RNAase III-Type Enzyme Dicer Regulates Mitochondrial Fatty Acid Oxidative Metabolism in Cardiac Mesenchymal Stem Cells


Abstract
1. Introduction
2. Results
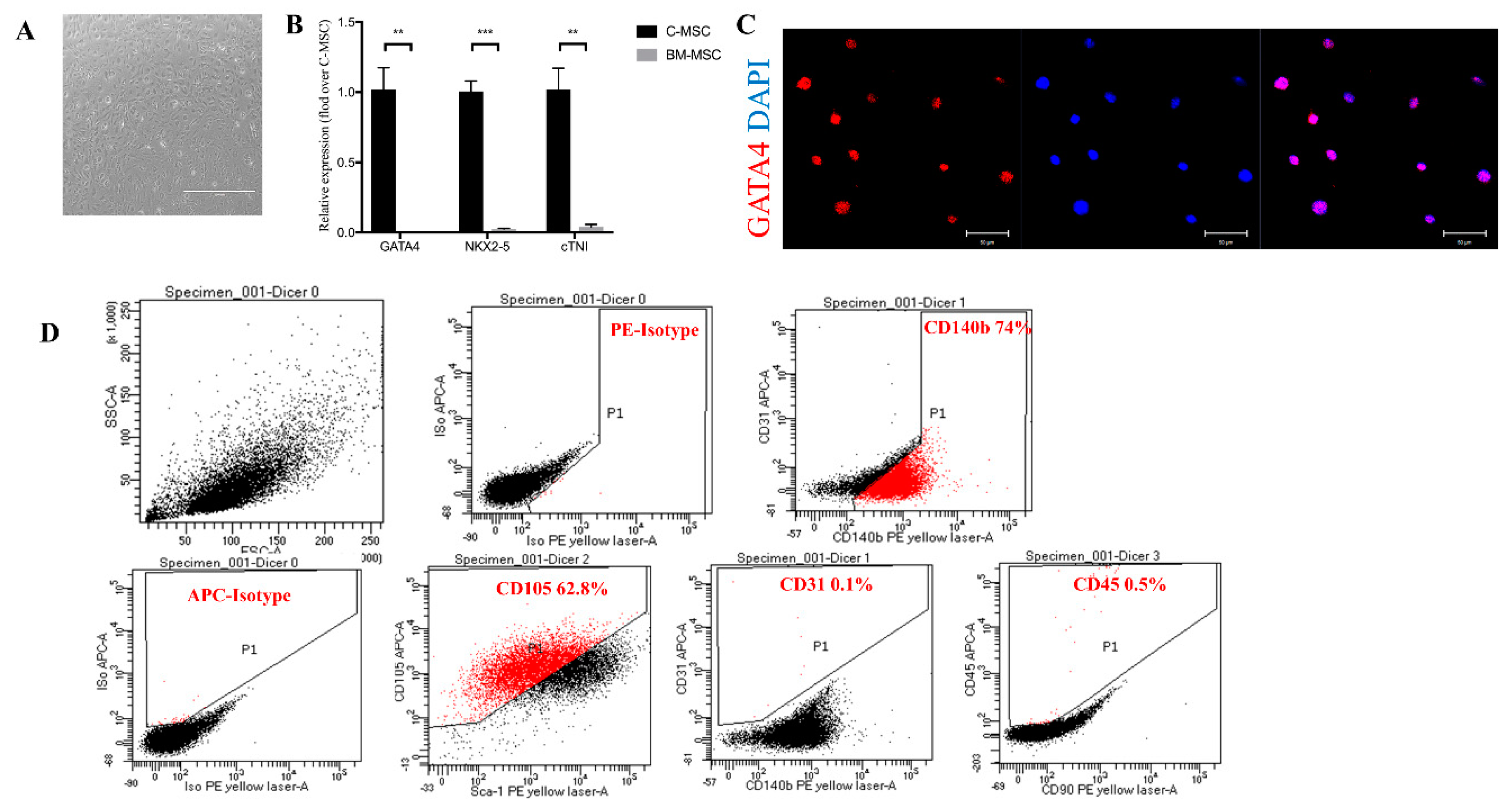
2.1. Characterization of C-MSC
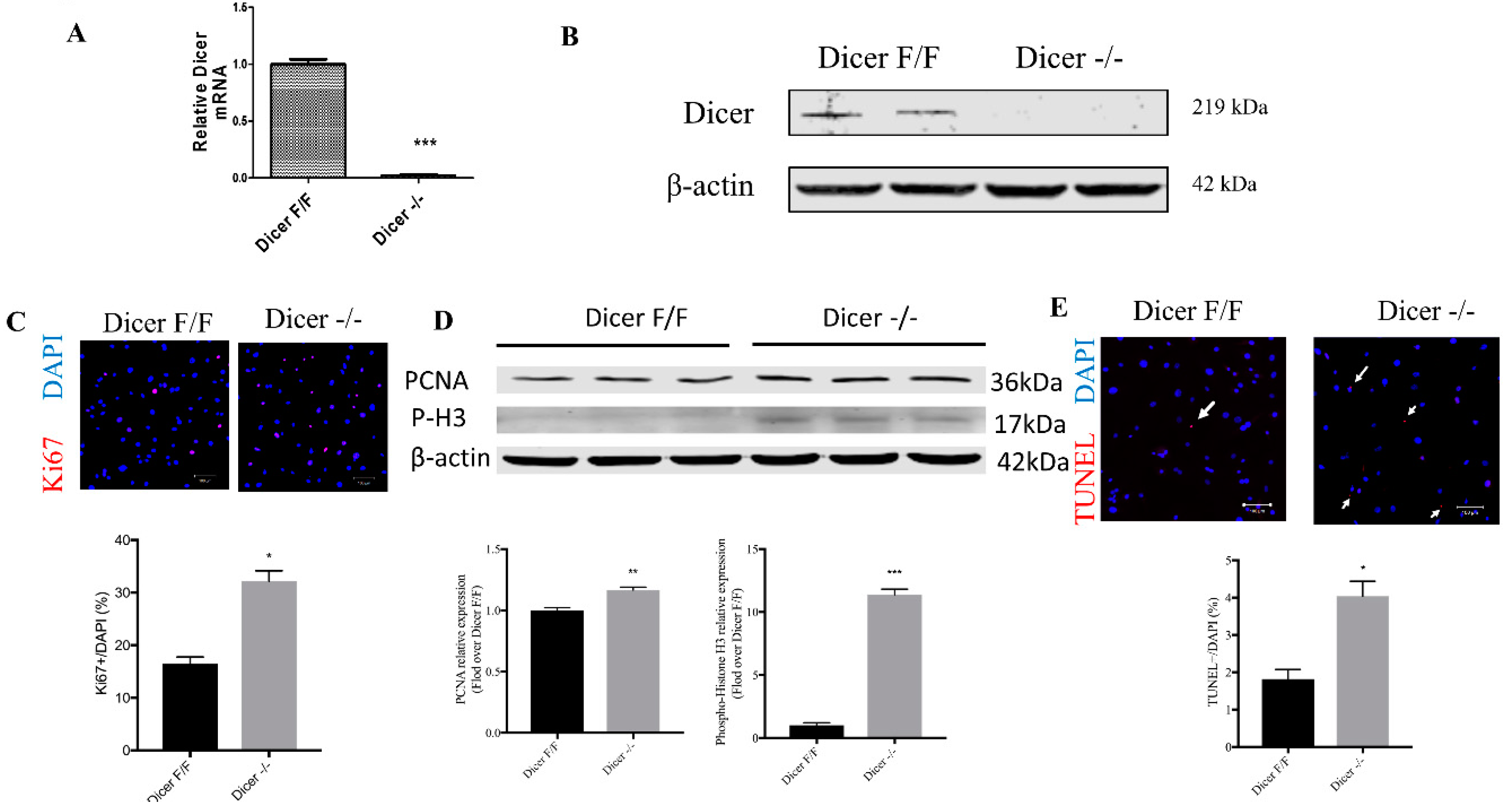
2.2. Adenovirus-Mediated Cre Recombinase Enzyme (CRE) Deletion of Dicer in C-MSC
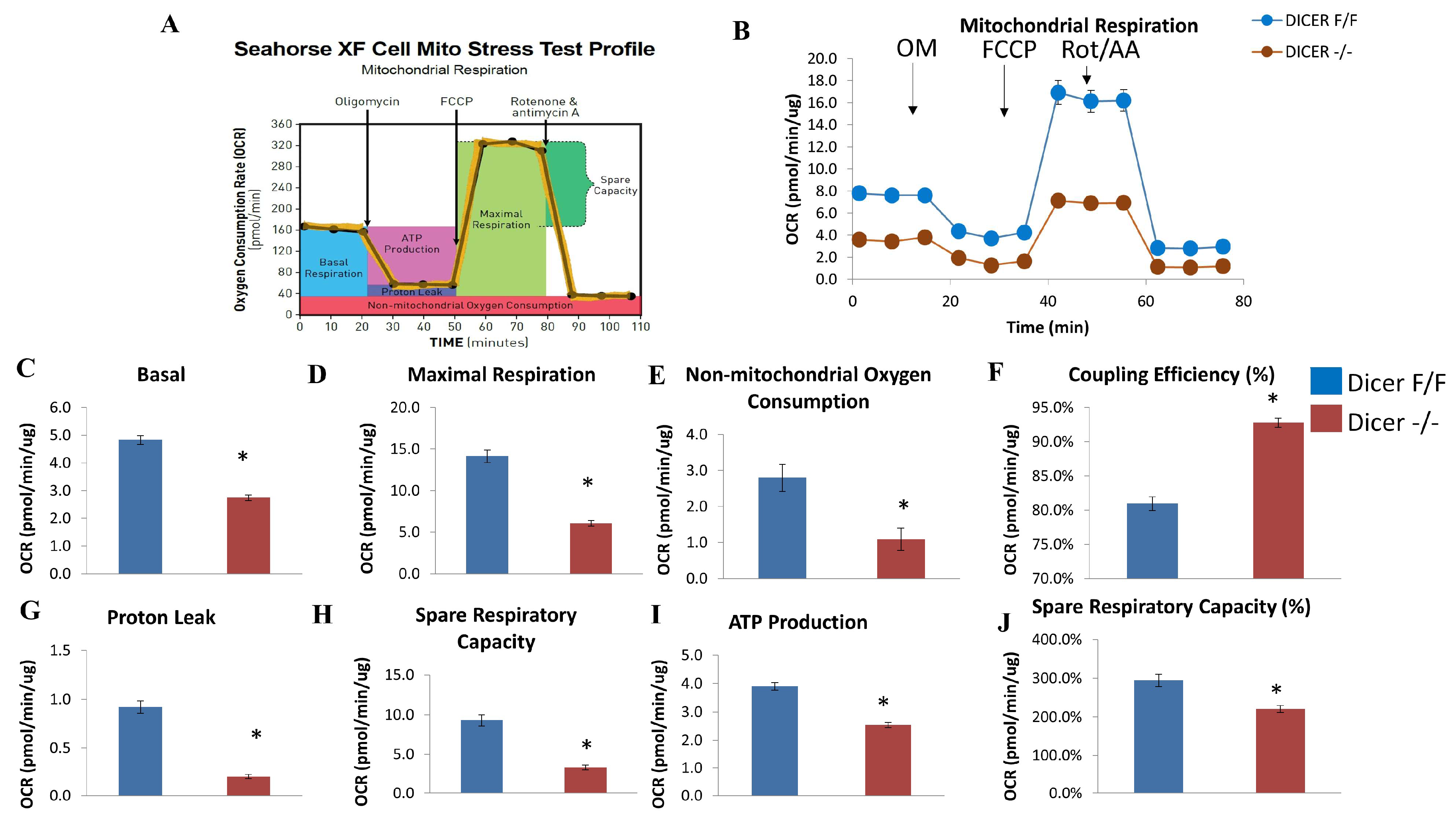
2.3. Dicer Deletion Impairs Mitochondrial Respiration in C-MSC
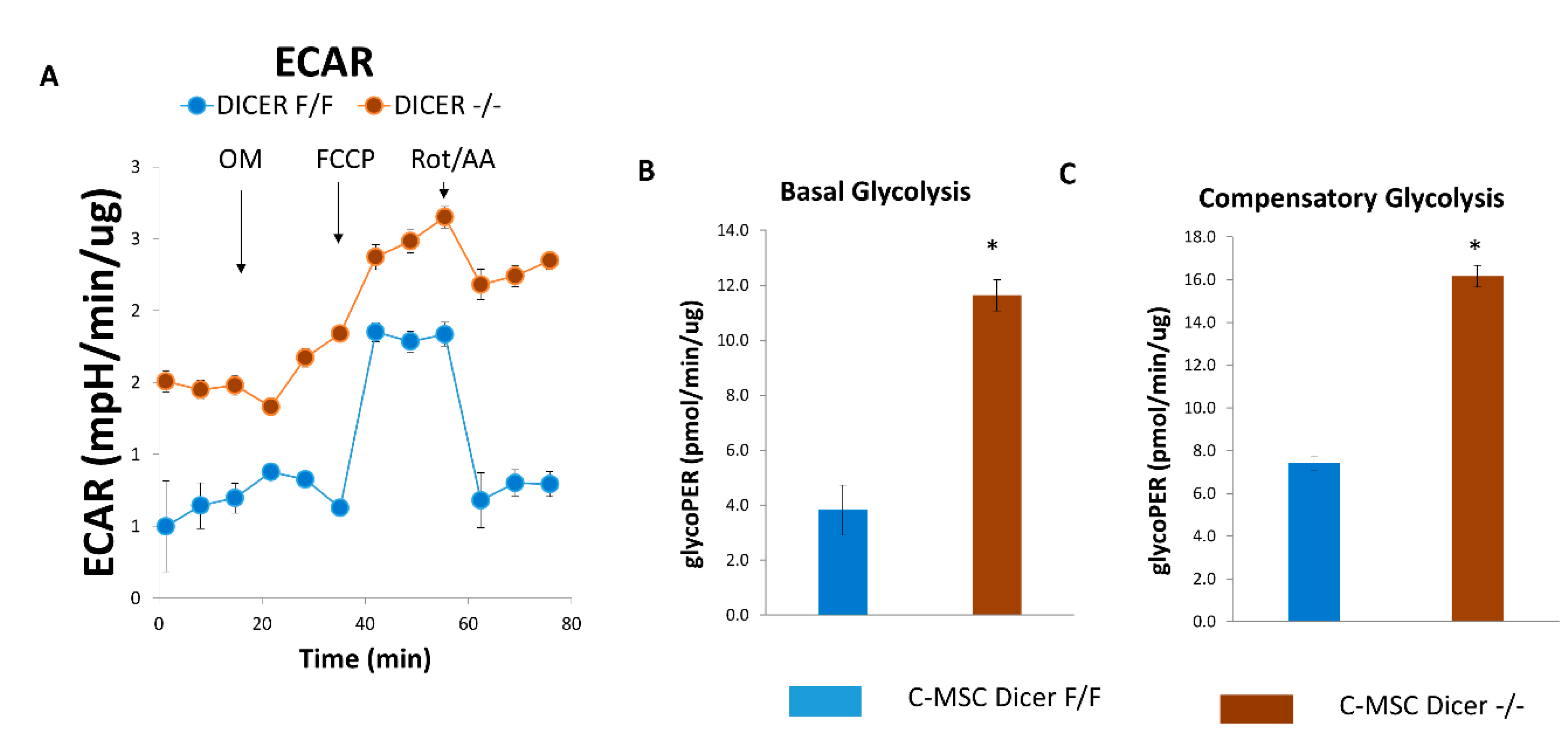
2.4. Dicer Gene Deletion Increases Glycolysis in C-MSC
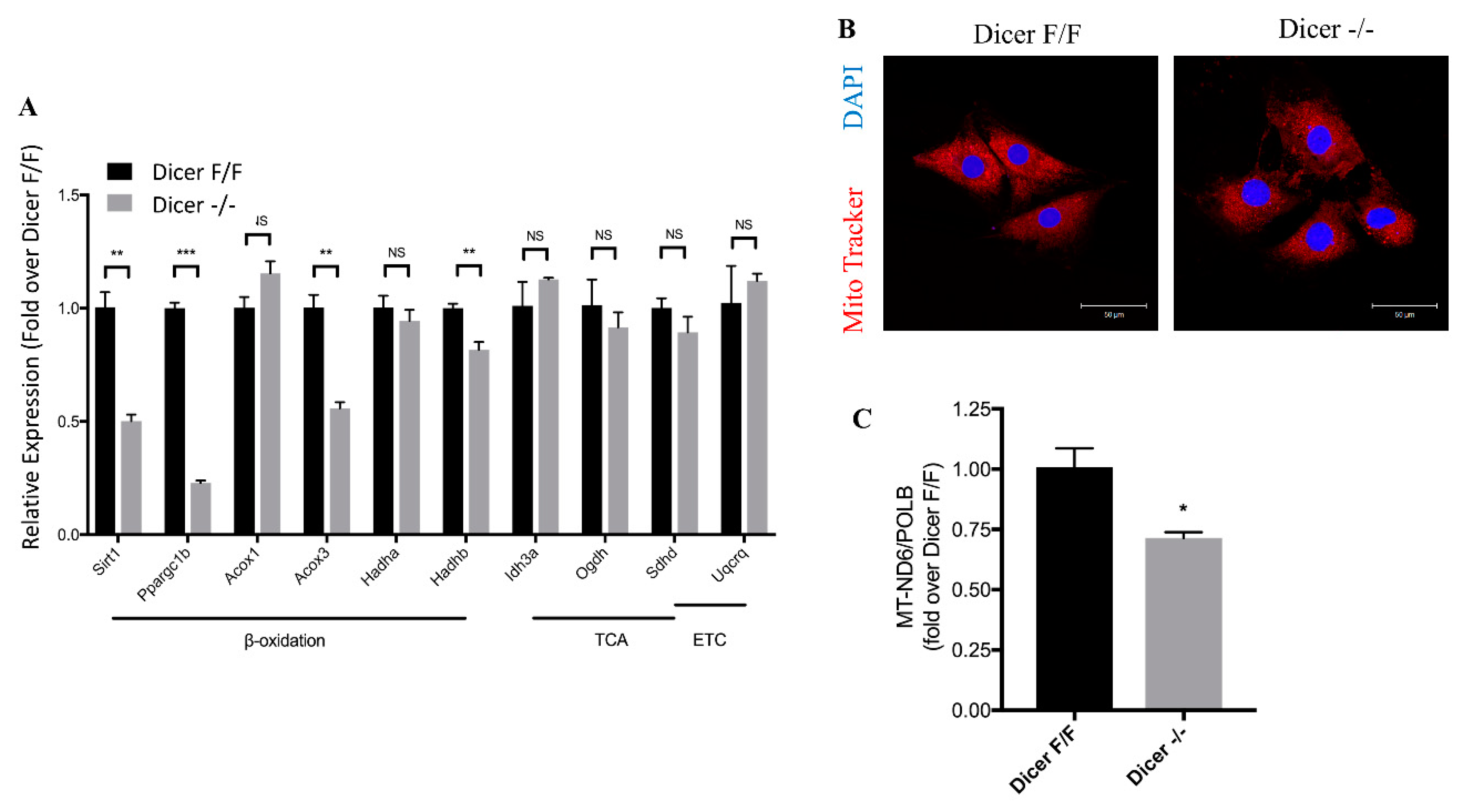
2.5. Dicer Deletion Decreases Mitochondrial Fatty Acid β-Oxidation in C-MSC
3. Discussion
4. Materials and Methods
4.1. Isolation of Mouse and Human C-MSC
4.2. siRNA Transfection
4.3. Flow Cytometry
4.4. Immunofluorescent Staining
4.5. Plasmid Construct and Generation of Recombinant Adenoviruses
4.6. Isolation and Quantification of Genomic DNA and Messenger RNA
4.7. Western Blotting Assay
4.8. Seahorse Analysis of Mitochondrial Respirometry
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BM-MSC | Bone marrow mesenchymal stem cells |
| Nkx2.5 | NK2 homeobox 5 |
| cTNI | Cardiac troponin-I |
| MT-ND6 | mitochondrially encoded NADH: ubiquinone oxidoreductase core subunit 6 |
| POLB | DNA polymerase beta |
| TUNEL | Terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling |
| TCA | Tricarboxylic acid |
| ETC | Electron transport chain |
| C-MSC | Cardiac mesenchymal stem cells |
| miRNAs | microRNAs |
| OCR | Oxygen consumption rate |
| ECAR | Extracellular acidification rate |
| Sirt1 | Sirtuin 1 |
| Ppargc1b | Peroxisome proliferator-activated receptor gamma coactivator 1 beta |
| Acox1 | Acyl-CoA oxidase 1 |
| Acox3 | Acyl-CoA oxidase 3 |
| Hadha | Hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha |
