.; Tolmachev, V. Site-Specific Radiometal Labeling and Improved Biodistribution Using ABY-027, A Novel HER2-Targeting Affibody Molecule-Albumin-Binding Domain Fusion Protein. J. Nucl. Med. 2013, 54, 961–968. [Google Scholar] [CrossRef]
- Frost, S.H.; Back, T.; Chouin, N.; Hultborn, R.; Jacobsson, L.; Elgqvist, J.; Jensen, H.; Albertsson, P.; Lindegren, S. Comparison of At-211-PRIT and At-211 RIT of ovarian microtumors in a nude mouse model. Cancer Biother. Radiopharm. 2013, 28, 108–114. [Google Scholar] [CrossRef]
- Suzuki, M.; Kato, C.; Kato, A. Therapeutic antibodies: Their mechanisms of action and the pathological findings they induce in toxicity studies. J. Toxicol. Pathol. 2015, 28, 133–139. [Google Scholar] [CrossRef]
- Hudis, C.A. Trastuzumab—Mechanism of Action and Use in Clinical Practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef]
- Weiner, G.J. Rituximab: Mechanism of Action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Dhodapkar, M.V.; Ferrone, S. Monoclonal antibodies for cancer immunotherapy. Lancet 2009, 373, 1033–1040. [Google Scholar] [CrossRef]
- Cavallo, F.; Calogero, R.A.; Forni, G. Are oncoantigens suitable targets for anti-tumour therapy? Nat. Rev. Cancer 2007, 7, 707. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-Y.; Racila, E.; Taylor, R.P.; Weiner, G.J. NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood 2008, 111, 1456–1463. [Google Scholar] [CrossRef]
- Gül, N.; van Egmond, M. Antibody-Dependent Phagocytosis of Tumor Cells by Macrophages: A Potent Effector Mechanism of Monoclonal Antibody Therapy of Cancer. Cancer Res. 2015, 75, 5008–5013. [Google Scholar] [CrossRef]
- Lopez, J.A.; Susanto, O.; Jenkins, M.R.; Lukoyanova, N.; Sutton, V.R.; Law, R.H.P.; Johnston, A.; Bird, C.H.; Bird, P.I.; Whisstock, J.C.; et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood 2013, 121, 2659–2668. [Google Scholar] [CrossRef]
- Shresta, S.; MacIvor, D.M.; Heusel, J.W.; Russell, J.H.; Ley, T.J. Natural killer and lymphokine-activated killer cells require granzyme B for the rapid induction of apoptosis in susceptible target cells. Proc. Natl. Acad. Sci. USA 1995, 92, 5679–5683. [Google Scholar] [CrossRef]
- Patrick, M.; Glassman, J.P.B. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol. Med. 2014, 11, 20–33. [Google Scholar]
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2. [Google Scholar] [CrossRef]
- Ellis, L.M. Mechanisms of Action of Bevacizumab as a Component of Therapy for Metastatic Colorectal Cancer. Semin. Oncol. 2006, 33, S1–S7. [Google Scholar] [CrossRef]
- Hecht, J.R.; Mitchell, E.; Chidiac, T.; Scroggin, C.; Hagenstad, C.; Spigel, D.; Marshall, J.; Cohn, A.; McCollum, D.; Stella, P.; et al. A Randomized Phase IIIB Trial of Chemotherapy, Bevacizumab, and Panitumumab Compared with Chemotherapy and Bevacizumab Alone for Metastatic Colorectal Cancer. J. Clin. Oncol. 2009, 27, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Tol, J.; Koopman, M.; Cats, A.; Rodenburg, C.J.; Creemers, G.J.M.; Schrama, J.G.; Erdkamp, F.L.G.; Vos, A.H.; van Groeningen, C.J.; Sinnige, H.A.M.; et al. Chemotherapy, Bevacizumab, and Cetuximab in Metastatic Colorectal Cancer. N. Engl. J. Med. 2009, 360, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, L.E.; Golijanin, D.; Itin, A.; Pode, D.; Keshet, E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J. Clin. Investig. 1999, 103, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Inai, T.; Mancuso, M.; Hashizume, H.; Baffert, F.; Haskell, A.; Baluk, P.; Hu-Lowe, D.D.; Shalinsky, D.R.; Thurston, G.; Yancopoulos, G.D.; et al. Inhibition of Vascular Endothelial Growth Factor (VEGF) Signaling in Cancer Causes Loss of Endothelial Fenestrations, Regression of Tumor Vessels, and Appearance of Basement Membrane Ghosts. Am. J. Pathol. 2004, 165, 35–52. [Google Scholar] [CrossRef]
