PFKFB3 Inhibition Attenuates Oxaliplatin-Induced Autophagy and Enhances Its Cytotoxicity in Colon Cancer Cells

 ,
,
Abstract
:1. Introduction
2. Results
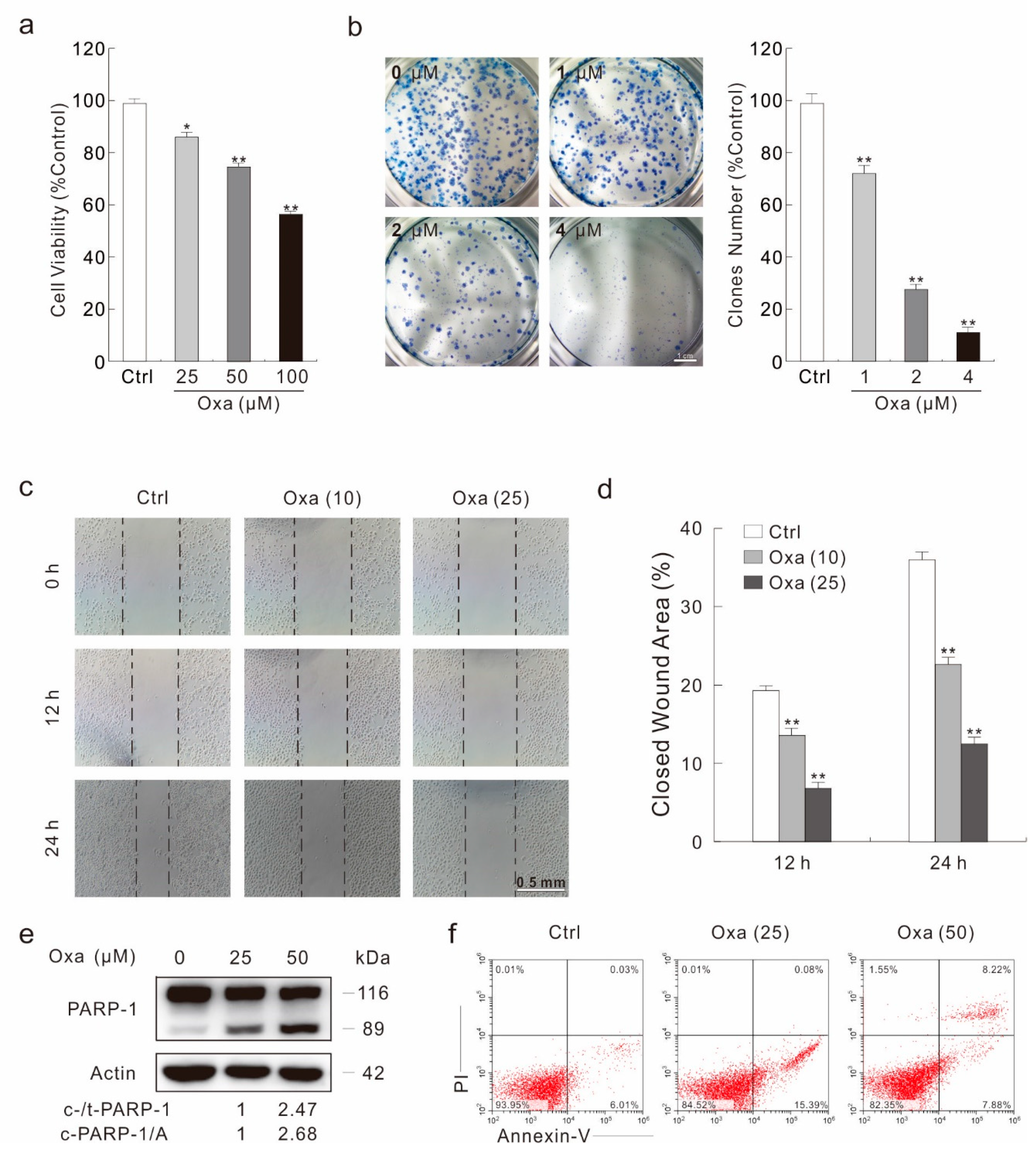
2.1. Oxa Shows Effectively Cytotoxic Effect in SW480 Cells
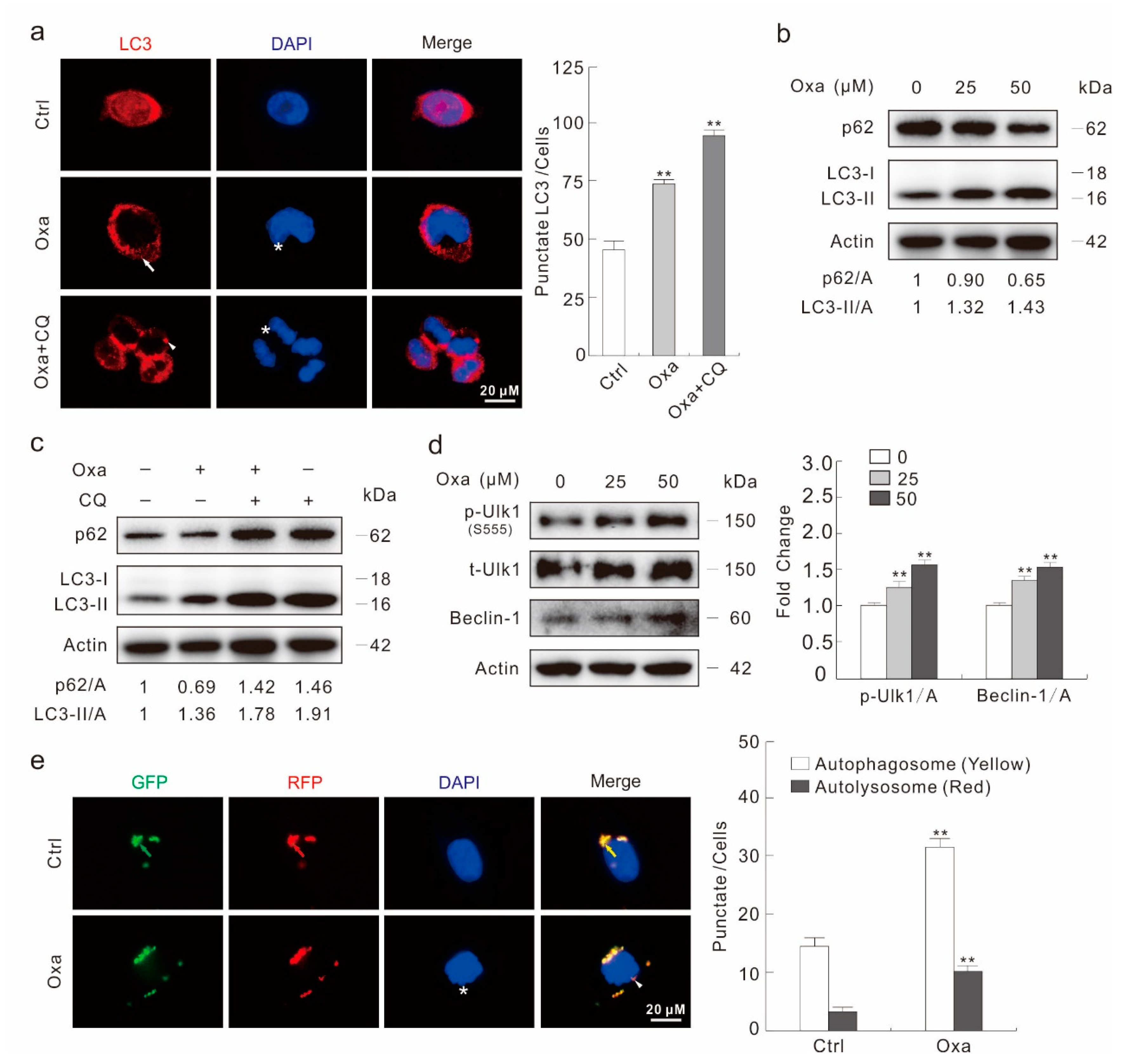
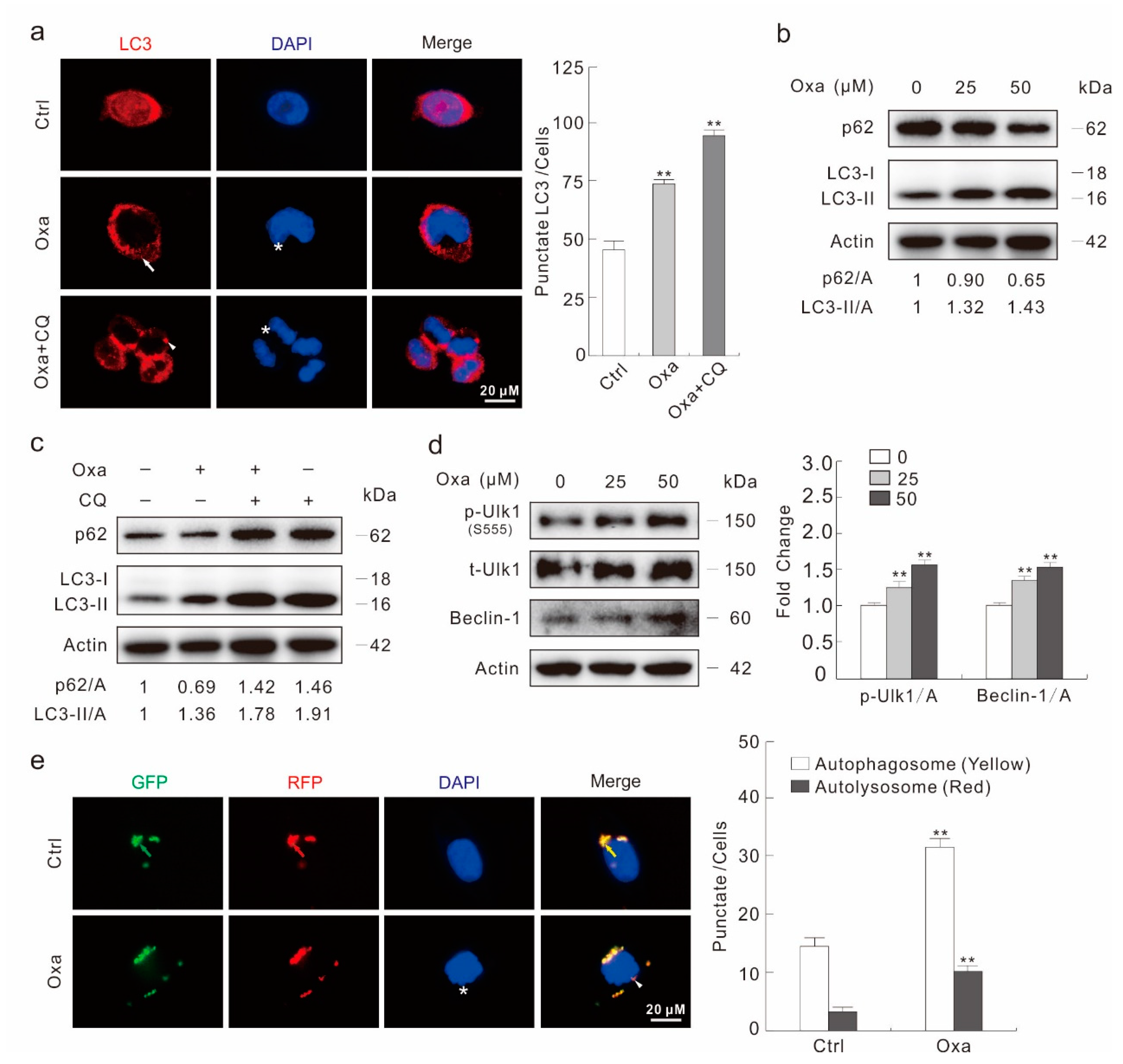
2.2. Oxa Enhances Autophagic Flux
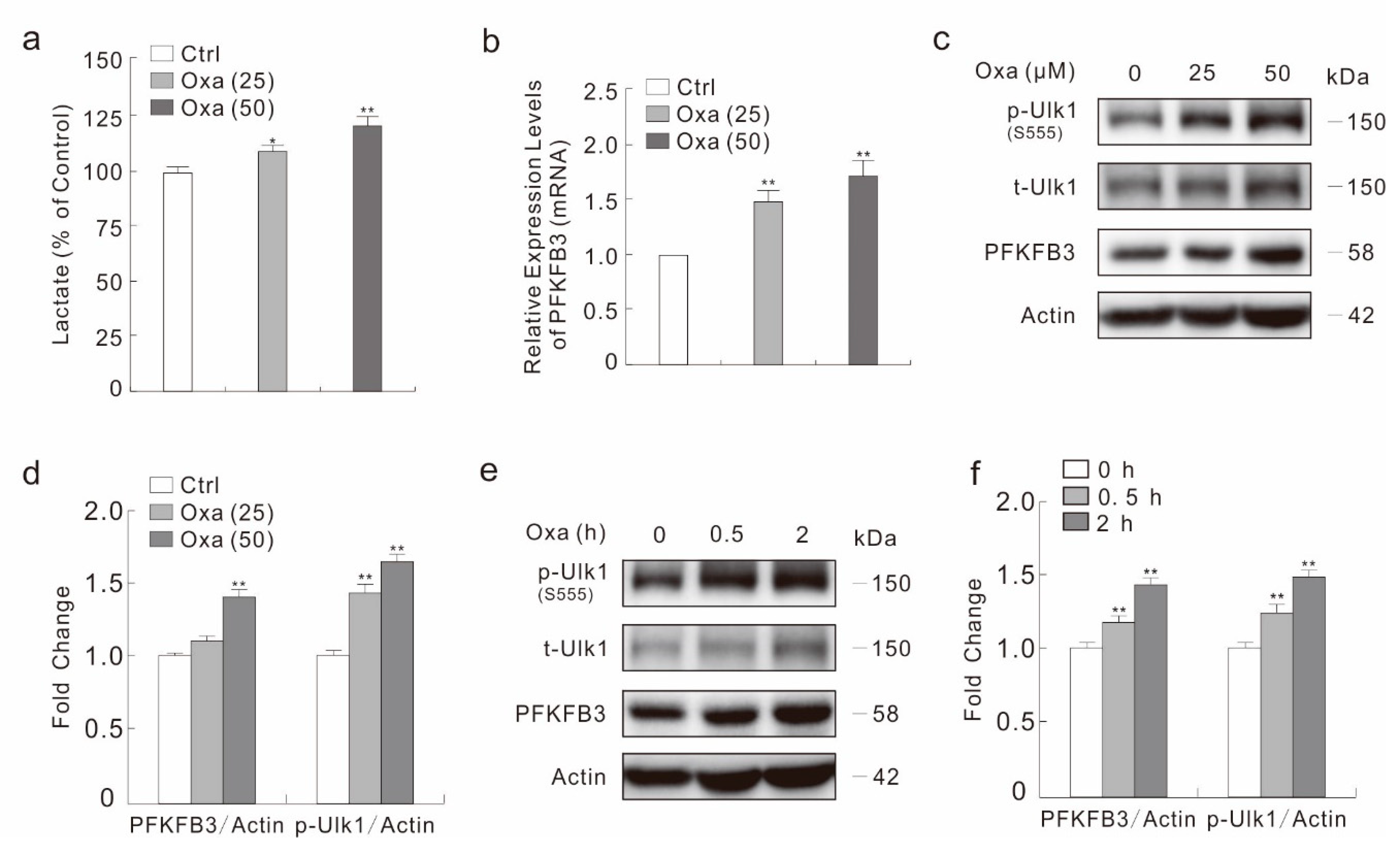
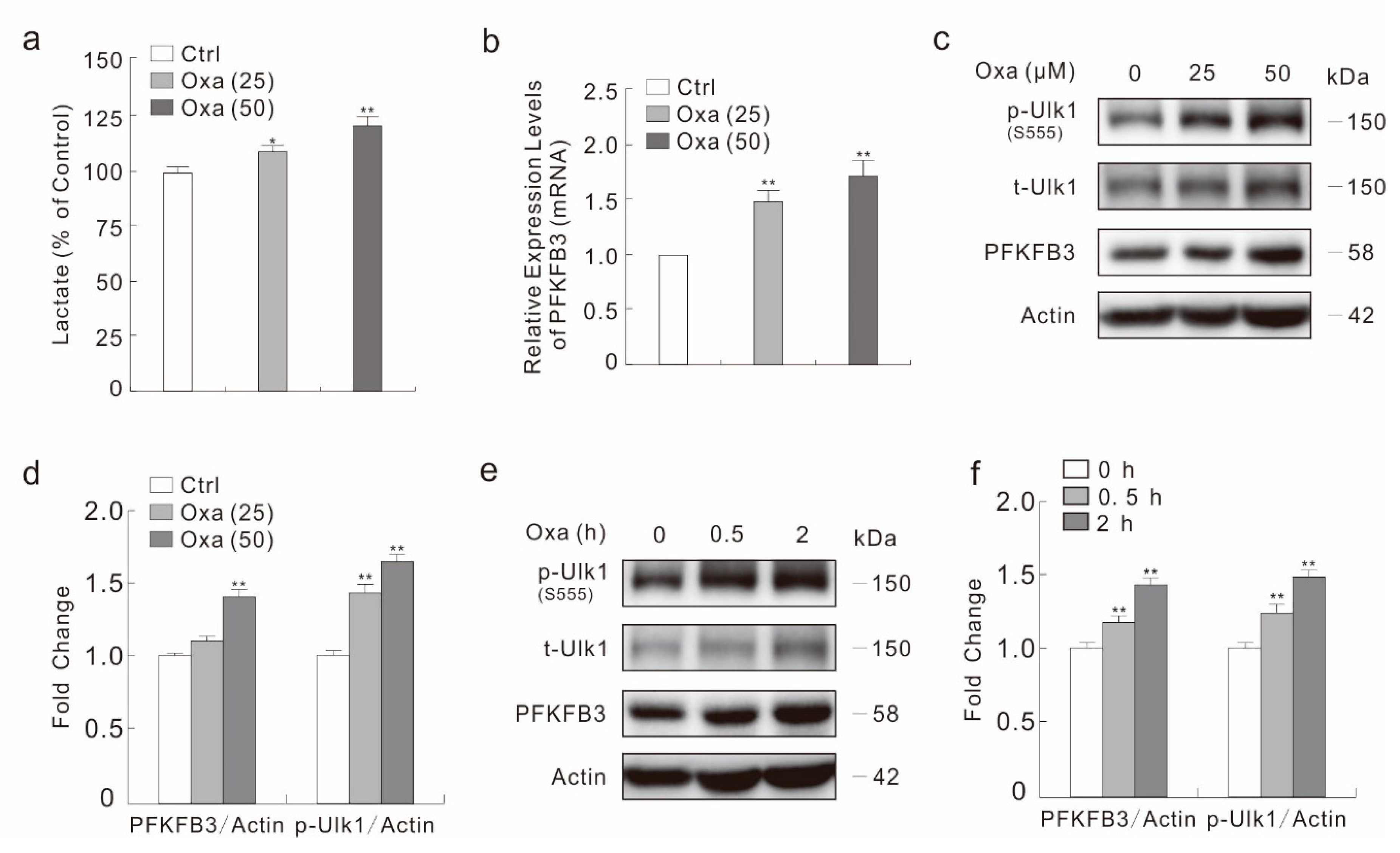
2.3. Oxa Enhances the Expression of PFKFB3
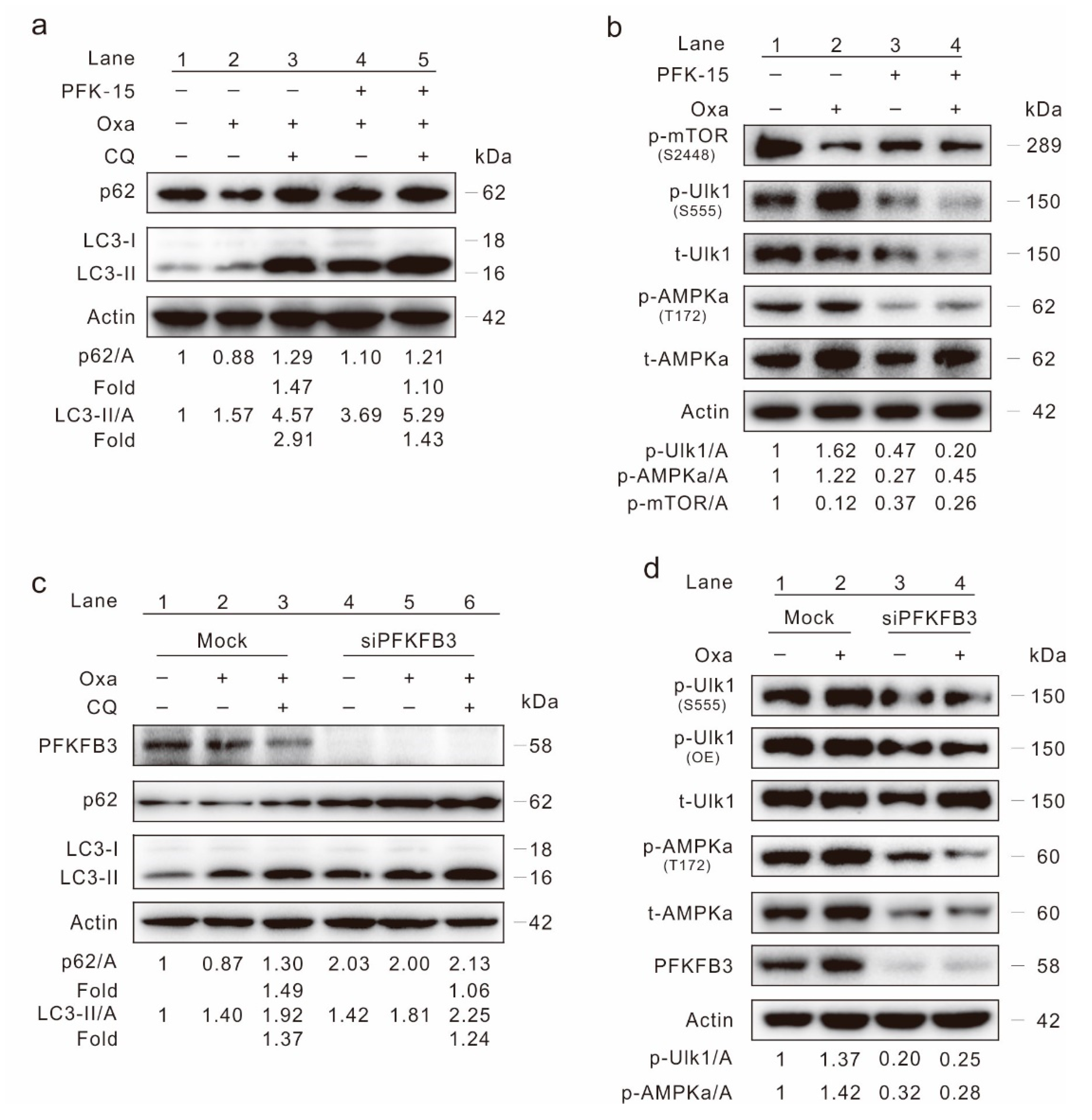
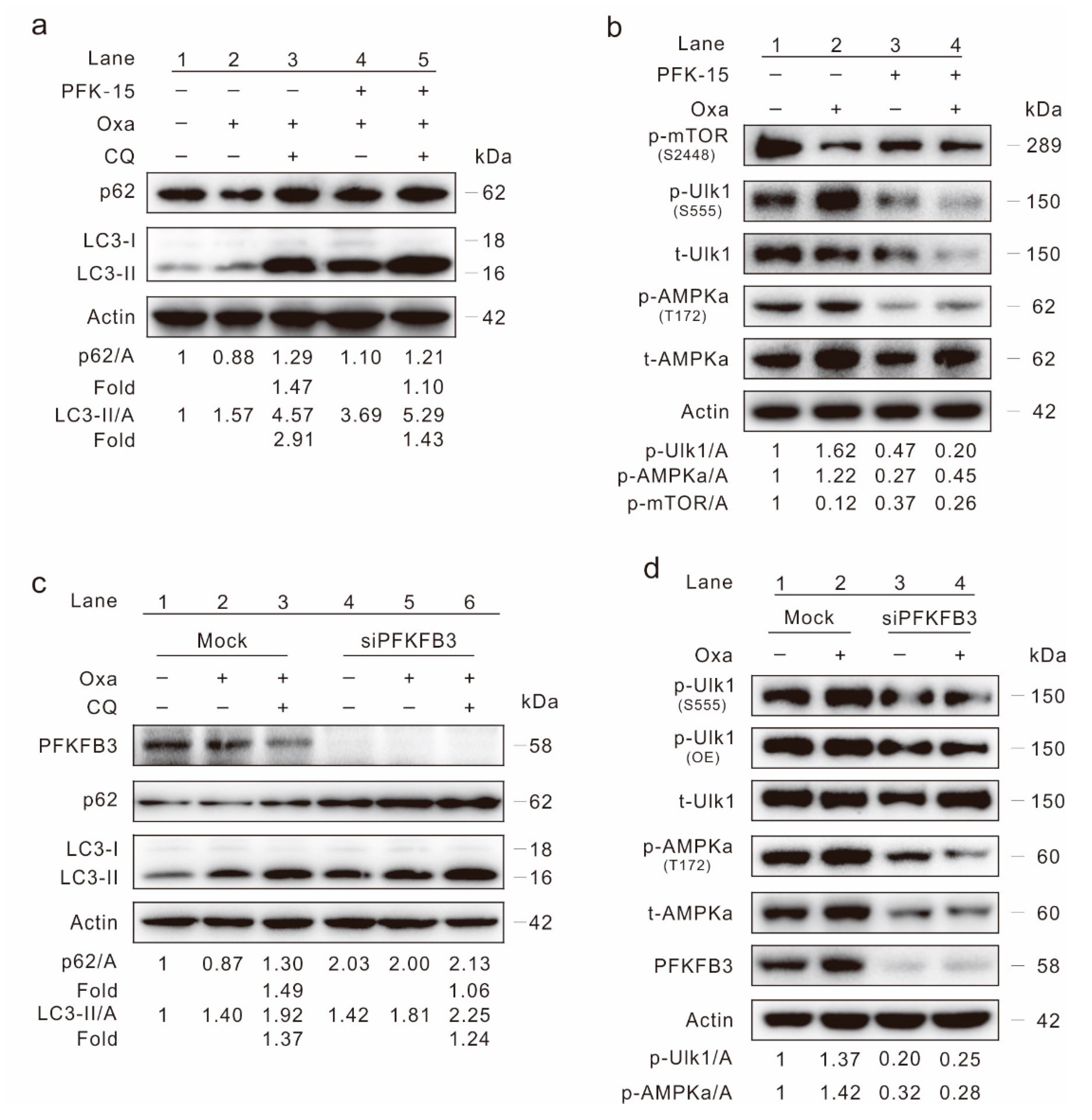
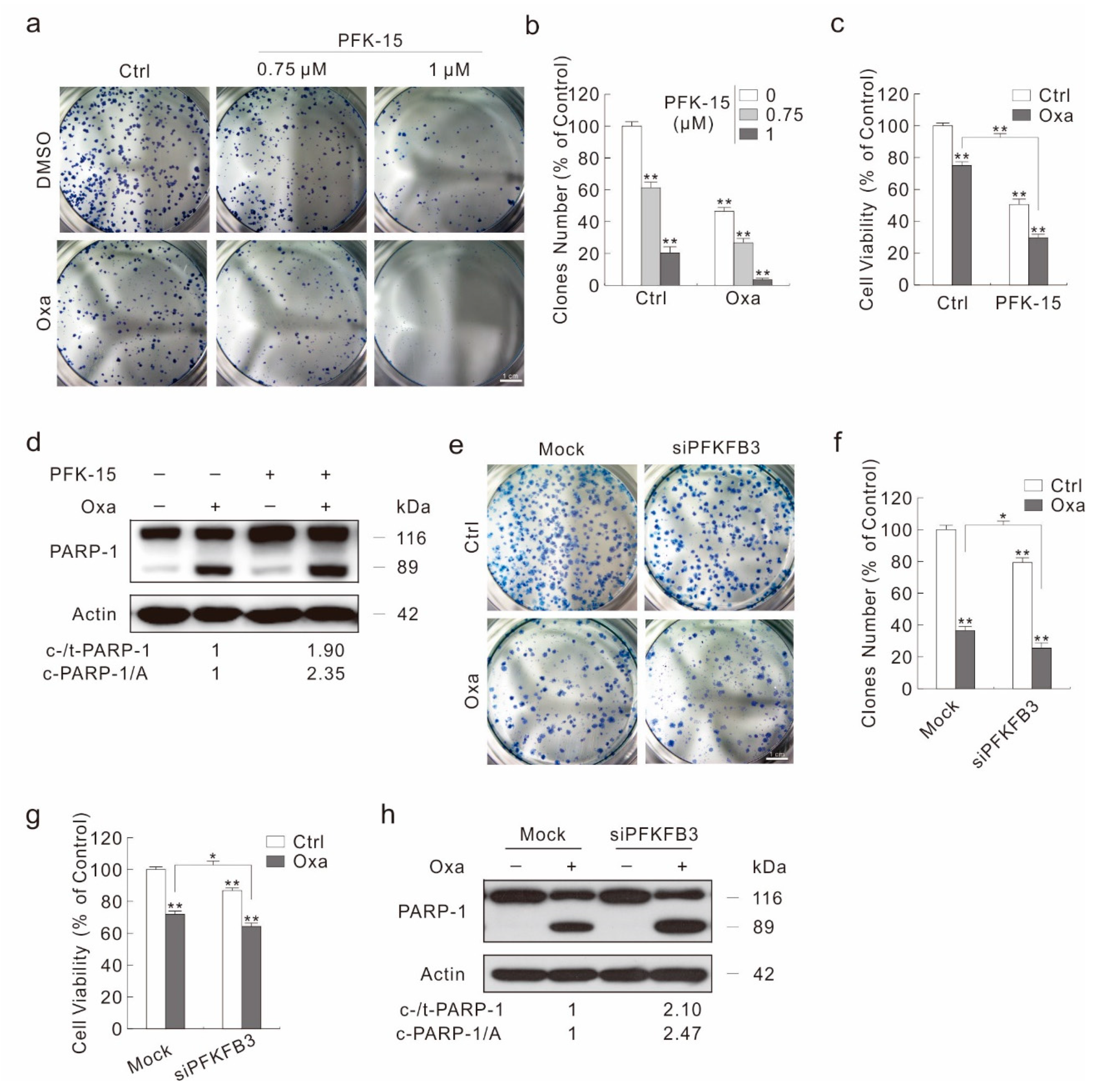
2.4. Inhibition of PFKFB3 Suppresses Oxa-Induced Autophagy
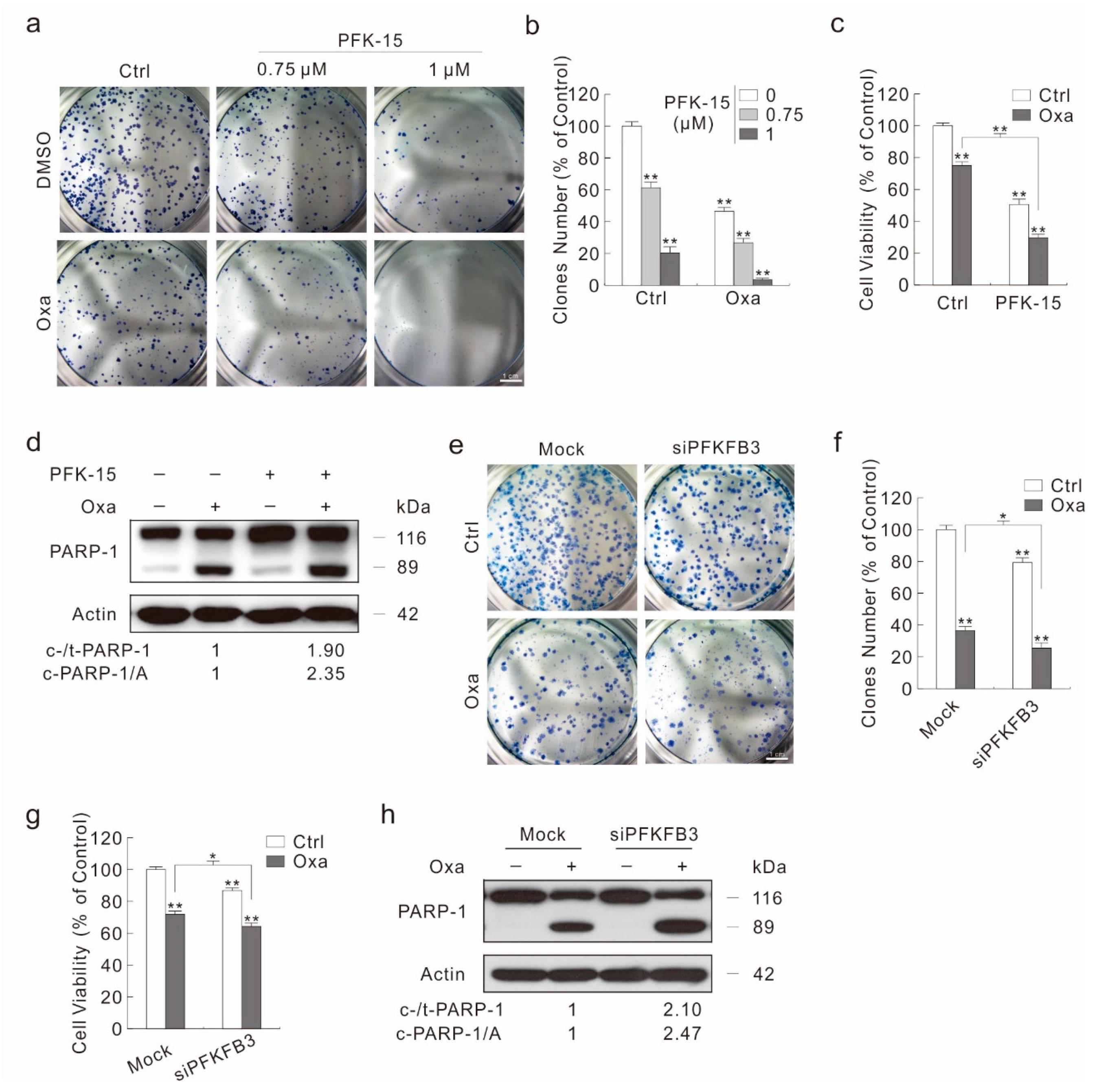
2.5. Inhibition of PFKFB3 Enhances the Cytotoxic Effect of Oxa
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Plasmids and siRNAs
4.3. Cell Culture, Plasmid Transfection and siRNA Interference
4.4. Immunoblotting Analysis
4.5. Cell Viability Assay (MTS)
4.6. Colony Growth Assay
4.7. Wound Healing Assay
4.8. Lactate Assay
4.9. Fluorescence Microscopy
4.10. Flow Cytometry Assay