| Hadhb | Hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit beta |
| Idh3a | Isocitrate dehydrogenase (NAD(+)) 3 alpha |
| Ogdh | Oxoglutarate dehydrogenase |
| Sdhd | Succinate dehydrogenase complex subunit D |
| Uqcrq | Ubiquinol-cytochrome C reductase complex III subunit VII |
References
- Smith, S.C., Jr.; Collins, A.; Ferrari, R.; Holmes, D.R., Jr.; Logstrup, S.; McGhie, D.V.; Ralston, J.; Sacco, R.L.; Stam, H.; Taubert, K.; et al. Our time: A call to save preventable death from cardiovascular disease (heart disease and stroke). J. Am.Coll. Cardiol. 2012, 60, 2343–2348. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol 2018, 15, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, V.; Stanley, W.C.; Recchia, F.A. Modulating fatty acid oxidation in heart failure. Cardiovasc. Res. 2011, 90, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Phys. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Liu, Q.; Docherty, J.C.; Rendell, J.C.; Clanachan, A.S.; Lopaschuk, G.D. High levels of fatty acids delay the recovery of intracellular pH and cardiac efficiency in post-ischemic hearts by inhibiting glucose oxidation. J. Am. Coll. Cardiol. 2002, 39, 718–725. [Google Scholar] [CrossRef]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Yun, U.J.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005, 146, 5341–5349. [Google Scholar] [CrossRef]
- Ruan, X.F.; Ju, C.W.; Shen, Y.; Liu, Y.T.; Kim, I.M.; Yu, H.; Weintraub, N.; Wang, X.L.; Tang, Y. Suxiao Jiuxin pill promotes exosome secretion from mouse cardiac mesenchymal stem cells in vitro. Acta Pharm. Sin. 2018, 39, 569–578. [Google Scholar] [CrossRef]
