Isoquinolinamine FX-9 Exhibits Anti-Mitotic Activity in Human and Canine Prostate Carcinoma Cell Lines

 , , ,
, , ,  , ,
, ,
Abstract
1. Introduction
2. Results
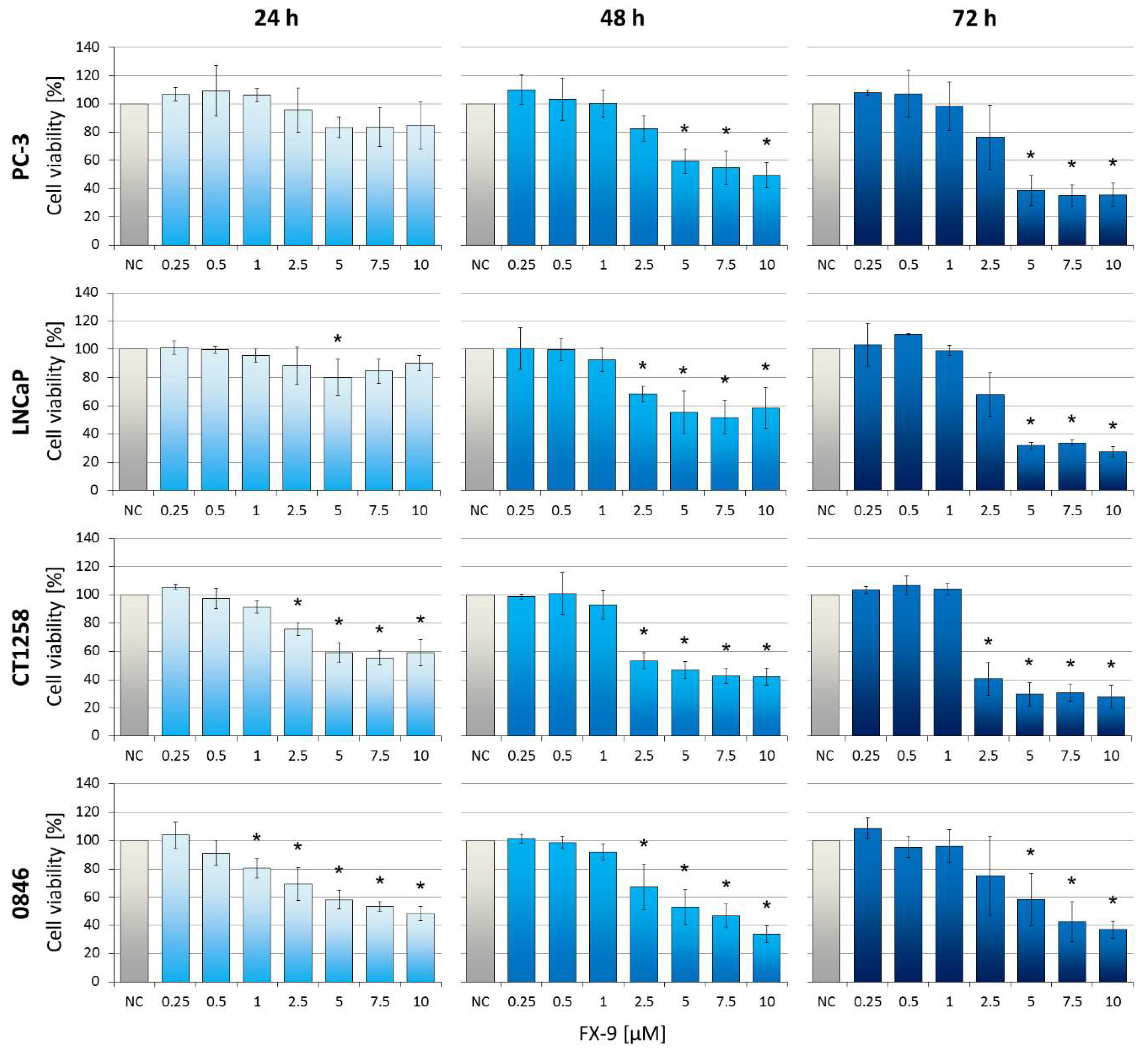
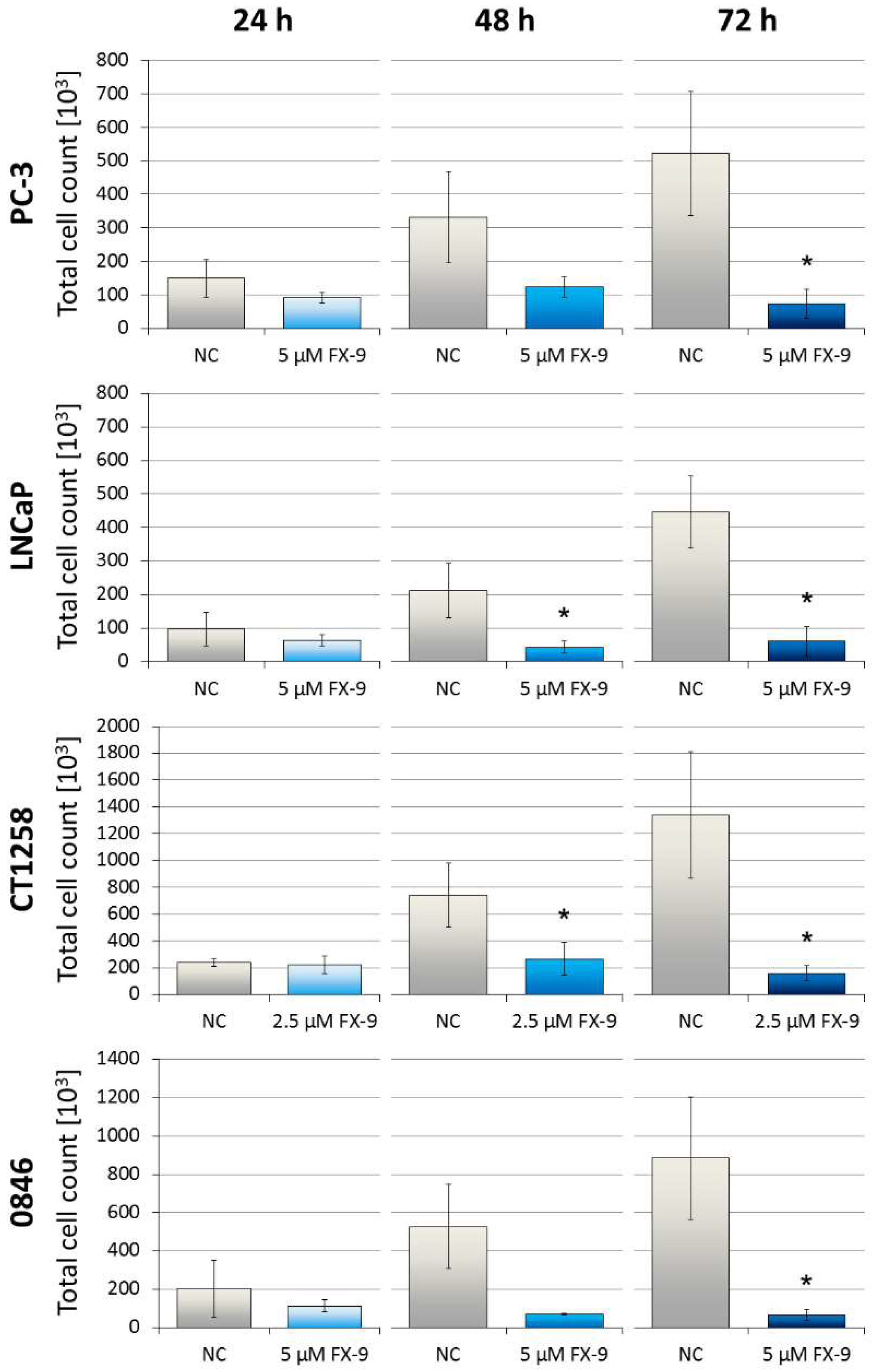
2.1. Decrease of Cell Viability in Prostate Carcinoma Cells
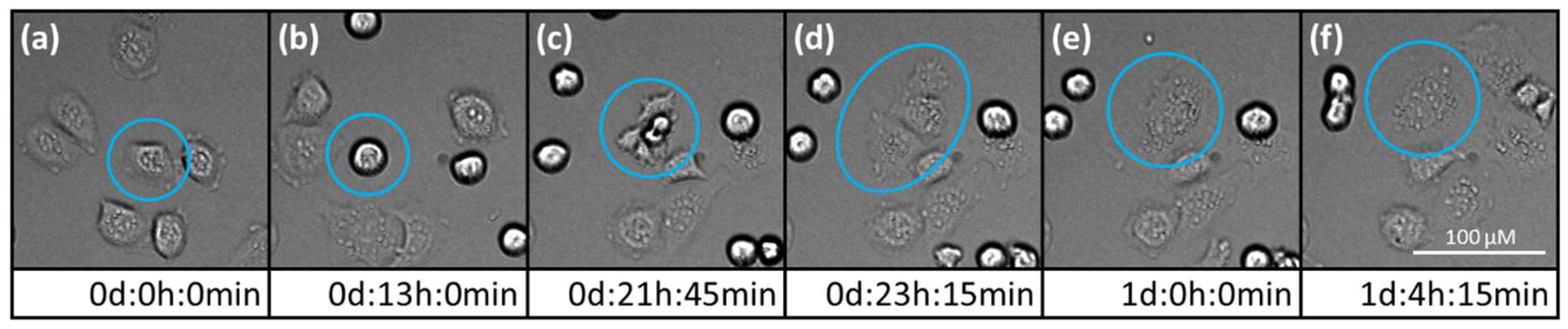
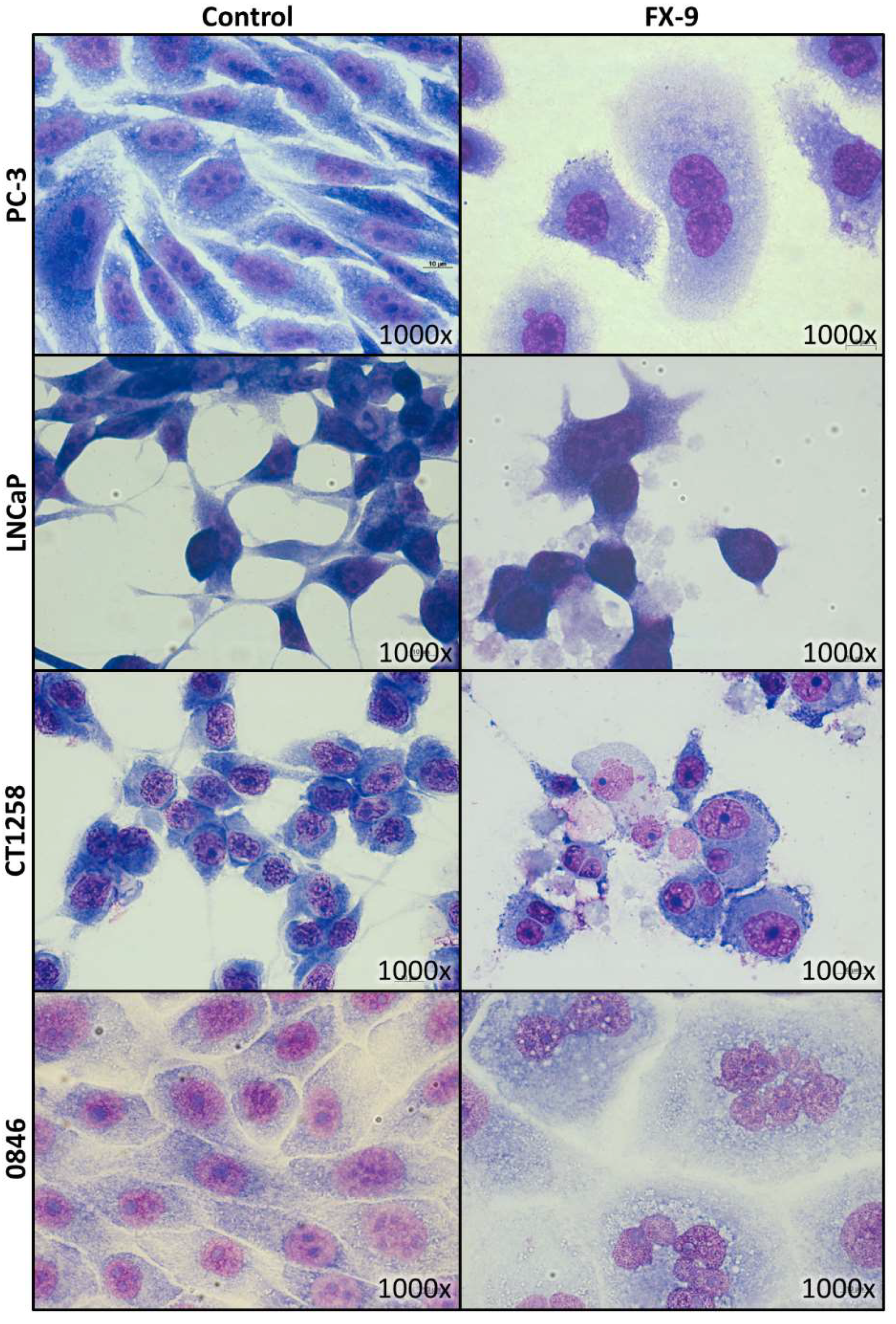
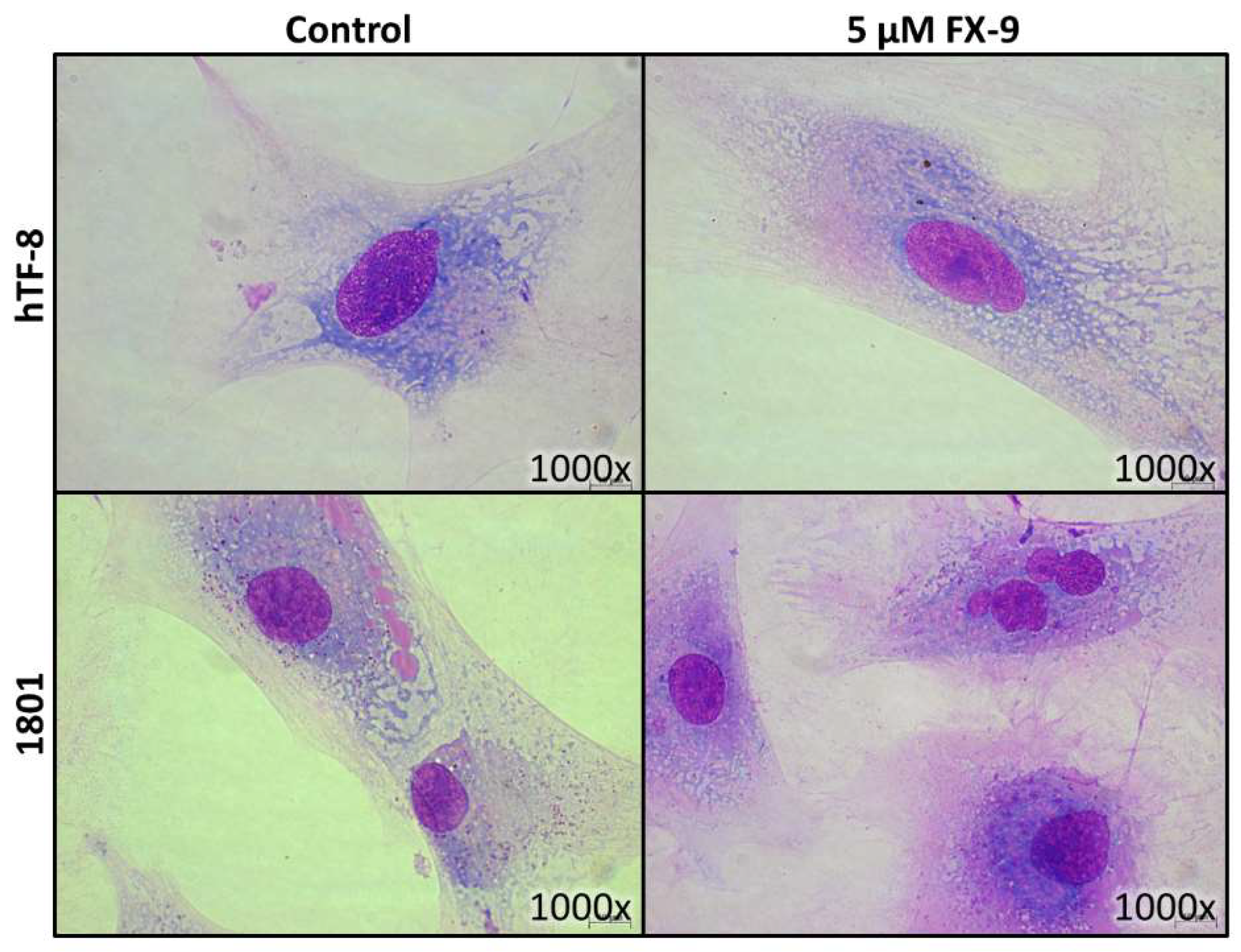
2.2. Morphological Changes in Prostate Carcinoma Cells
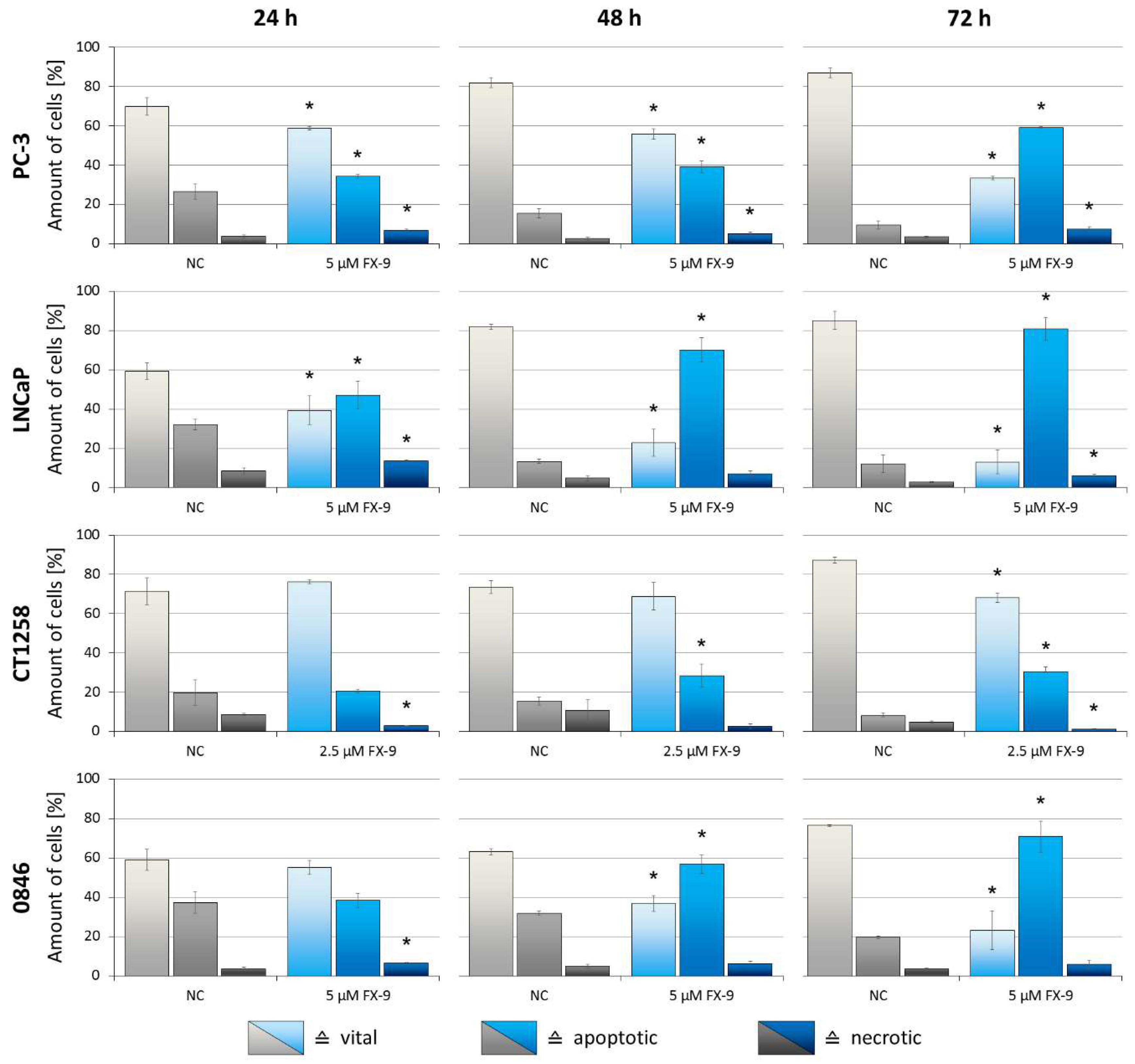
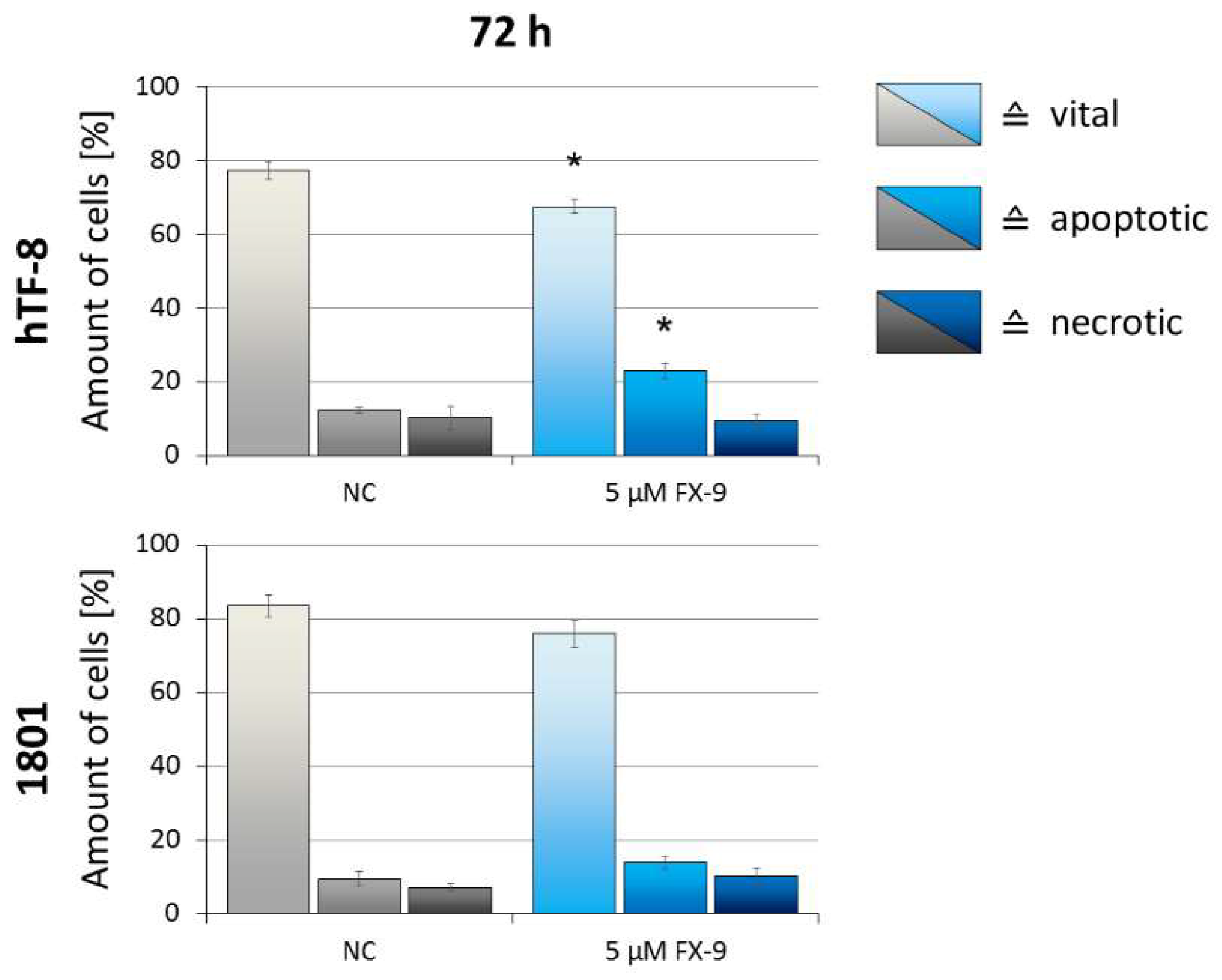
2.3. Induction of Apoptosis in Prostate Carcinoma Cells
2.4. Cell Cycle Arrest
2.5. Effects on Benign Cells
3. Discussion
4. Materials and Methods
4.1. Isoquinolinamine FX-9
4.2. Cell Lines and Cultivation
4.3. MTS Assay
4.4. Cell Count Analysis
4.5. Live Cell Imaging
4.6. May-Grünwald-Giemsa Staining
4.7. Analysis of Apoptosis
4.8. Cell Cycle Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PCa | prostate cancer |
| ALL | acute lymphoblastic leukemia |
| DMSO | dimethyl sulfoxide |
| SD | standard deviation |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Quinn, D.I.; Sandler, H.M.; Horvath, L.G.; Goldkorn, A.; Eastham, J.A. The evolution of chemotherapy for the treatment of prostate cancer. Ann. Oncol. 2017, 28, 2658–2669. [Google Scholar] [CrossRef] [PubMed]
- Kregel, S.; Chen, J.L.; Tom, W.; Krishnan, V.; Kach, J.; Brechka, H.; Fessenden, T.B.; Isikbay, M.; Paner, G.P.; Szmulewitz, R.Z.; et al. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget 2016, 7, 26259–26274. [Google Scholar] [CrossRef]
- Chong, J.T.; Oh, W.K.; Liaw, B.C. Profile of apalutamide in the treatment of metastatic castration-resistant prostate cancer: Evidence to date. Onco Targets Ther. 2018, 11, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef]
