Transcriptome Analysis Implicates Involvement of Long Noncoding RNAs in Cytoplasmic Male Sterility and Fertility Restoration in Cotton

 , ,
, ,
Abstract
1. Introduction
2. Results
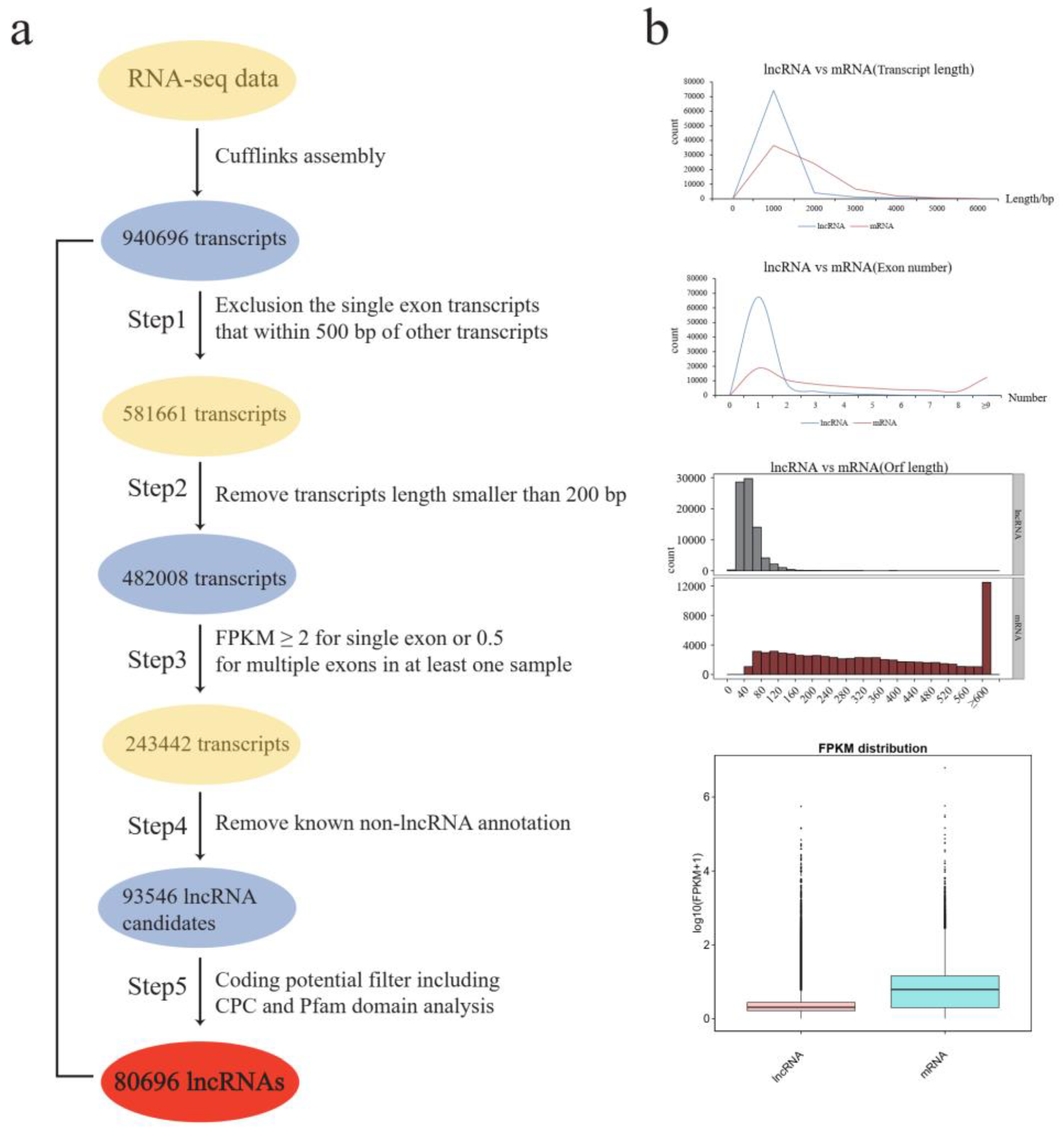
2.1. Genome-Wide Identification and Characterization of LncRNAs during Anther Development of Three-Line Hybrid Cotton
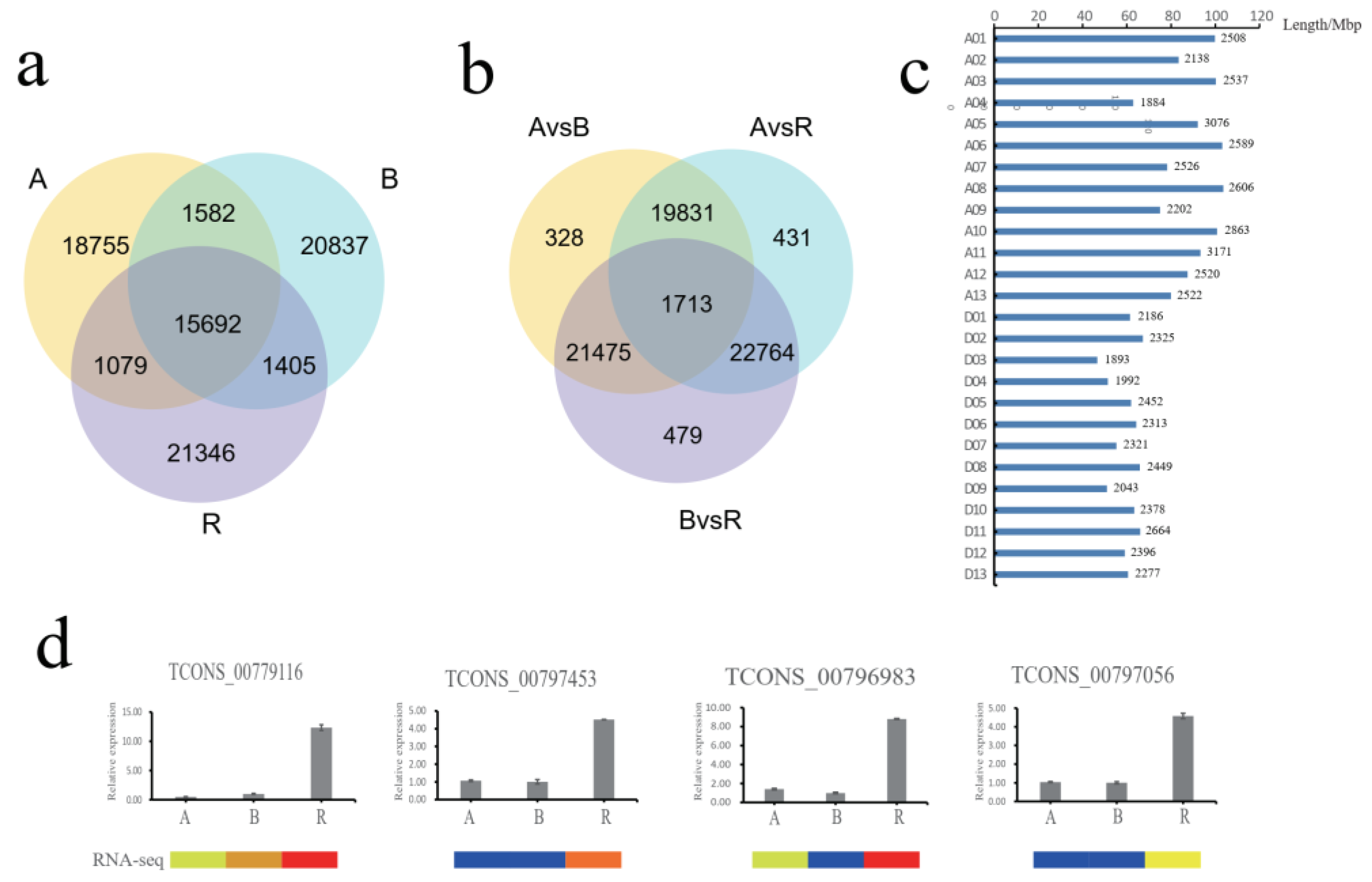
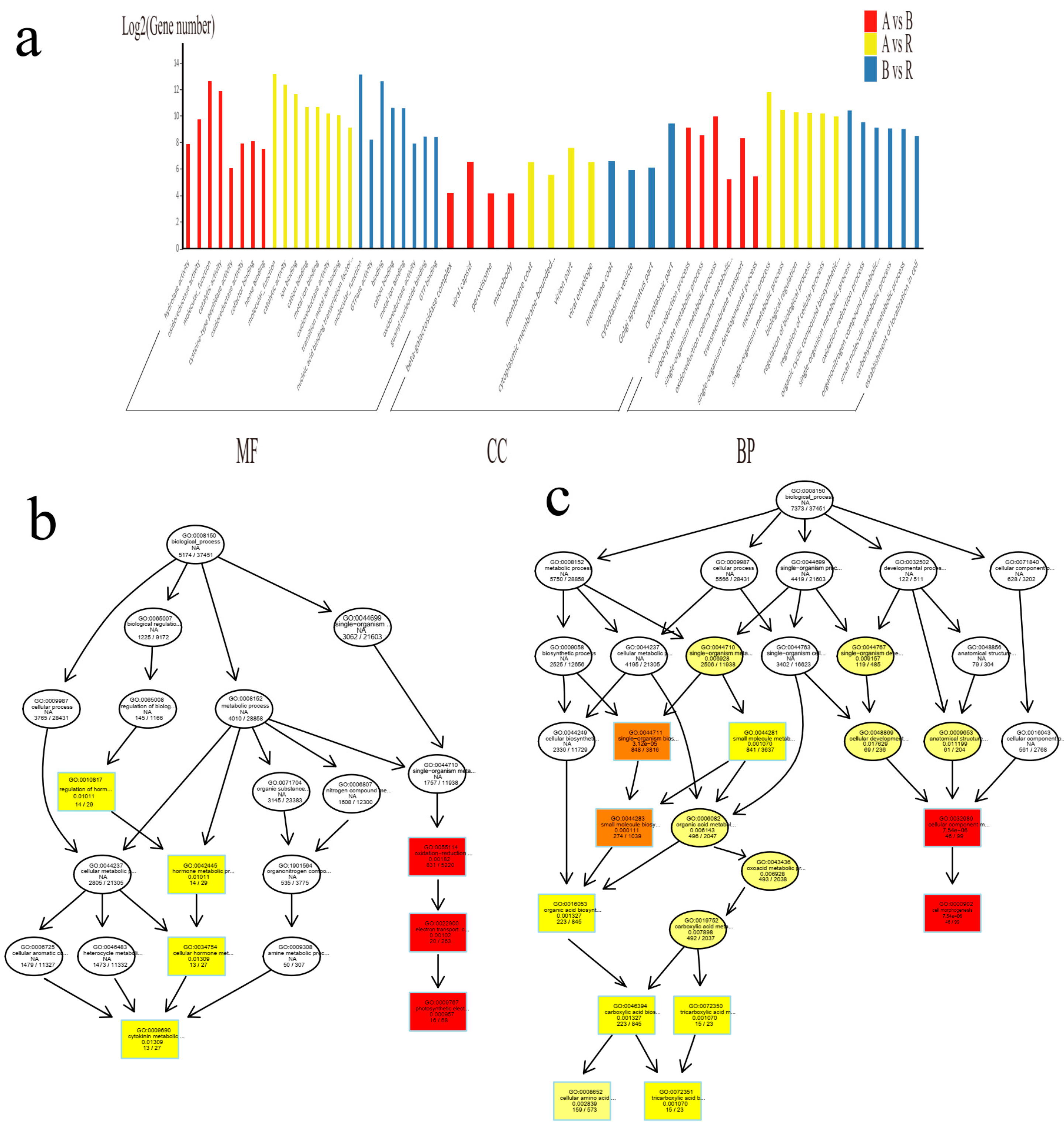
2.2. Identification and Functional Analysis of Differentially Expressed LncRNAs in A, B, and R Lines
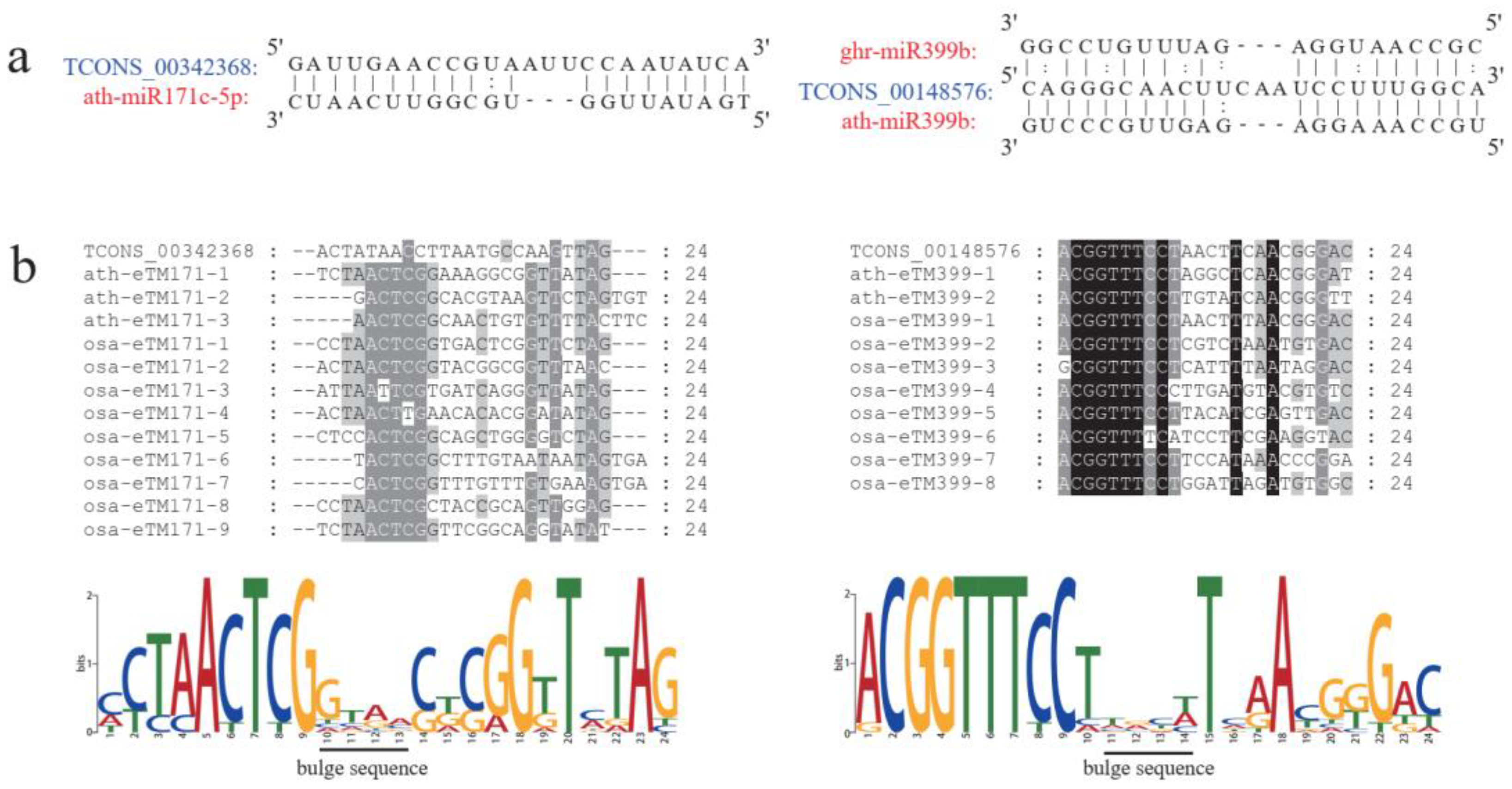
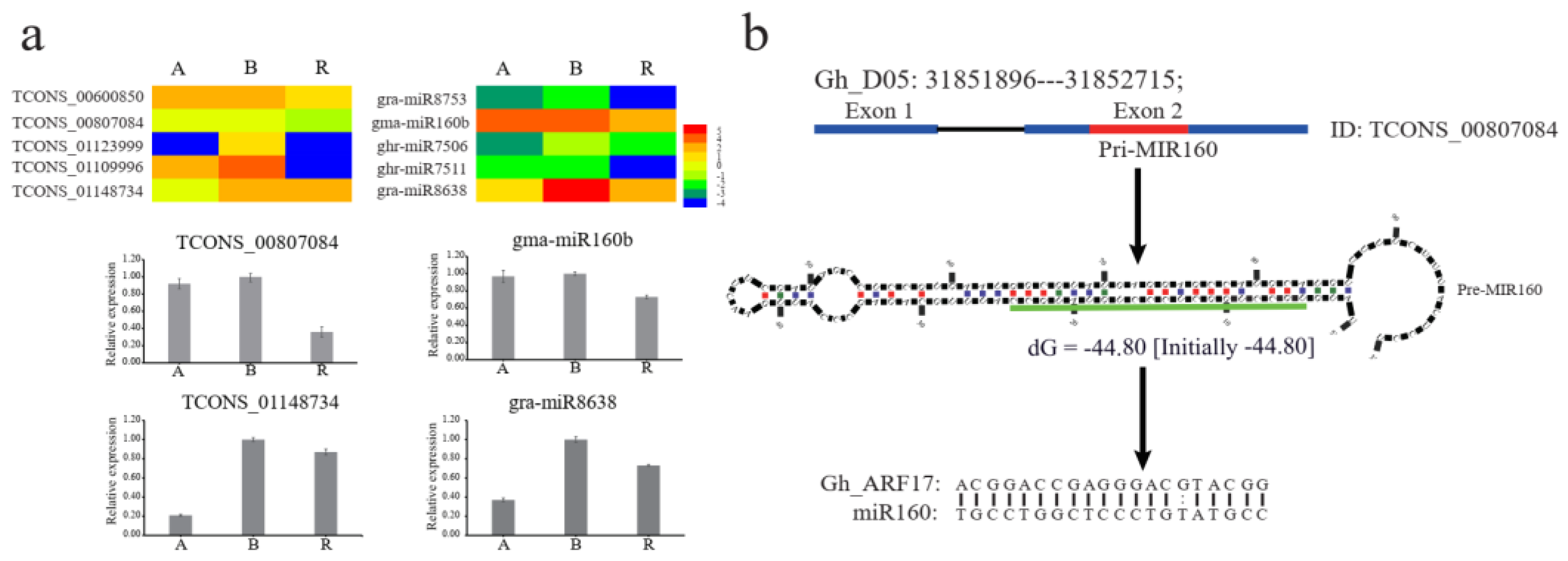
2.3. Predicted Interactions between LncRNAs and MiRNAs during Anther Development
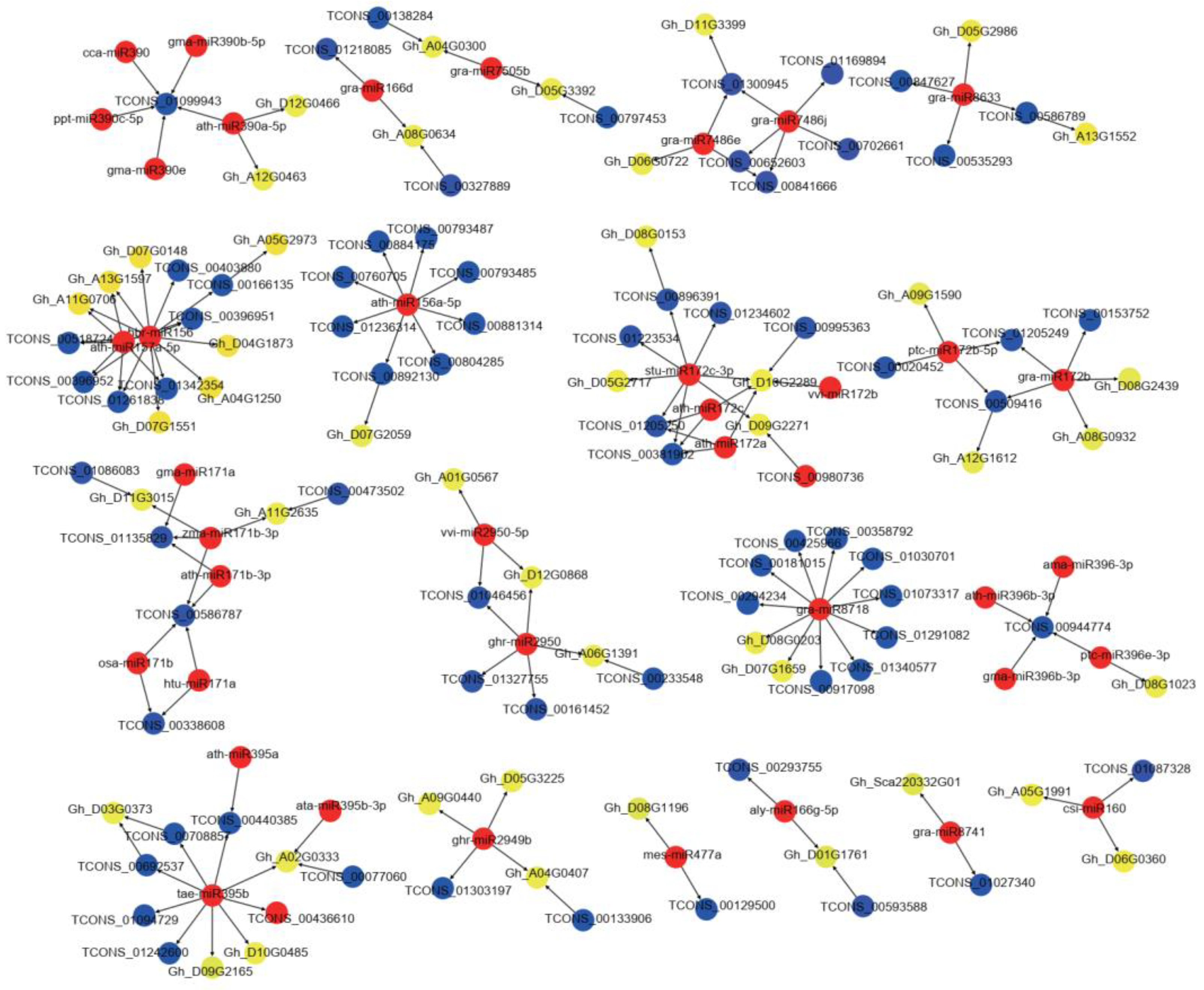
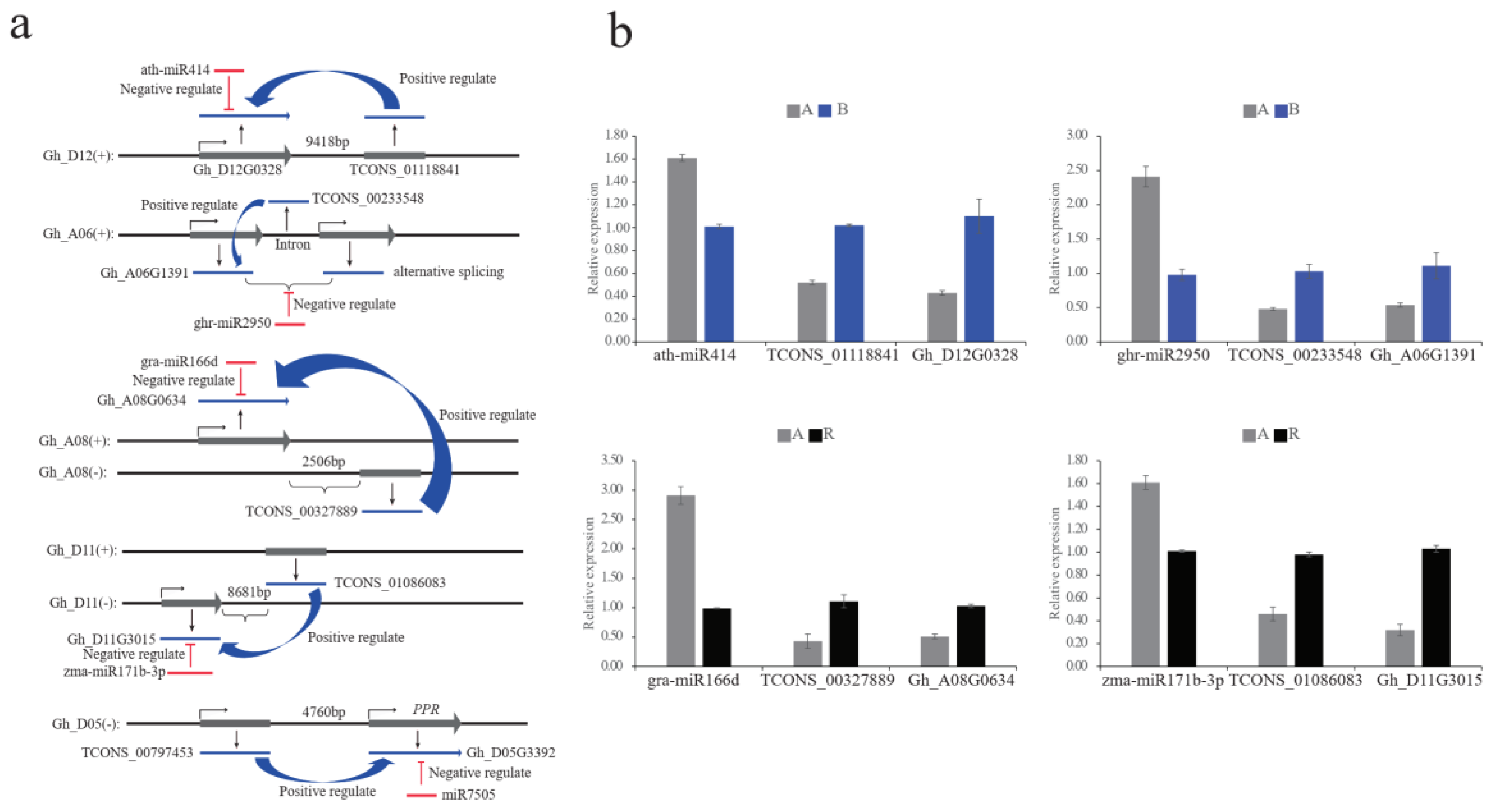
2.4. The miRNA–LncRNA–mRNA Regulatory Networks between A, B, and R Lines
3. Discussion
3.1. Overview of LncRNAs Identification and Function in Anther Development
3.2. Relationship between LncRNAs and MiRNAs in Anther Development of Cotton
4. Materials and Methods
4.1. Plant Materials and Transcriptome Sequence
4.2. Annotation of Transcripts and Identification of Long Noncoding RNAs
4.3. Expression and Target Gene Analysis of LncRNAs
4.4. Prediction of Putative Targets and Endogenous Target Mimics for MiRNAs
4.5. Construction of MiRNA-lncRNA-mRNA Regulatory Networks
4.6. Quantitative RT-PCR Validation of LncRNAs, MiRNAs, and mRNAs Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CMS | Cytoplasmic male sterility |
| Rf gene | Restorer-of-fertility gene |
| LncRNAs | Long noncoding RNAs |
| A | CMS line |