- Ruan, X.F.; Li, Y.J.; Ju, C.W.; Shen, Y.; Lei, W.; Chen, C.; Li, Y.; Yu, H.; Liu, Y.T.; Kim, I.M.; et al. Exosomes from Suxiao Jiuxin pill-treated cardiac mesenchymal stem cells decrease H3K27 demethylase UTX expression in mouse cardiomyocytes in vitro. Acta Pharm. Sin. 2018, 39, 579–586. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.; Pan, Y.; Zhang, L.; Shen, C.; Qin, G.; Ashraf, M.; Weintraub, N.; Ma, G.; Tang, Y. Cardiac progenitor-derived exosomes protect ischemic myocardium from acute ischemia/reperfusion injury. Biochem. Biophys. Res. Commun. 2013, 431, 566–571. [Google Scholar] [CrossRef]
- Tang, Y.L.; Zhu, W.; Cheng, M.; Chen, L.; Zhang, J.; Sun, T.; Kishore, R.; Phillips, M.I.; Losordo, D.W.; Qin, G. Hypoxic preconditioning enhances the benefit of cardiac progenitor cell therapy for treatment of myocardial infarction by inducing CXCR4 expression. Circ. Res. 2009, 104, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nature reviews. Mol. Cell. Biol. 2014, 15, 509–524. [Google Scholar]
- Demkes, C.J.; Van Rooij, E. MicroRNA-146a as a Regulator of Cardiac Energy Metabolism. Circulation 2017, 136, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Kohr, M.; Dunkerly-Eyring, B.; Lee, D.I.; Bedja, D.; Kent, O.A.; Leung, A.K.; Henao-Mejia, J.; Flavell, R.A.; Steenbergen, C. Divergent Effects of miR-181 Family Members on Myocardial Function Through Protective Cytosolic and Detrimental Mitochondrial microRNA Targets. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Pinti, M.V.; Hathaway, Q.A.; Hollander, J.M. Role of microRNA in metabolic shift during heart failure. American journal of physiology. Heart Circ. Phys. 2017, 312, H33–H45. [Google Scholar] [CrossRef]
- Kurzynska-Kokorniak, A.; Koralewska, N.; Pokornowska, M.; Urbanowicz, A.; Tworak, A.; Mickiewicz, A.; Figlerowicz, M. The many faces of Dicer: The complexity of the mechanisms regulating Dicer gene expression and enzyme activities. Nucleic Acids Res. 2015, 43, 4365–4380. [Google Scholar] [CrossRef]