- Hoang, D.T.; Iczkowski, K.A.; Kilari, D.; See, W.; Nevalainen, M.T. Androgen receptor-dependent and -independent mechanisms driving prostate cancer progression: Opportunities for therapeutic targeting from multiple angles. Oncotarget 2017, 8, 3724–3745. [Google Scholar] [CrossRef]
- Leroy, B.E.; Northrup, N. Prostate cancer in dogs: Comparative and clinical aspects. Vet. J. 2009, 180, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Reimann-Berg, N.; Willenbrock, S.; Murua Escobar, H.; Eberle, N.; Gerhauser, I.; Mischke, R.; Bullerdiek, J.; Nolte, I. Two new cases of polysomy 13 in canine prostate cancer. Cytogenet. Genome Res. 2011, 132, 16–21. [Google Scholar] [CrossRef]
- Vail, D.M.; MacEwen, E.G. Spontaneously occurring tumors of companion animals as models for human cancer. Cancer Investig. 2000, 18, 781–792. [Google Scholar] [CrossRef]
- Park, J.S.; Withers, S.S.; Modiano, J.F.; Kent, M.S.; Chen, M.; Luna, J.I.; Culp, W.T.; Sparger, E.E.; Rebhun, R.B.; Monjazeb, A.M.; et al. Canine cancer immunotherapy studies: Linking mouse and human. J. Immunother. Cancer 2016, 4, 97. [Google Scholar] [CrossRef] [PubMed]
- Cornell, K.K.; Bostwick, D.G.; Cooley, D.M.; Hall, G.; Harvey, H.J.; Hendrick, M.J.; Pauli, B.U.; Render, J.A.; Stoica, G.; Sweet, D.C.; et al. Clinical and pathologic aspects of spontaneous canine prostate carcinoma: A retrospective analysis of 76 cases. Prostate 2000, 45, 173–183. [Google Scholar] [CrossRef]