| B | Maintainer line |
| R | Restorer-of-fertility line |
| GO | Gene ontology |
| FPKM | Fragments per kilobase of exon per million fragments |
References
- Costa, F.F. Non-coding RNAs: New players in eukaryotic biology. Gene 2005, 357, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Eun-Deok, K.; Sibum, S. Long noncoding RNA: Unveiling hidden layer of gene regulatory networks. Trends Plant. Sci. 2012, 17, 16–21. [Google Scholar]
- Hangauer, M.J.; Vaughn, I.W.; Mcmanus, M.T. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013, 9, e1003569. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.T.; Zhu, D.; Chen, W.; Deng, W.; He, H.; He, G.; Bai, B.; Qi, Y.; Chen, R.; Deng, X.W. A global identification and analysis of small nucleolar RNAs and possible intermediate-sized non-coding RNAs in Oryza sativa. Mol. Plant 2013, 6, 830–846. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Julie, D.; Carey, B.W.; Manuel, G.; Grenier, J.K.; Glen, M.; Geneva, Y.; Anne Bergstrom, L.; Robert, A.; Laurakay, B. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295. [Google Scholar]
- Guil, S.; Esteller, M. Cis-acting noncoding RNAs: Friends and foes. Nat. Struct. Mol. Biol. 2012, 19, 1068–1075. [Google Scholar] [CrossRef]
- Jun, L.; Choonkyun, J.; Jun, X.; Huan, W.; Shulin, D.; Lucia, B.; Catalina, A.H.; Nam-Hai, C. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant. Cell 2012, 24, 4333–4345. [Google Scholar]
- Lin, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.T.; Wei, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar]