4.11. RNA Extraction and Real-Time PCR Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Fan, C.; Wang, C.; Huang, Q.; Meng, W.; Yu, Y.; Yang, L.; Hu, J.; Li, Y.; Mo, X.; et al. Prognostic value of CD133+ CD54+ CD44+ circulating tumor cells in colorectal cancer with liver metastasis. Cancer Med. 2017, 6, 2850–2857. [Google Scholar] [CrossRef]
- Edwards, B.K.; Noone, A.M.; Mariotto, A.B.; Simard, E.P.; Boscoe, F.P.; Henley, S.J.; Jemal, A.; Cho, H.; Anderson, R.N.; Kohler, B.A.; et al. Annual Report to the Nation on the status of cancer, 1975–2010, featuring prevalence of comorbidity and impact on survival among persons with lung, colorectal, breast, or prostate cancer. Cancer 2014, 120, 1290–1314. [Google Scholar] [CrossRef]
- Du, Z.; Liu, X.; Chen, T.; Gao, W.; Wu, Z.; Hu, Z.; Wei, D.; Gao, C.; Li, Q. Targeting a Sirt5-Positive Subpopulation Overcomes Multidrug Resistance in Wild-Type Kras Colorectal Carcinomas. Cell Rep. 2018, 22, 2677–2689. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.-L.; Ting, H.-J.; Tseng, T.-C.; Juang, V.; Lo, Y.-L. Modulation of the mRNA-binding protein HuR as a novel reversal mechanism of epirubicin-triggered multidrug resistance in colorectal cancer cells. PLoS ONE 2017, 12, e0185625. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Cho, H.J.; Paul, S.; Jakhar, R.; Khan, I.; Lee, S.J.; Kim, B.Y.; Krishnan, M.; Khaket, T.P.; Lee, H.G.; et al. Vitexin induces apoptosis by suppressing autophagy in multi-drug resistant colorectal cancer cells. Oncotarget 2018, 9, 3278–3291. [Google Scholar] [CrossRef]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Kuwano, H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur. J. Cancer 2010, 46, 1900–1909. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Boil. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Yan, S.; Yang, X.; Chen, T.; Xi, Z.; Jiang, X. The PPARgamma agonist Troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther. 2014, 21, 188–193. [Google Scholar] [CrossRef]
- Kumar, P.; Zhang, D.-M.; Degenhardt, K.; Chen, Z.-S. Autophagy and Transporter-Based Multi-Drug Resistance. Cells 2012, 1, 558–575. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Méry, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Brown, E.B.; Campbell, R.B.; Tsuzuki, Y.; Xu, L.; Carmeliet, P.; Fukumura, D.; Jain, R.K. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nat. Med. 2001, 7, 864–868. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Ros, S.; Schulze, A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef]
- Li, F.-L.; Liu, J.-P.; Bao, R.-X.; Yan, G.; Feng, X.; Xu, Y.-P.; Sun, Y.-P.; Yan, W.; Ling, Z.-Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 508. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Wei, X.; Xu, S.; Sun, H.; Wang, W.; Liu, L.; Jiang, X.; Zhang, Y.; Che, Y. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 3 spatially mediates autophagy through the AMPK signaling pathway. Oncotarget 2017, 8, 80909–80922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalcin, A.; Clem, B.F.; Simmons, A.; Lane, A.; Nelson, K.; Clem, A.L.; Brock, E.; Siow, D.; Wattenberg, B.; Telang, S.; et al. Nuclear Targeting of 6-Phosphofructo-2-kinase (PFKFB3) Increases Proliferation via Cyclin-dependent Kinases. J. Boil. Chem. 2009, 284, 24223–24232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Qu, J.; Yan, S.; Gao, Q.; Hao, S.; Zhou, D. PFK15, a PFKFB3 antagonist, inhibits autophagy and proliferation in rhabdomyosarcoma cells. Int. J. Mol. Med. 2018, 42, 359–367. [Google Scholar] [CrossRef]
- Sarkar Bhattacharya, S.; Thirusangu, P.; Jin, L.; Roy, D.; Jung, D.; Xiao, Y.; Staub, J.; Roy, B.; Molina, J.R.; Shridhar, V. PFKFB3 inhibition reprograms malignant pleural mesothelioma to nutrient stress-induced macropinocytosis and ER stress as independent binary adaptive responses. Cell Death Dis. 2019, 10, 725. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Roy, D.; Sarkar Bhattacharya, S.; Jin, L.; Jung, D.; Zhang, S.; Kalogera, E.; Staub, J.; Wang, Y.; Chien, J.; et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. J. Int. Cancer 2019, 144, 178–189. [Google Scholar] [CrossRef]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquière, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and Transient Reduction of Glycolysis by PFKFB3 Blockade Reduces Pathological Angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Cantelmo, A.R.; Conradi, L.-C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.-A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Dai, W.; Mo, W.; Li, J.; Feng, J.; Wu, L.; Liu, T.; Yu, Q.; Xu, S.; Wang, W.; et al. By inhibiting PFKFB3, aspirin overcomes sorafenib resistance in hepatocellular carcinoma. Int. J. Cancer 2017, 141, 2571–2584. [Google Scholar] [CrossRef]
- Mou, J.-J.; Peng, J.; Shi, Y.-Y.; Li, N.; Wang, Y.; Ke, Y.; Zhou, Y.-F.; Zhou, F.-X. Mitochondrial DNA content reduction induces aerobic glycolysis and reversible resistance to drug-induced apoptosis in SW480 colorectal cancer cells. Biomed. Pharm. 2018, 103, 729–737. [Google Scholar] [CrossRef]
- Lu, W.-Q.; Hu, Y.-Y.; Lin, X.-P.; Fan, W. Knockdown of PKM2 and GLS1 expression can significantly reverse oxaliplatin-resistance in colorectal cancer cells. Oncotarget 2017, 8, 44171–44185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekert, P.G.; Silke, J.; Vaux, D.L. Caspase inhibitors. Cell Death Differ. 1999, 6, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Yan, S.; Sun, H.; Wang, W.; Li, Y.; Yang, X.; Jiang, X.; Che, Y.; Xi, Z. Akt inhibition attenuates rasfonin-induced autophagy and apoptosis through the glycolytic pathway in renal cancer cells. Cell Death Dis. 2015, 6, e2005. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Fujii, H.; Mohan, S.V.; Goronzy, J.J.; Weyand, C.M. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J. Exp. Med. 2013, 210, 2119–2134. [Google Scholar] [CrossRef] [Green Version]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in Cancer Therapy: A Double-Edged Sword of Autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar] [CrossRef]
- El-Khattouti, A.; Selimovic, D.; Haikel, Y.; Hassan, M. Crosstalk between Apoptosis and Autophagy: Molecular Mechanisms and Therapeutic Strategies in Cancer. J. Cell Death 2013, 6, 37–55. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [Green Version]