- Bennett, T.C.; Matz, B.M.; Henderson, R.A.; Straw, R.C.; Liptak, J.M.; Selmic, L.E.; Collivignarelli, F.; Buracco, P. Total prostatectomy as a treatment for prostatic carcinoma in 25 dogs. Vet. Surg. 2018, 47, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Cunto, M.; Mariani, E.; Anicito Guido, E.; Ballotta, G.; Zambelli, D. Clinical approach to prostatic diseases in the dog. Reprod. Domest. Anim. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ravicini, S.; Baines, S.J.; Taylor, A.; Amores-Fuster, I.; Mason, S.L.; Treggiari, E. Outcome and prognostic factors in medically treated canine prostatic carcinomas: A multi-institutional study. Vet. Comp. Oncol. 2018, 16, 450–458. [Google Scholar] [CrossRef]
- Walji, A.M.; Hostetler, E.D.; Selnick, H.; Zeng, Z.; Miller, P.; Bennacef, I.; Salinas, C.; Connolly, B.; Gantert, L.; Holahan, M.; et al. Discovery of 6-(Fluoro-(18)F)-3-(1H-pyrrolo[2,3-c]pyridin-1-yl)isoquinolin-5-amine ([(18)F]-MK-6240): A Positron Emission Tomography (PET) Imaging Agent for Quantification of Neurofibrillary Tangles (NFTs). J. Med. Chem. 2016, 59, 4778–4789. [Google Scholar] [CrossRef] [PubMed]
- Lohith, T.G.; Bennacef, I.; Vandenberghe, R.; Vandenbulcke, M.; Salinas, C.A.; Declercq, R.; Reynders, T.; Telan-Choing, N.F.; Riffel, K.; Celen, S.; et al. Brain Imaging of Alzheimer Dementia Patients and Elderly Controls with (18)F-MK-6240, a PET Tracer Targeting Neurofibrillary Tangles. J. Nucl. Med. 2019, 60, 107–114. [Google Scholar] [CrossRef]
- Rombouts, F.J.R.; Declercq, L.; Andres, J.I.; Bottelbergs, A.; Chen, L.; Iturrino, L.; Leenaerts, J.E.; Marien, J.; Song, F.; Wintmolders, C.; et al. Discovery of N-(4-[(18)F]Fluoro-5-methylpyridin-2-yl)isoquinolin-6-amine (JNJ-64326067), a New Promising Tau Positron Emission Tomography Imaging Tracer. J. Med. Chem. 2019, 62, 2974–2987. [Google Scholar] [CrossRef] [PubMed]