- Zhang, Y.-C.; Liao, J.-Y.; Li, Z.-Y.; Yu, Y.; Zhang, J.-P.; Li, Q.-F.; Qu, L.-H.; Shu, W.-S.; Chen, Y.-Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef]
- Fuquan, L.; Sebastian, M.; Clare, L.; Szymon, S.; Caroline, D. Targeted 3′ processing of antisense transcripts triggers Arabidopsis FLC chromatin silencing. Science 2010, 327, 94–97. [Google Scholar]
- Jae Bok, H.; Sibum, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef] [PubMed]
- Boerner, S.; McGinnis, K.M. Computational Identification and Functional Predictions of Long Noncoding RNA in Zea mays. PLoS ONE 2012, 7, e43047. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yuan, D.; Tu, L.; Gao, W.; He, Y.; Hu, H.; Wang, P.; Liu, N.; Lindsey, K.; Zhang, X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef]
- Cagirici, H.B.; Alptekin, B.; Budak, H. RNA Sequencing and co-expressed long non-coding RNA in modern and wild wheats. Sci. Rep. 2017, 7, 10670. [Google Scholar] [CrossRef]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; Garcia, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Wu, H.-J.; Wang, Z.M.; Wang, M.; Wang, X.J. Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol. 2013, 161, 1875–1884. [Google Scholar] [CrossRef]
- Huang, L.; Dong, H.; Zhou, D.; Li, M.; Cao, J. Systematic identification of long non-coding RNAs during pollen development and fertilization in Brassica rapa. Plant J. 2018, 96, 203–222. [Google Scholar] [CrossRef]
- Zou, C.; Wang, Q.; Lu, C.; Yang, W.; Zhang, Y.; Cheng, H.; Feng, X.; Prosper, M.A.; Song, G. Transcriptome analysis reveals long noncoding RNAs involved in fiber development in cotton (Gossypium arboreum). Sci. China (Life Sci.) 2016, 59, 164–171. [Google Scholar] [CrossRef]