- Cho, D.-H.; Jo, Y.K.; Hwang, J.J.; Lee, Y.M.; Roh, S.A.; Kim, J.C. Caspase-mediated cleavage of ATG6/Beclin-1 links apoptosis to autophagy in HeLa cells. Cancer Lett. 2009, 274, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Takano, N.; Ishiwata, K.; Ohmura, M.; Nagahata, Y.; Matsuura, T.; Kamata, A.; Sakamoto, K.; Nakanishi, T.; Kubo, A.; et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat. Commun. 2014, 5, 3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; An, X.; Guo, X.; Habtetsion, T.G.; Wang, Y.; Xu, X.; Kandala, S.; Li, Q.; Li, H.; Zhang, C.; et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler. Thromb. Vasc. Boil. 2014, 34, 1231–1239. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward/Reverse | Nucleotides |
|---|---|---|
| PFKFB3 | Forward (5′→3′) | GTGCCTTAGCTGCCTTGAGA |
| Reverse (5′→3′) | CCGACTCGATGAAAAACGCC | |
| LC3 | Forward (5′→3′) | TAGAAGGCGCTTACAGCTCAAT |
| Reverse (5′→3′) | ACTGACAATTTCATCCCGAACG | |
| p62 | Forward (5′→3′) | CATCGGAGGATCCGAGTGTG |
| Reverse (5′→3′) | TTCTTTTCCCTCCGTGCTCC | |
| β-Actin | Forward (5′→3′) | GCCTGACGGCCAGGTCATCAC |