- Beaton, H.; Hamley, P.; Nicholls, D.J.; Tinker, A.C.; Wallace, A.V. 3,4-Dihydro-1-isoquinolinamines: A novel class of nitric oxide synthase inhibitors with a range of isoform selectivity and potency. Bioorganic Med. Chem. Lett. 2001, 11, 1023–1026. [Google Scholar] [CrossRef]
- Gossnitzer, E.; Punkenhofer, A.; Amon, A.; Favre, B. Novel high energy intermediate analogues with triazasterol-related structures as inhibitors of ergosterol biosynthesis. III. Synthesis and antifungal activity of N4-alkyl-1,6,7,11b-tetrahydro-2H-pyrimido[4,3-a]isoquinolin-4-amine salts. Eur. J. Pharm. Sci. 2003, 19, 151–164. [Google Scholar] [CrossRef]
- Khadka, D.B.; Woo, H.; Yang, S.H.; Zhao, C.; Jin, Y.; Le, T.N.; Kwon, Y.; Cho, W.J. Modification of 3-arylisoquinolines into 3,4-diarylisoquinolines and assessment of their cytotoxicity and topoisomerase inhibition. Eur. J. Med. Chem. 2015, 92, 583–607. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, C.E.; Hoffman, M.M.; Bhattacharjee, A.K.; Milhous, W.K.; Gerena, L. In vitro efficacy of 7-benzylamino-1-isoquinolinamines against Plasmodium falciparum related to the efficacy of chalcones. Bioorganic Med. Chem. Lett. 2011, 21, 786–789. [Google Scholar] [CrossRef]
- Yang, S.H.; Van, H.T.; Le, T.N.; Khadka, D.B.; Cho, S.H.; Lee, K.T.; Lee, E.S.; Lee, Y.B.; Ahn, C.H.; Cho, W.J. Development of 3-aryl-1-isoquinolinamines as potent antitumor agents based on CoMFA. Eur. J. Med. Chem. 2010, 45, 5493–5497. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.S.; Choi, H.E.; Shin, J.S.; Cho, Y.W.; Choi, J.H.; Cho, W.J.; Lee, K.T. 6,7-Dimethoxy-3-(3-methoxyphenyl)isoquinolin-1-amine induces mitotic arrest and apoptotic cell death through the activation of spindle assembly checkpoint in human cervical cancer cells. Carcinogenesis 2013, 34, 1852–1860. [Google Scholar] [CrossRef]
