Alignment-Free Method to Predict Enzyme Classes and Subclasses



Abstract
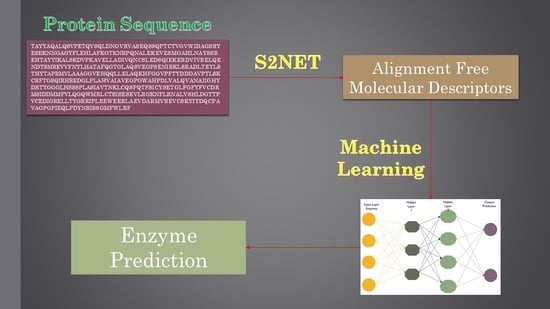
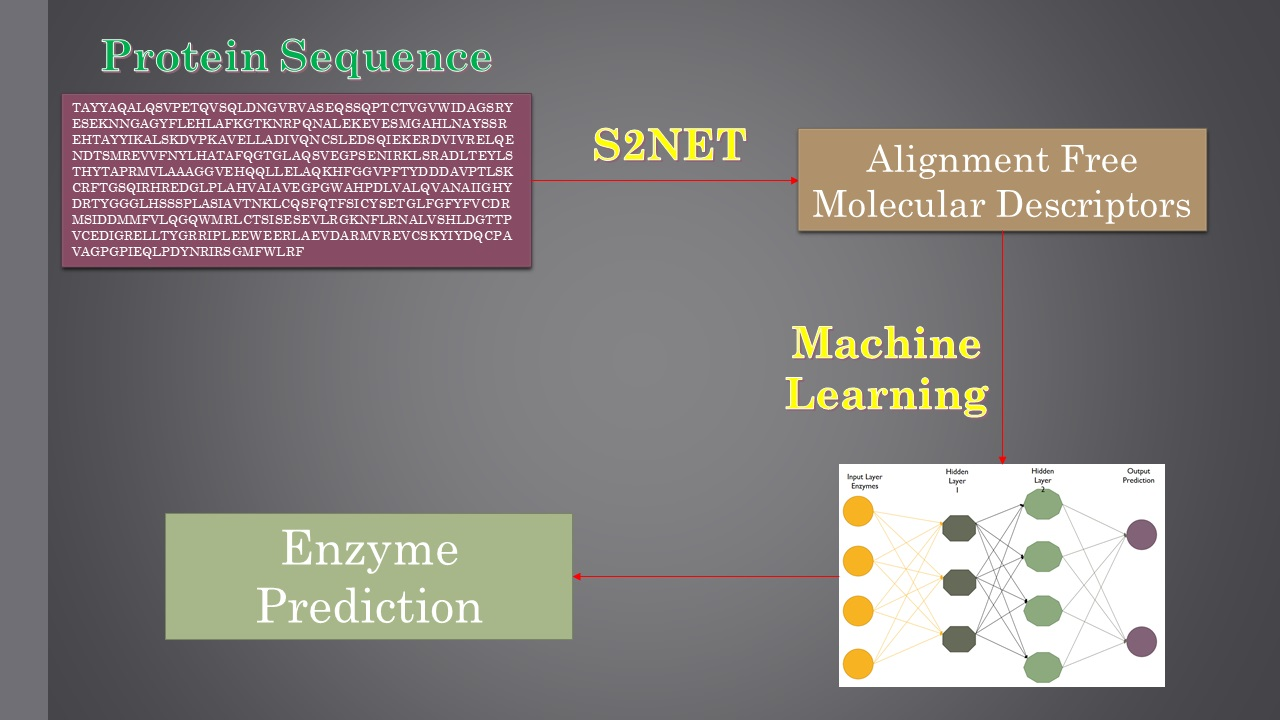
1. Introduction
2. Results
2.1. LDA Model
2.2. ANN models
3. Discussion
4. Materials and Methods
4.1. Dataset
4.2. Molecular Descriptor Calculation
4.3. Multi-Target Linear model
4.4. Non-Linear Models
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nomenclature, E. Enzyme nomenclature: Recommendations (1972) of the international union of pure and applied chemistry and the international union of biochemistry: Supplement i: Corrections & additions (1975). Biochim. Et Biophys. Acta (BBA) Enzymol. 1976, 429, 1–45. [Google Scholar]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Di Costanzo, L.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2016, 45, D271–D281. [Google Scholar] [PubMed]
- Jensen, L.J.; Gupta, R.; Blom, N.S.; Devos, D.; Tamames, J.; Kesmir, C.; Nielsen, H.; Stærfeldt, H.; Rapacki, K.; Workman, C.; et al. Prediction of Human Protein Function from Post-translational Modifications and Localization Features. J. Mol. Biol. 2002, 319, 1257–1265. [Google Scholar] [CrossRef]
- Davidson, N.J.; Wang, X. Non-Alignment Features based Enzyme/Non-Enzyme Classification Using an Ensemble Method. In Proceedings of the Ninth International Conference on Machine Learning and Applications, Washington, DC, USA, 12–14 December 2010; pp. 546–551. [Google Scholar]
- Wang, Y.C.; Wang, X.B.; Yang, Z.X.; Deng, N.Y. Prediction of enzyme subfamily class via pseudo amino acid composition by incorporating the conjoint triad feature. Protein Pept. Lett. 2010, 17, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Concu, R.; Dias Soeiro Cordeiro, M.; Munteanu, C.R.; Gonzalez-Diaz, H. Ptml model of enzyme subclasses for mining the proteome of bio-fuel producing microorganisms. J. Proteome Res. 2019, 18, 2735–2746. [Google Scholar] [CrossRef]
- Dobson, P.D.; Doig, A.J. Distinguishing enzyme structures from non-enzymes without alignments. J. Mol. Biol. 2003, 330, 771–783. [Google Scholar] [CrossRef]
- Che, Y.; Ju, Y.; Xuan, P.; Long, R.; Xing, F. Identification of Multi-Functional Enzyme with Multi-Label Classifier. PLoS ONE 2016, 11, e0153503. [Google Scholar] [CrossRef]
- Amidi, A.; Amidi, S.; Vlachakis, D.; Megalooikonomou, V.; Paragios, N.; Zacharaki, E.I. EnzyNet: Enzyme classification using 3D convolutional neural networks on spatial representation. PeerJ 2018, 6, e4750. [Google Scholar] [CrossRef]
- Hu, Q.N.; Zhu, H.; Li, X.; Zhang, M.; Deng, Z.; Yang, X.; Deng, Z. Assignment of EC Numbers to Enzymatic Reactions with Reaction Difference Fingerprints. PLoS ONE 2012, 7, e52901. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Chilton, J.M.; Grüning, B.; Johnson, J.E.; Soranzo, N. Ncbi blast integrated into galaxy. Gigascience 2015, 4, 39. [Google Scholar] [CrossRef]
- Todd, A.E.; Orengo, C.A.; Thornton, J.M. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 2001, 307, 1113–1143. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Skolnick, J. How Well is Enzyme Function Conserved as a Function of Pairwise Sequence Identity? J. Mol. Biol. 2003, 333, 863–882. [Google Scholar] [CrossRef] [PubMed]