- Hu, H.; Wang, M.; Ding, Y.; Zhu, S.; Zhao, G.; Tu, L.; Zhang, X. Transcriptomic repertoires depict the initiation of lint and fuzz fibers in cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2017, 16, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Chen, X.; Mu, M.; Wang, J.; Wang, X.; Wang, D.; Yin, Z.; Fan, W.; Wang, S.; Guo, L. Genome-wide analysis of long noncoding RNAs and their responses to drought stress in cotton (Gossypium hirsutum L.). PLoS ONE 2016, 11, e0156723. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Zhang, X.; Wang, W.; Yuan, R.; Shen, F. Identification of gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant Biol. 2018, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, M.; Zhang, B.; Zhang, X.; Guo, L.; Qi, T.; Wang, H.; Zhang, J.; Xing, C. Genome-wide comparative transcriptome analysis of CMS-D2 and its maintainer and restorer lines in upland cotton. BMC Genom. 2017, 18, 454. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, X.; Liu, G.; Guo, L.; Qi, T.; Zhang, M.; Li, X.; Wang, H.; Tang, H.; Qiao, X. A combined small RNA and transcriptome sequencing analysis reveal regulatory roles of miRNAs during another development of Upland cotton carrying cytoplasmic male sterile Gossypium harknessii (D2) cytoplasm. BMC Plant Biol. 2018, 18, 242. [Google Scholar] [CrossRef]
- Wang, F.; Stewart, J.M.; Zhang, J. Molecular markers linked to the Rf2 fertility restorer gene in cotton. Genome 2007, 50, 818–824. [Google Scholar] [CrossRef]