- Singh, M.K.; Lu, M.M.; Massera, D.; Epstein, J.A. MicroRNA-processing enzyme Dicer is required in epicardium for coronary vasculature development. J. Biol. Chem. 2011, 286, 41036–41045. [Google Scholar] [CrossRef]
- Hu, Q.; Tanasa, B.; Trabucchi, M.; Li, W.; Zhang, J.; Ohgi, K.A.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. DICER- and AGO3-dependent generation of retinoic acid-induced DR2 Alu RNAs regulates human stem cell proliferation. Nat. Struct. Mol. Biol. 2012, 19, 1168–1175. [Google Scholar] [CrossRef]
- Martin, E.C.; Qureshi, A.T.; Llamas, C.B.; Burow, M.E.; King, A.G.; Lee, O.C.; Dasa, V.; Freitas, M.A.; Forsberg, J.A.; Elster, E.A.; et al. Mirna biogenesis pathway is differentially regulated during adipose derived stromal/stem cell differentiation. Adipocyte 2018, 7, 96–105. [Google Scholar] [CrossRef]
- Chen, J.F.; Murchison, E.P.; Tang, R.; Callis, T.E.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.M.; Schneider, M.D.; Selzman, C.H.; et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2008, 105, 2111–2116. [Google Scholar] [CrossRef]
- Da Costa Martins, P.A.; Bourajjaj, M.; Gladka, M.; Kortland, M.; van Oort, R.J.; Pinto, Y.M.; Molkentin, J.D.; De Windt, L.J. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation 2008, 118, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, J.K.; Bruneau, B.G. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature 2009, 459, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Nery, A.A.; Nascimento, I.C.; Glaser, T.; Bassaneze, V.; Krieger, J.E.; Ulrich, H. Human mesenchymal stem cells: From immunophenotyping by flow cytometry to clinical applications. Cytom. Part A J. Int. Soc. Anal. Cytol. 