- Belzacq-Casagrande, A.S.; Bachelot, F.; De Oliveira, C.; Coutadeur, S.; Maurier-Mahe, F.; Throo, E.; Chauvignac, C.; Pognante, L.; Petibon, A.; Taverne, T.; et al. Vascular disrupting activity and the mechanism of action of EHT 6706, a novel anticancer tubulin polymerization inhibitor. Investig. New Drugs 2013, 31, 304–319. [Google Scholar] [CrossRef]
- Sobel, R.E.; Sadar, M.D. Cell lines used in prostate cancer research: A compendium of old and new lines--part 2. J. Urol. 2005, 173, 360–372. [Google Scholar] [CrossRef]
- Cunningham, D.; You, Z. In vitro and in vivo model systems used in prostate cancer research. J. Biol. Methods 2015, 2. [Google Scholar] [CrossRef]
- Winkler, S.; Murua Escobar, H.; Eberle, N.; Reimann-Berg, N.; Nolte, I.; Bullerdiek, J. Establishment of a cell line derived from a canine prostate carcinoma with a highly rearranged karyotype. J. Hered. 2005, 96, 782–785. [Google Scholar] [CrossRef]
- Fork, M.A.; Murua Escobar, H.; Soller, J.T.; Sterenczak, K.A.; Willenbrock, S.; Winkler, S.; Dorsch, M.; Reimann-Berg, N.; Hedrich, H.J.; Bullerdiek, J.; et al. Establishing an in vivo model of canine prostate carcinoma using the new cell line CT1258. BMC Cancer 2008, 8, 240. [Google Scholar] [CrossRef]
- Willenbrock, S.; Wagner, S.; Reimann-Berg, N.; Moulay, M.; Hewicker-Trautwein, M.; Nolte, I.; Murua Escobar, H. Generation and characterisation of a canine EGFP-HMGA2 prostate cancer in vitro model. PLoS ONE 2014, 9, e98788. [Google Scholar] [CrossRef]
- Liu, W.; Moulay, M.; Willenbrock, S.; Roolf, C.; Junghanss, C.; Ngenazahayo, A.; Nolte, I.; Murua Escobar, H. Comparative characterization of stem cell marker expression, metabolic activity and resistance to doxorubicin in adherent and spheroid cells derived from the canine prostate adenocarcinoma cell line CT1258. Anticancer Res. 2015, 35, 1917–1927. [Google Scholar]