- Rost, B.; Liu, J.; Nair, R.; Wrzeszczynski, K.O.; Ofran, Y. Automatic prediction of protein function. Cell. Mol. Life Sci. CMLS 2003, 60, 2637–2650. [Google Scholar] [PubMed]
- Nagao, C.; Nagano, N.; Mizuguchi, K. Prediction of Detailed Enzyme Functions and Identification of Specificity Determining Residues by Random Forests. PLoS ONE 2014, 9, 84623. [Google Scholar] [CrossRef] [PubMed]
- Quester, S.; Schomburg, D. EnzymeDetector: An integrated enzyme function prediction tool and database. BMC Bioinform. 2011, 12, 376. [Google Scholar] [CrossRef]
- The UniProt, C. Ongoing and future developments at the universal protein resource. Nucleic Acids Res. 2011, 39, D214–D219. [Google Scholar] [CrossRef]
- Kanehisa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Frishman, D.; Mokrejs, M.; Kosykh, D.; Kastenmüller, G.; Kolesov, G.; Zubrzycki, I.; Gruber, C.; Geier, B.; Kaps, A.; Albermann, K.; et al. The pedant genome database. Nucleic Acids Res. 2003, 31, 207–211. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Umarov, R.; Xie, B.; Fan, M.; Li, L.; Gao, X. Deepre: Sequence-based enzyme ec number prediction by deep learning. Bioinformatics 2018, 34, 760–769. [Google Scholar] [CrossRef]
- Dalkiran, A.; Rifaioglu, A.S.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. ECPred: A tool for the prediction of the enzymatic functions of protein sequences based on the EC nomenclature. BMC Bioinform. 2018, 19, 334. [Google Scholar] [CrossRef]
- Shen, H.B.; Chou, K.C. EzyPred: A top–down approach for predicting enzyme functional classes and subclasses. Biochem. Biophys. Res. Commun. 2007, 364, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A. The enzyme data bank. Nucleic Acids Res. 1993, 21, 3155–3156. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.; Choudhary, A. A top-down approach to classify enzyme functional classes and sub-classes using random forest. EURASIP J. Bioinform. Syst. Biol. 2012, 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Matsuta, Y.; Ito, M.; Tohsato, Y. Ecoh: An enzyme commission number predictor using mutual information and a support vector machine. Bioinformatics 2013, 29, 365–372. [Google Scholar] [CrossRef]
- Agüero-Chapin, G.; González-Díaz, H.; Molina, R.; Varona-Santos, J.; Uriarte, E.; González-Díaz, Y. Novel 2D maps and coupling numbers for protein sequences. The first QSAR study of polygalacturonases; isolation and prediction of a novel sequence fromPsidium guajavaL. FEBS Lett. 2006, 580, 723–730. [Google Scholar] [CrossRef]
- Concu, R.; Dea-Ayuela, M.; Pérez-Montoto, L.G.; Prado-Prado, F.J.; Uriarte, E.; Fernandez, F.B.; Podda, G.; Pazos, A.; Munteanu, C.-R.; Ubeira, F.; et al. 3D entropy and moments prediction of enzyme classes and experimental-theoretic study of peptide fingerprints in Leishmania parasites. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2009, 1794, 1784–1794. [Google Scholar] [CrossRef]