2013, 83, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Au-Van den Bossche, J.; Au-Baardman, J.; Au-de Winther, M.P.J. Metabolic Characterization of Polarized M1 and M2 Bone Marrow-derived Macrophages Using Real-time Extracellular Flux Analysis. JoVE 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, B.; Valdez, J.M.; Wang, F.; Ittmann, M.; Xin, L. Dicer ablation impairs prostate stem cell activity and causes prostate atrophy. St. Cell. 2010, 28, 1260–1269. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wanders, R.J.; Ruiter, J.P.; L, I.J.; Waterham, H.R.; Houten, S.M. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J. Inherit. Metab. Dis. 2010, 33, 479–494. [Google Scholar] [CrossRef]
- Akram, M. Citric acid cycle and role of its intermediates in metabolism. Cell. Biochem. Biophys. 2014, 68, 475–478. [Google Scholar] [CrossRef]
- Wei, Y.; Corbalan-Campos, J.; Gurung, R.; Natarelli, L.; Zhu, M.; Exner, N.; Erhard, F.; Greulich, F.; Geissler, C.; Uhlenhaut, N.H.; et al. Dicer in Macrophages Prevents Atherosclerosis by Promoting Mitochondrial Oxidative Metabolism. Circulation 2018, 138, 2007–2020. [Google Scholar] [CrossRef]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef]
- Lim, J.H.; Gerhart-Hines, Z.; Dominy, J.E.; Lee, Y.; Kim, S.; Tabata, M.; Xiang, Y.K.; Puigserver, P. Oleic acid stimulates complete oxidation of fatty acids through protein kinase A-dependent activation of SIRT1-PGC1alpha complex. J. Biol. Chem. 2013, 288, 7117–7126. [Google Scholar] [CrossRef]
- Ding, R.B.; Bao, J.; Deng, C.X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.F.; Tian, M.X.; Sun, R.Q.; Zhang, M.L.; Zhou, L.S.; Jin, L.; Chen, L.L.; Zhou, W.J.; Duan, K.L.; Chen, Y.J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Zha, S.; Ferdinandusse, S.; Hicks, J.L.; Denis, S.; Dunn, T.A.; Wanders, R.J.; Luo, J.; De Marzo, A.M.; Isaacs, W.B. Peroxisomal branched chain fatty acid beta-oxidation pathway is upregulated in prostate cancer. Prostate 2005, 63, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Schon, U.; Horvath, R.; Schwartz, O.; Holinski-Feder, E.; Kolbel, H.; Abicht, A. HADHA and HADHB gene associated phenotypes-Identification of rare variants in a patient cohort by Next Generation Sequencing. Mol. Cell. Probe 2019, 44, 14–20. [Google Scholar] [CrossRef]
- Vasilescu, C.; Ojala, T.H.; Brilhante, V.; Ojanen, S.; Hinterding, H.M.; Palin, E.; Alastalo, T.P.; Koskenvuo, J.; Hiippala, A.; Jokinen, E.; et al. Genetic Basis of Severe Childhood-Onset Cardiomyopathies. J. Am. Coll. Cardiol. 2018, 72, 2324–2338. [Google Scholar] [CrossRef]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef]
- Malandraki-Miller, S.; Lopez, C.A.; Al-Siddiqi, H.; Carr, C.A. Changing Metabolism in Differentiating Cardiac Progenitor Cells-Can Stem Cells Become Metabolically Flexible Cardiomyocytes? Front Cardiovasc. Med. 2018, 5, 119. [Google Scholar] [CrossRef]
- Riester, M.; Xu, Q.; Moreira, A.; Zheng, J.; Michor, F.; Downey, R.J. The Warburg effect: Persistence of stem-cell metabolism in cancers as a failure of differentiation. Ann. Oncol. 2018, 29, 264–270. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Ann. Rev. Cell. Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Chinchore, Y.; Begaj, T.; Wu, D.; Drokhlyansky, E.; Cepko, C.L. Glycolytic reliance promotes anabolism in photoreceptors. eLife 2017, 6. [Google Scholar] [CrossRef]
- Ju, C.; Shen, Y.; Ma, G.; Liu, Y.; Cai, J.; Kim, I.M.; Weintraub, N.L.; Liu, N.; Tang, Y. Transplantation of Cardiac Mesenchymal Stem Cell-Derived Exosomes Promotes Repair in Ischemic Myocardium. J. Cardiovasc. Transl. Res. 2018, 11, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Su, X.; Shen, Y.; Jin, Y.; Luo, T.; Kim, I.M.; Weintraub, N.L.; Tang, Y. MiR322 mediates cardioprotection against ischemia/reperfusion injury via FBXW7/notch pathway. J. Mol. Cell. Cardiol. 