- Jang, B.S.; Lee, S.M.; Kim, H.S.; Shin, I.S.; Razjouyan, F.; Wang, S.; Yao, Z.; Pastan, I.; Dreher, M.R.; Paik, C.H. Combined-modality radioimmunotherapy: Synergistic effect of paclitaxel and additive effect of bevacizumab. Nucl. Med. Biol. 2012, 39, 472–483. [Google Scholar] [CrossRef][Green Version]
- Arjaans, M.; Oude Munnink, T.H.; Oosting, S.F.; Terwisscha van Scheltinga, A.G.T.; Gietema, J.A.; Garbacik, E.T.; Timmer-Bosscha, H.; Lub-de Hooge, M.N.; Schröder, C.P.; de Vries, E.G.E. Bevacizumab-Induced Normalization of Blood Vessels in Tumors Hampers Antibody Uptake. Cancer Res. 2013, 73, 3347–3355. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Zhuang, H.; Xue, Z.; Wang, L.; Li, X.; Zhang, N.; Zhang, R. Efficacy and Immune Mechanisms of Cetuximab for the Treatment of Metastatic Colorectal Cancer. Clin. Oncol. Cancer Res. 2011, 8, 207–214. [Google Scholar] [CrossRef]
- Vincenzi, B.; Santini, D.; Tonini, G. New Issues on Cetuximab Mechanism of Action in Epidermal Growth Factor Receptor–Negative Colorectal Cancer: The Role of Vascular Endothelial Growth Factor. J. Clin. Oncol. 2006, 24, 1957–1965. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, M. Immune Checkpoint Blockade: A CommonDenominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Andrade de Mello, R.; Flávia Veloso, A.; Esrom Catarina, P.; Nadine, S.; Antoniou, G. Potential role of immunotherapy in advanced non-small-cell lung cancer. Oncol. Targets Ther. 2016, 10, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Khaibullina, A.; Jang, B.S.; Sun, H.; Le, N.; Yu, S.; Frenkel, V.; Carrasquillo, J.A.; Pastan, I.; Li, K.C.; Paik, C.H. Pulsed high-intensity focused ultrasound enhances uptake of radiolabeled monoclonal antibody to human epidermoid tumor in nude mice. J. Nucl. Med. 2008, 49, 295–302. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 2014, 1243–1253. [Google Scholar] [CrossRef]
- González-Mariscal, L.; Betanzos, A.; Nava, P.; Jaramillo, B.E. Tight junction proteins. Prog. Biophys. Mol. Biol. 2003, 81, 1–44. [Google Scholar] [CrossRef]
- Alain, P.; Boucher, Y.; Saroja, R.; McKee, T.D.; Gohongi, T.; Emmanuelle di, T.; Brown, E.B.; Yotaro, I.; Campbell, R.B.; Berk, D.A.; et al. Role of tumor–host interactions in interstitial diffusion of macromolecules: Cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 4628–4633. [Google Scholar]
- Navarro-Teulon, I.; Lozza, C.; Pèlegrin, A.; Vivès, E.; Pouget, J.P. General overview of radioimmunotherapy of solid tumors. Immunotherapy 2013, 5, 467–487. [Google Scholar] [CrossRef]
- Marc, Y.; David, A.; Cathy, M.; Patrick, F.; François, Q.; Bernard, S.-A.; Philippe, R.; Monique, P.; Caroline, B.-M.; Dominique, G.; et al. Adjuvant Radioimmunotherapy Trialwith Iodine-131^ Labeled Anti Carcinoembryonic AntigenMonoclonal Antibody F6 F(ab¶)2 after Resection of LiverMetastases from Colorectal Cancer. Clin. Cancer Res. 2008, 14, 3487–3493. [Google Scholar]
- Crittenden, M.; Kohrt, H.; Levy, R.; Jones, J.; Camphausen, K.; Dicker, A.; Demaria, S.; Formenti, S. Current Clinical Trials Testing Combinations of Immunotherapy and Radiation. Semin. Radiat. Oncol. 2015, 25, 54–64. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2018, 196, 395–406. [Google Scholar] [CrossRef]
- Hu, G.; Li, L.; Xu, W. Extracellular matrix in mammary gland development and breast cancer progression. Front. Lab. Med. 2017, 1, 36–39. [Google Scholar] [CrossRef]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of Extracellular Matrix Assembly in Interstitial Transport in Solid Tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar] [PubMed]