| Reverse (5′→3′) | CGGATGTCCACGTCACACTTC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, S.; Zhou, N.; Zhang, D.; Zhang, K.; Zheng, W.; Bao, Y.; Yang, W. PFKFB3 Inhibition Attenuates Oxaliplatin-Induced Autophagy and Enhances Its Cytotoxicity in Colon Cancer Cells. Int. J. Mol. Sci. 2019, 20, 5415. https://doi.org/10.3390/ijms20215415
Yan S, Zhou N, Zhang D, Zhang K, Zheng W, Bao Y, Yang W. PFKFB3 Inhibition Attenuates Oxaliplatin-Induced Autophagy and Enhances Its Cytotoxicity in Colon Cancer Cells. International Journal of Molecular Sciences. 2019; 20(21):5415. https://doi.org/10.3390/ijms20215415
Chicago/Turabian StyleYan, Siyuan, Nan Zhou, Deru Zhang, Kaile Zhang, Wenao Zheng, Yonghua Bao, and Wancai Yang. 2019. "PFKFB3 Inhibition Attenuates Oxaliplatin-Induced Autophagy and Enhances Its Cytotoxicity in Colon Cancer Cells" International Journal of Molecular Sciences 20, no. 21: 5415. https://doi.org/10.3390/ijms20215415
APA StyleYan, S., Zhou, N., Zhang, D., Zhang, K., Zheng, W., Bao, Y., & Yang, W. (2019). PFKFB3 Inhibition Attenuates Oxaliplatin-Induced Autophagy and Enhances Its Cytotoxicity in Colon Cancer Cells. International Journal of Molecular Sciences, 20(21), 5415. https://doi.org/10.3390/ijms20215415