2019, 133, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Deng, Z.L.; Luo, X.; Tang, N.; Song, W.X.; Chen, J.; Sharff, K.A.; Luu, H.H.; Haydon, R.C.; Kinzler, K.W.; et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat. Protoc. 2007, 2, 1236–1247. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence (5′–3′) |
|---|---|
| β-actin FWD (mouse) | AGAGCATAGCCCTCGTAGAT |
| β-actin REV (mouse) | GCTGTGCTGTCCCTGTATG |
| Dicer FWD (mouse) | TTACCAGCGCTTAGAATTCCTGGG |
| Dicer REV (mouse) | GTTATTGACAAGGGCAGAGCGCAA |
| Sirt1 FWD (mouse) | GTTGGTGGCAACTCTGATAAATG |
| Sirt1 REV (mouse) | GTCATAGGCTAGGTGGTGAATATG |
| Ppargc1b FWD (mouse) | AGGTGTGAGGGAAGCATAGA |
| Ppargc1b REV (mouse) | CAAAGCCTTCTGGACTGAGTT |
| Acox1 FWD (mouse) | CCTTGGCCAATGCTCTCATTA |
| Acox1 REV (mouse) | CGCAGCAGTATAAACTCTTCCC |
| Acox3 FWD (mouse) | CCCTAGAGAAGCTACGAGAACT |
| Acox3 REV (mouse) | CAGGCAGTTAATCAGCACTAGAA |
| Hadha FWD (mouse) | CCATGTCGGCCTTCTCAAA |
| Hadha REV (mouse) | AGTGAAGAAGAAAGCTCTCACAT |
| Hadhb FWD (mouse) | AGACCATGGGCCACTCT |
| Hadhb REV (mouse) | CTTCTTGGCCAGACTATGAGAAC |
| Idh3a FWD (mouse) | GGCCATCCATCTATGAATCTGT |
| Idh3a REV (mouse) | GTATTCTCCTTCCGTGTTCTCTC |
| Ogdh FWD (mouse) | CATGTATCACCGCAGGATCAA |
| Ogdh REV (mouse) | GGTCTTTCCCATCACGACAG |
| Sdhd FWD (mouse) | GATGCCGACATCGTGGTAAT |
| Sdhd REV (mouse) | GTTACCGACTACGTTCATGGG |
| Uqcrq FWD (mouse) | CTTTGCTGAAATAGCTTGGGAAG |
| Uqcrq REV (mouse) | GAACCTGGCGCGGATAC |
| GATA4 FWD (mouse) | GAGGGTGAGCCTGTATGTAATG |
| GATA4 REV (mouse) | CCTGCTGGCGTCTTAGATTTAT |
| NKX2-5 FWD (mouse) | GCAGTGGAGCTGGACAAA |
| NKX2-5 REV (mouse) | GGTACCGCTGTTGCTTGA |
| cTNI FWD (mouse) | CACCTCAAGCAGGTGAAGAA |
| cTNI REV (mouse) | GCCACTCAGTGCATCGATATT |
| MT-ND6 FWD (mouse) | CACCCAGCTACTACCATCATTC |
| MT-ND6 REV (mouse) | GTTTGGGAGATTGGTTGATGTATG |