- Live Eikenes, Ø.S.B.; Christian, B.; Catharina de Lange, D. Collagenase Increases the Transcapillary Pressure Gradient and Improves the Uptake and Distribution of Monoclonal Antibodies in Human Osteosarcoma Xenografts. Cancer Res. 2004, 64, 4768–4773. [Google Scholar] [CrossRef] [PubMed]
- Váradi, T.; Mersich, T.; Auvinen, P.; Tammi, R.; Tammi, M.; Salamon, F.; Besznyák, I., Jr.; Jakab, F.; Baranyai, Z.; Szöllősi, J.; et al. Binding of trastuzumab to ErbB2 is inhibited by a high pericellular density of hyaluronan. J. Histochem. Cytochem. 2012, 60, 567–575. [Google Scholar] [CrossRef]
- Pan, A.; Wang, Z.; Chen, B.; Dai, W.; Zhang, H.; He, B.; Wang, X.; Wang, Y.; Zhang, Q. Localized co-delivery of collagenase and trastuzumab by thermosensitive hydrogels for enhanced antitumor efficacy in human breast xenograft. Drug Deliv. 2018, 25, 1495–1503. [Google Scholar] [CrossRef]
- Chon, H.J.; Lee, W.S.; Yang, H.; Kong, S.J.; Lee, N.K.; Moon, E.S.; Choi, J.; Han, E.C.; Kim, J.H.; Ahn, J.B.; et al. Tumor Microenvironment Remodeling by Intratumoral Oncolytic Vaccinia Virus Enhances the Efficacy of Immune-Checkpoint Blockade. Clin. Cancer Res. 2019, 25, 1612–1623. [Google Scholar] [CrossRef]
- Anderson, J.M.; Van Itallie, C.M. Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 2009, 1, a002584. [Google Scholar] [CrossRef]
- Ines, B.; Hua, C.; Jonas, P.; Roma, Y.; Andre, L. A New Epithelial Junction Opener for Cancer Therapy. Mol. Ther. 2013, 21, S78–S79. [Google Scholar]
- Wang, H.; Li, Z.-Y.; Liu, Y.; Persson, J.; Beyer, I.; Möller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.-B.; et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 2010, 17, 96. [Google Scholar] [CrossRef]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High Interstitial Fluid Pressure—An Obstacle In Cancer Therapy. Nature 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.; Schwartz, J.N.; Hagerty, K.L.; Allred, D.C.; Cote, R.J.; Dowsett, M.; Fitzgibbons, P.L.; Hanna, W.M.; Langer, A.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J. Clin. Oncol. 2007, 25, 118–145. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Tumor Vasculature: Improving Drug Delivery and Efficacy. Trends Cancer 2018, 4, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Transport of Molecules in the Tumor Interstitium: A Review. Cancer Res. 1987, 47, 3039–3051. [Google Scholar] [PubMed]
- Young, J.S.; Llumsden, C.E.; Stalker, A.L. The significance of the “tissue pressure” of normal testicular and of neoplastic (Brown-Pearce carcinoma) tissue in the rabbit. J. Pathol. Bacteriol. 1950, 62, 313–333. [Google Scholar] [CrossRef]
- Li, T.; Wang, Y.N.; Khokhlova, T.D.; D’Andrea, S.; Starr, F.; Chen, H.; McCune, J.S.; Risler, L.J.; Mashadi-Hossein, A.; Hingorani, S.R.; et al. Pulsed high intensity focused ultrasound (pHIFU) enhances delivery of doxorubicin in a preclinical model of pancreatic cancer. Cancer Res. 2015, 75, 3738–3746. [Google Scholar] [CrossRef]
- Wang, S.; Shin, I.S.; Hancock, H.; Jang, B.-S.; Kim, H.-S.; Lee, S.M.; Zderic, V.; Frenkel, V.; Pastan, I.; Paik, C.H.; et al. Pulsed high intensity focused ultrasound increases penetration and therapeutic efficacy of monoclonal antibodies in murine xenograft tumors. J. Control. Release 2012, 162, 218–224. [Google Scholar] [CrossRef]
- Griffon-Etienne, G.; Boucher, Y.; Brekken, C.; Suit, H.D.; Jain, R.K. Taxane-induced Apoptosis Decompresses Blood Vessels and Lowers Interstitial Fluid Pressure in Solid Tumors. Clin. Implic. 1999, 59, 3776–3782. [Google Scholar]