- Harting, T.; Stubbendorff, M.; Willenbrock, S.; Wagner, S.; Schadzek, P.; Ngezahayo, A.; Murua Escobar, H.M.; Nolte, I. The effect of dichloroacetate in canine prostate adenocarcinomas and transitional cell carcinomas in vitro. Int. J. Oncol. 2016, 49, 2341–2350. [Google Scholar] [CrossRef][Green Version]
- Hammer, S.C.; Nagel, S.; Junginger, J.; Hewicker-Trautwein, M.; Wagner, S.; Heisterkamp, A.; Ngezahayo, A.; Nolte, I.; Murua Escobar, H. Claudin-1, -3, -4 and -7 gene expression analyses in canine prostate carcinoma and mammary tissue derived cell lines. Neoplasma 2016, 63, 231–238. [Google Scholar] [CrossRef]
- Roolf, C.; Saleweski, J.N.; Stein, A.; Richter, A.; Maletzki, C.; Sekora, A.; Escobar, H.M.; Wu, X.F.; Beller, M.; Junghanss, C. Novel Isoquinolinamine and Isoindoloquinazolinone Compounds Exhibit Antiproliferative Activity in Acute Lymphoblastic Leukemia Cells. Biomol. Ther. 2019. [Google Scholar] [CrossRef]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Werfel, T.A.; Cook, R.S. Efferocytosis in the tumor microenvironment. Semin. Immunopathol. 2018, 40, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef]
- Mc Gee, M.M. Targeting the Mitotic Catastrophe Signaling Pathway in Cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef]
- Sinha, D.; Duijf, P.H.G.; Khanna, K.K. Mitotic slippage: An old tale with a new twist. Cell Cycle 2019, 18, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Santaguida, S.; Amon, A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol. 2015, 16, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Inoue, T. Antiproliferative Fate of the Tetraploid Formed after Mitotic Slippage and Its Promotion; A Novel Target for Cancer Therapy Based on Microtubule Poisons. Molecules 2016, 21, 663. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.-B.; Wu, X.-F. Potassium tert-Butoxide-Promoted Synthesis of 1-Aminoisoquinolines from 2-Methylbenzonitriles and Benzonitriles under Catalyst-Free Conditions. Adv. Synth. Catal. 2016, 358, 2179–2185. [Google Scholar] [CrossRef]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investig. Urol. 1979, 17, 16–23. [Google Scholar]
- Webber, M.M.; Bello, D.; Quader, S. Immortalized and tumorigenic adult human prostatic epithelial cell lines: Characteristics and applications Part 2. Tumorigenic cell lines. Prostate 1997, 30, 58–64. [Google Scholar] [CrossRef]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar]
- Windhovel, C.; Harder, L.; Bach, J.P.; Teske, M.; Grabow, N.; Eickner, T.; Hinze, U.; Chichkov, B.; Nolte, I. Comparison of Six Different Silicones In Vitro for Application as Glaucoma Drainage Device. Materials 2018, 11, 341. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | PC-3 | 0846 | ||
|---|---|---|---|---|
| Application | Control | 12 h of 5 µM FX-9 | Control | 12 h of 5 µM FX-9 |
| G0/G1 phase [%] | 36.2 | 31.5 * | 39.5 | 32.3 * |
| S phase [%] | 24.4 | 19.5 * | 21.7 | 17.9 * |
| G2/M phase [%] | 39.4 | 49.0 * | 38.8 | 49.8 * |
| Cellular Analysis | Malignant Cell Lines | Benign Cells | ||||
|---|---|---|---|---|---|---|
| PC-3 | LNCaP | CT1258 | 0846 | hTF-8 | 1801 | |
| Cell viability (5 µM FX-9) [%] | 38.7 * | 31.8 * | 29.6 * | 58.4 | 77.8 | 78.8 |
| Cell viability (7.5 µM FX-9) [%] | 35.2 * | 33.9 * | 30.7 * | 42.5 * | 74.5 | 69.8 |