- Wu, J.; Cao, X.; Guo, L.; Qi, T.; Wang, H.; Tang, H.; Zhang, J.; Xing, C. Development of a candidate gene marker for Rf 1 based on a PPR gene in cytoplasmic male sterile CMS-D2 Upland cotton. Mol. Breed. 2014, 34, 231–240. [Google Scholar] [CrossRef]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef]
- Luo, S.; Lu, J.Y.; Liu, L.; Yin, Y.; Chen, C.; Han, X.; Wu, B.; Xu, R.; Liu, W.; Yan, P. Divergent lncRNAs Regulate Gene Expression and Lineage Differentiation in Pluripotent Cells. Cell Stem Cell 2016, 18, 637–652. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, X.; Sun, F.; Hu, J.; Zha, X.; Su, W.; Yang, J. Overexpressing lncRNA LAIR increases grain yield and regulates neighbouring gene cluster expression in rice. Nat. Commun. 2018, 9, 3516. [Google Scholar] [CrossRef]
- Akpinar, B.A.; Kantar, M.; Budak, H. Root precursors of microRNAs in wild emmer and modern wheats show major differences in response to drought stress. Funct. Integr. Genom. 2015, 15, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant. Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, M.; Li, N.; Wang, H.; Qiu, P.; Pei, L.; Xu, Z.; Wang, T.; Gao, E.; Liu, J. Long non-coding RNAs involve in resistance to Verticillium dahliae, a fungal disease in cotton. Plant Biotechnol. J. 2018, 16, 1172–1185. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, W.; Yang, Y.; Li, X.; Chen, T.; Liu, T.; Ma, N.; Yang, X.; Liu, R.; Zhang, B. Corrigendum: Genome-wide analysis of tomato long non-coding RNAs and identification as endogenous target mimic for microRNA in response to TYLCV infection. Sci. Rep. 2015, 5, 16946. [Google Scholar] [CrossRef]