- De Souza, A.P.; Bonorino, C. Tumor immunosuppressive environment: Effects on tumor-specific and nontumor antigen immune responses. Expert Rev. Anticancer Ther. 2009, 9, 1317–1332. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Principles of Tumor Suppression. Cell 2004, 116, 235–246. [Google Scholar] [CrossRef]
- Azad, A.; Yin Lim, S.; D’Costa, Z.; Jones, K.; Diana, A.; Sansom, O.J.; Kruger, P.; Liu, S.; McKenna, W.G.; Dushek, O.; et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol. Med. 2017, 9, 167–180. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Formenti, S.C.; Demaria, S. Toward Precision Radiotherapy for Use with Immune Checkpoint Blockers. Clin. Cancer Res. 2018, 24, 259–265. [Google Scholar] [CrossRef]
- Rockwell, S.; Dobrucki, I.T.; Kim, E.Y.; Marrison, S.T.; Vu, V.T. Hypoxia and radiation therapy: Past history, ongoing research, and future promise. Curr. Mol. Med. 2009, 9, 442–458. [Google Scholar] [CrossRef]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2010; pp. 92–93. [Google Scholar]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Hashimoto, T.; Shibasaki, F. Hypoxia-inducible factor as an angiogenic master switch. Front. Pediatr. 2015, 3, 33. [Google Scholar] [CrossRef]
- Harada, H.; Kizaka-Kondoh, S.; Li, G.; Itasaka, S.; Shibuya, K.; Inoue, M.; Hiraoka, M. Significance of HIF-1-active cells in angiogenesis and radioresistance. Oncogene 2007, 26, 7508–7516. [Google Scholar] [CrossRef]
- Xu, S.; Tang, Y.Y.; Yu, Y.X.; Yun, Q.; Yang, J.P.; Zhang, H.; Peng, Q.; Sun, X.; Yang, L.L.; Fu, S.; et al. Novel composite drug delivery system as a novel radio sensitizer for the local treatment of cervical carcinoma. Drug Deliv. 2017, 24, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.; Kramer, E.; Liebes, L.; Wasserheit, C.; Hochster, H.; Blank, E.; Ceriani, R.; Furmanski, P. Radiosensitization of Tumor-targeted Radioimmunotherapy with Prolonged Topotecan Infusion in Human Breast Cancer Xenografts. Cancer Res. 2001, 61, 2996–3001. [Google Scholar] [PubMed]
- Milenic, D.E.; Garmestani, K.; Brady, E.D.; Albert, P.S.; Abdulla, A.; Flynn, J.; Brechbiel, M.W. Potentiation of High-LET Radiation by Gemcitabine:Targeting HER2 withTrastuzumab toTreat Disseminated Peritoneal Disease. Clin. Cancer Res. 2007, 13, 1926–1935. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.J.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W. Impact of a-Targeted Radiation Therapy on Gene Expression in a Pre-Clinical Model for Disseminated Peritoneal Disease when Combined with Paclitaxel. PLoS ONE 2014, 9, e108511. [Google Scholar] [CrossRef]
- Cividalli, A.; Arcangeli, G.; Cruciani, G.; Livdi, E.; Cordelli, E.; Danesi, D.T. Enhancement of Radiation Response by Paclitaxel in Mice According to Different Treatment Schedules. Int. J. Radiat. Oncol. Biol. Phys. 1998, 40, 1163–1170. [Google Scholar] [CrossRef]
- Kelly, M.P.; Lee, F.T.; Smyth, F.E.; Brechbiel, M.W.; Scott, A.M. Enhanced Efficacy of 90Y-Radiolabeled Anti-Lewis Y Humanized Monoclonal Antibody hu3S193 and Paclitaxel Combined-Modality Radioimmunotherapy in a Breast Cancer Model. J. Nucl. Med. 2018, 47, 716–725. [Google Scholar]
- Kurizaki, T.; Okazaki, S.; Sanderson, S.D.; Colcher, D.; Enke, C.A.; Tempero, M.A.; Baranowska-Kortylewicz, J. Potentiation of Radioimmunotherapy with Response-Selective Peptide Agonist of Human C5a. J. Nucl. Med. 2002, 43, 957–967. [Google Scholar]
- Koppe, M.J.; Oyen, W.J.; Bleichrodt, R.P.; Hendriks, T.; Verhofstad, A.A.; Goldenberg, D.M.; Boerman, O.C. Combination therapy using the cyclooxygenase-2 inhibitor Parecoxib and radioimmunotherapy in nude mice with small peritoneal metastases of colonic origin. Cancer Immunol. Immunother. 2006, 55, 47–55. [Google Scholar] [CrossRef]