| Cell viability (10 µM FX-9) [%] | 35.6 1 | 27.4 * | 27.8 * | 37.0 1 | 66.9 | 60.1 |
| Total cell count (5 µM FX-9) [%] | 14.1 * | 13.4 * | − | 7.6 * | 95.2 | 74.4 |
| Vital cells (5 µM FX-9) [%] | 33.3 * | 13.0 * | − | 23.2 * | 67.5 | 75.9 |
| Apoptotic cells (5 µM FX-9) [%] | 59.1 * | 80.8 * | − | 71.0 * | 23.0 | 13.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schille, J.T.; Nolte, I.; Packeiser, E.-M.; Wiesner, L.; Hein, J.I.; Weiner, F.; Wu, X.-F.; Beller, M.; Junghanss, C.; Murua Escobar, H. Isoquinolinamine FX-9 Exhibits Anti-Mitotic Activity in Human and Canine Prostate Carcinoma Cell Lines. Int. J. Mol. Sci. 2019, 20, 5567. https://doi.org/10.3390/ijms20225567
Schille JT, Nolte I, Packeiser E-M, Wiesner L, Hein JI, Weiner F, Wu X-F, Beller M, Junghanss C, Murua Escobar H. Isoquinolinamine FX-9 Exhibits Anti-Mitotic Activity in Human and Canine Prostate Carcinoma Cell Lines. International Journal of Molecular Sciences. 2019; 20(22):5567. https://doi.org/10.3390/ijms20225567
Chicago/Turabian StyleSchille, Jan Torben, Ingo Nolte, Eva-Maria Packeiser, Laura Wiesner, Jens Ingo Hein, Franziska Weiner, Xiao-Feng Wu, Matthias Beller, Christian Junghanss, and Hugo Murua Escobar. 2019. "Isoquinolinamine FX-9 Exhibits Anti-Mitotic Activity in Human and Canine Prostate Carcinoma Cell Lines" International Journal of Molecular Sciences 20, no. 22: 5567. https://doi.org/10.3390/ijms20225567
APA StyleSchille, J. T., Nolte, I., Packeiser, E.-M., Wiesner, L., Hein, J. I., Weiner, F., Wu, X.-F., Beller, M., Junghanss, C., & Murua Escobar, H. (2019). Isoquinolinamine FX-9 Exhibits Anti-Mitotic Activity in Human and Canine Prostate Carcinoma Cell Lines. International Journal of Molecular Sciences, 20(22), 5567. https://doi.org/10.3390/ijms20225567