- Fan, C.; Hao, Z.; Yan, J.; Li, G. Genome-wide identification and functional analysis of lincRNAs acting as miRNA targets or decoys in maize. BMC Genom. 2015, 16, 793. [Google Scholar] [CrossRef]
- Mallory, A.; Bartel, D.; Bartel, B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 2005, 17, 1360–1375. [Google Scholar] [CrossRef]
- Yang, J.; Tian, L.; Sun, M.X.; Huang, X.Y.; Zhu, J.; Guan, Y.F.; Jia, Q.S.; Yang, Z.N. AUXIN RESPONSE FACTOR17 is essential for pollen wall pattern formation in Arabidopsis. Plant Physiol. 2013, 162, 720–731. [Google Scholar] [CrossRef]
- Ding, Y.; Ma, Y.; Liu, N.; Xu, J.; Hu, Q.; Li, Y.; Wu, Y.; Xie, S.; Zhu, L.; Min, L. microRNAs involved in auxin signalling modulate male sterility under high-temperature stress in cotton (Gossypium hirsutum). Plant J. 2017, 91, 977–994. [Google Scholar] [CrossRef]
- Reina, K.; Hajime, O.; Mitsuru, N.; Toshiaki, W.; Mutsuko, N.; Nori, K.; Ken-Ichi, N. Rice germline-specific Argonaute MEL1 protein binds to phasiRNAs generated from more than 700 lincRNAs. Plant J. 2014, 78, 385–397. [Google Scholar]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef]
- Fan, Y.; Yang, J.; Mathioni, S.M.; Yu, J.; Shen, J.; Yang, X.; Wang, L.; Zhang, Q.; Cai, Z.; Xu, C. PMS1T, producing phased small-interfering RNAs, regulates photoperiod-sensitive male sterility in rice. Proc. Natl. Acad. Sci. USA 2016, 113, 15144–15149. [Google Scholar] [CrossRef]