- Kosaka, A.; Ohkuri, T.; Okada, H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol. Immunother. 2014, 63, 847–857. [Google Scholar] [CrossRef]
- Reilly, R.M. Radioimmunotherapy of Solid Tumors: The Promise of Pretargeting Strategies Using Bispecific Antibodies and Radiolabeled Haptens. J. Nucl. Med. 2006, 47, 196–199. [Google Scholar]
- Paganelli, G.; Bartolomei, M.; Ferrari, M.; Cremonesi, M.; Broggi, G.; Maira, C.; Sturiale, C.; Grana, C.; Prisco, G.; Gatti, M.; et al. Pre-Targeted Locoregional Radioimmunotherapy with 90Y-biotin in Glioma Patients: Phase I Study and Preliminary Therapeutic Results. Cancer Biother. Radiopharm. 2001, 16, 227–235. [Google Scholar] [CrossRef]
- Axworthy, D.B.; Reno, J.M.; Hylarides, M.D.; Mallett, R.W.; Theodore, L.J.; Gustavson, L.M.; Su, F.; Hobson, L.J.; Beaumier, P.L.; Fritzberg, A.R. Cure of human carcinoma xenografts by a single dose of pretargeted Y-90 with negligible toxicity. Proc. Natl. Acad. Sci. USA 2000, 97, 1802–1807. [Google Scholar] [CrossRef] [PubMed]
- Graves, S.S.; Dearstyne, E.; Lin, Y.; Zuo, Y.; Sanderson, J.; Schultz, J.; Pantalias, A.; Gray, D.; Axworthy, D.; Jones, H.M.; et al. Combination Therapy with Pretarget CC49 Radioimmunotherapy and Gemcitabine Prolongs Tumor Doubling Time in a Murine Xenograft Model of Colon Cancer More Effectively Than Either Monotherapy. Clin. Cancer Res. 2003, 9, 3712–3721. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radioisotopes | Max Range (in Water) | Half-Life | Max Energy (keV) |
|---|---|---|---|
| β-emitter | |||
| 67Cu | 2.1 mm | 61.9 h | 575 |
| 90Y | 11.3 mm | 64.1 h | 2284 |
| 131I | 2.3 mm | 8.0 days | 606 |
| 177Lu | 1.8 mm | 6.7 days | 497 |
| α-emitter | |||
| 211At | <50 μm | 7.2 h | 586 |
| 213Bi | <50 μm | 45.6 min | 5870 |
| 225Ac | <50 μm | 240 h | 5830 |
| 223Ra | <100 μm | 11.4 days | 5979 |
| Monoclonal Antibody | Targeted Site |
|---|---|
| Trastuzumab (Herceptin, Roche, Basel, Switzerland) | HER2 |
| Bevacizumab (Avastin, Genentech, CA, USA) | VEGF-A |
| Cetuximab (Erbitux, Kenilworth, NJ, USA) | EGFR |
| Rituximab (Rituxan, Genentech, CA, USA) | CD20 |
| Tositumomab (Bexxar, Genentech, CA, USA) | CD20 |
| Ibritumomab tiuxetan (Zevalin, Biogen Idec, Cambridge, MA, USA) | CD20 |
| Aim | Radionuclide | Cancer Type | Targeted Antigen | Subject | Combination | Reference |
|---|---|---|---|---|---|---|
| To determine maximum tolerated dose and antitumor efficacy of three-step pre-targeting method | 90Y | Glioma | Tenascin | Human | No | [92] |
| Safe and effective with negligible toxicity pre-targeting RIT | 90Y | LS-180, human carcinoma | Ep-CAM | BALB/c nude mice | No | [93] |
| Response to combination pre-targeted with less toxicity | 90Y | LS174T, human colon adenocarcinoma | Tag-72 (CC49) | BALB/c nude mice | Gemcitabine | [94] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaheer, J.; Kim, H.; Lee, Y.-J.; Kim, J.S.; Lim, S.M. Combination Radioimmunotherapy Strategies for Solid Tumors. Int. J. Mol. Sci. 2019, 20, 5579. https://doi.org/10.3390/ijms20225579
Zaheer J, Kim H, Lee Y-J, Kim JS, Lim SM. Combination Radioimmunotherapy Strategies for Solid Tumors. International Journal of Molecular Sciences. 2019; 20(22):5579. https://doi.org/10.3390/ijms20225579
Chicago/Turabian StyleZaheer, Javeria, Hyeongi Kim, Yong-Jin Lee, Jin Su Kim, and Sang Moo Lim. 2019. "Combination Radioimmunotherapy Strategies for Solid Tumors" International Journal of Molecular Sciences 20, no. 22: 5579. https://doi.org/10.3390/ijms20225579
APA StyleZaheer, J., Kim, H., Lee, Y.-J., Kim, J. S., & Lim, S. M. (2019). Combination Radioimmunotherapy Strategies for Solid Tumors. International Journal of Molecular Sciences, 20(22), 5579. https://doi.org/10.3390/ijms20225579