| POLB FWD (mouse) | GGCGGATGGTGTACTCATT |
| POLB REV (mouse) | ACTGTGGTGTTCTCTACTTCAC |
| β-actin FWD (human) | CGTAGCACAGCTTCTCCTTAAT |
| β-actin REV (human) | GGACCTGACTGACTACCTCAT |
| Dicer FWD (human) | GGTTCCAGAACTCTGTGCTATAC |
| Dicer REV (human) | AGGCAGTGAAGGCGATAAAG |
| Sirt1 FWD (human) | CACCCACACCTCTTCATGTT |
| Sirt1 REV (human) | CATTACTCTTAGCTGCTTGGTCTA |
| Ppargc1b FWD (human) | GGTAAGGATCGCAGCTTCAC |
| Ppargc1b REV (human) | CCTCTTCCTCCTCCTCTTCTT |
| Acox3 FWD (human) | CCCACAGAGGAGGAGGAAATA |
| Acox3 REV (human) | GCGACTTGGAGAAATGGTCTAA |
| Hadhb FWD (human) | CCAAGAAGGCACAGGATGAA |
| Hadhb REV (human) | GGAAGGACGGATGCCATTAT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, X.; Jin, Y.; Shen, Y.; Kim, I.-m.; Weintraub, N.L.; Tang, Y. RNAase III-Type Enzyme Dicer Regulates Mitochondrial Fatty Acid Oxidative Metabolism in Cardiac Mesenchymal Stem Cells. Int. J. Mol. Sci. 2019, 20, 5554. https://doi.org/10.3390/ijms20225554
Su X, Jin Y, Shen Y, Kim I-m, Weintraub NL, Tang Y. RNAase III-Type Enzyme Dicer Regulates Mitochondrial Fatty Acid Oxidative Metabolism in Cardiac Mesenchymal Stem Cells. International Journal of Molecular Sciences. 2019; 20(22):5554. https://doi.org/10.3390/ijms20225554
Chicago/Turabian StyleSu, Xuan, Yue Jin, Yan Shen, Il-man Kim, Neal L. Weintraub, and Yaoliang Tang. 2019. "RNAase III-Type Enzyme Dicer Regulates Mitochondrial Fatty Acid Oxidative Metabolism in Cardiac Mesenchymal Stem Cells" International Journal of Molecular Sciences 20, no. 22: 5554. https://doi.org/10.3390/ijms20225554
APA StyleSu, X., Jin, Y., Shen, Y., Kim, I.-m., Weintraub, N. L., & Tang, Y. (2019). RNAase III-Type Enzyme Dicer Regulates Mitochondrial Fatty Acid Oxidative Metabolism in Cardiac Mesenchymal Stem Cells. International Journal of Molecular Sciences, 20(22), 5554. https://doi.org/10.3390/ijms20225554