- Liu, Y.; Ke, L.; Wu, G.; Xu, Y.; Wu, X.; Xia, R.; Deng, X.; Xu, Q. miR3954 is a trigger of phasiRNAs that affects flowering time in citrus. Plant J. 2017, 92, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Hideaki, S.; Laura, R.U.; Xu, J.; Zhang, J. Transcriptome analysis of cytoplasmic male sterility and restoration in CMS-D8 cotton. Plant Cell Rep. 2013, 32, 1531–1542. [Google Scholar]
- Wu, J.; Gong, Y.; Cui, M.; Qi, T.; Guo, L.; Zhang, J.; Xing, C. Molecular characterization of cytoplasmic male sterility conditioned by Gossypium harknessii cytoplasm (CMS-D2) in upland cotton. Euphytica 2011, 181, 17–29. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Cole, T.; Adam, R.; Loyal, G.; Geo, P.; Daehwan, K.; Kelley, D.R.; Harold, P.; Salzberg, S.L.; Rinn, J.L.; Lior, P. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, 222–230. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A1 | A2 | A3 | B1 | B2 | B3 | R1 | R2 | R3 | |
|---|---|---|---|---|---|---|---|---|---|
| Raw Reads | 81,937,808 | 91,168,084 | 121,573,148 | 110,284,226 | 100,663,168 | 104,176,106 | 100,740,046 | 105,585,106 | 104,618,514 |
| Clean Reads | 78,996,350 | 87,941,972 | 116,946,292 | 106,115,854 | 96,899,546 | 100,304,158 | 97,149,736 | 101,878,248 | 1008,58,264 |
| Total Mapped Reads | 69,087,555 | 76,275,950 | 101,026,262 | 92,535,847 | 84,731,402 | 87,778,855 | 85,862,554 | 90,725,753 | 89,536,452 |
| Mapped Unique Reads | 62,121,536 | 67,953,623 | 89,895,211 | 82,087,734 | 75,065,210 | 77,755,238 | 76,292,918 | 81,060,568 | 79,962,304 |
| Overall Mapping | 87.46% | 86.73% | 86.39% | 87.20% | 87.44% | 87.51% | 88.38% | 89.05% | 88.77% |
| Reads Mapped to mRNA | 21,490,343 | 24,321,071 | 31,839,943 | 29,301,509 | 27,050,077 | 27,548,951 | 27,405,358 | 29,115,198 | 28,012,425 |
| Total lncRNA | 80,695 | ||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Zhang, X.; Zhang, M.; Guo, L.; Qi, T.; Wang, H.; Tang, H.; Qiao, X.; Shahzad, K.; Xing, C.; et al. Transcriptome Analysis Implicates Involvement of Long Noncoding RNAs in Cytoplasmic Male Sterility and Fertility Restoration in Cotton. Int. J. Mol. Sci. 2019, 20, 5530. https://doi.org/10.3390/ijms20225530
Zhang B, Zhang X, Zhang M, Guo L, Qi T, Wang H, Tang H, Qiao X, Shahzad K, Xing C, et al. Transcriptome Analysis Implicates Involvement of Long Noncoding RNAs in Cytoplasmic Male Sterility and Fertility Restoration in Cotton. International Journal of Molecular Sciences. 2019; 20(22):5530. https://doi.org/10.3390/ijms20225530
Chicago/Turabian StyleZhang, Bingbing, Xuexian Zhang, Meng Zhang, Liping Guo, Tingxiang Qi, Hailin Wang, Huini Tang, Xiuqin Qiao, Kashif Shahzad, Chaozhu Xing, and et al. 2019. "Transcriptome Analysis Implicates Involvement of Long Noncoding RNAs in Cytoplasmic Male Sterility and Fertility Restoration in Cotton" International Journal of Molecular Sciences 20, no. 22: 5530. https://doi.org/10.3390/ijms20225530
APA StyleZhang, B., Zhang, X., Zhang, M., Guo, L., Qi, T., Wang, H., Tang, H., Qiao, X., Shahzad, K., Xing, C., & Wu, J. (2019). Transcriptome Analysis Implicates Involvement of Long Noncoding RNAs in Cytoplasmic Male Sterility and Fertility Restoration in Cotton. International Journal of Molecular Sciences, 20(22), 5530. https://doi.org/10.3390/ijms20225530