- Concu, R.; Dea-Ayuela, M.A.; Pérez-Montoto, L.G.; Bolas-Fernández, F.; Prado-Prado, F.J.; Podda, G.; Uriarte, E.; Ubeira, F.M.; González-Díaz, H. Prediction of Enzyme Classes from 3D Structure: A General Model and Examples of Experimental-Theoretic Scoring of Peptide Mass Fingerprints ofLeishmaniaProteins. J. Proteome Res. 2009, 8, 4372–4382. [Google Scholar]
- Bernardes, J.S.; E Pedreira, C. A review of protein function prediction under machine learning perspective. Recent Pat. Biotechnol. 2013, 7, 122–141. [Google Scholar] [CrossRef]
- Barigye, S.J.; Marrero-Ponce, Y.; Pérez-Giménez, F.; Bonchev, D. Trends in information theory-based chemical structure codification. Mol. Divers. 2014, 18, 673–686. [Google Scholar] [CrossRef]
- Graham, D.J.; Malarkey, C.; Schulmerich, M.V. Information Content in Organic Molecules: Quantification and Statistical Structure via Brownian Processing. J. Chem. Inf. Comput. Sci. 2004, 35, 44. [Google Scholar]
- Graham, D.J.; Schacht, D. Base information content in organic molecular formulae. J. Chem. Inf. Comput. Sci. 2000, 40, 942. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.J. Information content and organic molecules: Aggregation states and solvent effects. J. Chem. Inf. Modeling 2005, 45, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.J. Information Content in Organic Molecules: Brownian Processing at Low Levels. J. Chem. Inf. Modeling 2007, 38, 376–389. [Google Scholar] [CrossRef] [PubMed]
- González-Díaz, H.; Molina, R.; Uriarte, E. Markov entropy backbone electrostatic descriptors for predicting proteins biological activity. Bioorganic Med. Chem. Lett. 2004, 14, 4691–4695. [Google Scholar]
- González-Díaz, H.; Saíz-Urra, L.; Molina, R.; Santana, L.; Uriarte, E. A Model for the Recognition of Protein Kinases Based on the Entropy of 3D van der Waals Interactions. J. Proteome Res. 2007, 6, 904–908. [Google Scholar] [CrossRef]
- Riera-Fernandez, P.; Munteanu, C.-R.; Escobar, M.; Prado-Prado, F.J.; Martín-Romalde, R.; Pereira, D.; Villalba, K.; Duardo-Sánchez, A.; González-Díaz, H. New Markov–Shannon Entropy models to assess connectivity quality in complex networks: From molecular to cellular pathway, Parasite–Host, Neural, Industry, and Legal–Social networks. J. Theor. Biol. 2012, 293, 174–188. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef]
- Basak, S.C.; Natarajan, R.; Mills, D.; Hawkins, D.M.; Kraker, J.J. Quantitative Structure—Activity Relationship Modeling of Juvenile Hormone Mimetic Compounds for Culex pipiens Larvae, with a Discussion of Descriptor-Thinning Methods. J. Chem. Inf. Modeling 2006, 37, 65–77. [Google Scholar] [CrossRef]
- Hill, T.; Lewicki, P. Statistics Methods and Applications. In A Comprehensive Reference for Science, Industry and Data Mining; StatSoft: Tulsa, OK, USA, 2006; Volume 1, p. 813. [Google Scholar]
- Shahsavari, S.; Bagheri, G.; Mahjub, R.; Bagheri, R.; Radmehr, M.; Rafiee-Tehrani, M.; Dorkoosh, F.A. Application of artificial neural networks for optimization of preparation of insulin nanoparticles composed of quaternized aromatic derivatives of chitosan. Drug Res. 2014, 64, 151–158. [Google Scholar] [CrossRef][Green Version]
- Tenorio-Borroto, E.; Rivas, C.G.P.; Chagoyán, J.C.V.; Castañedo, N.; Prado-Prado, F.J.; Garcia-Mera, X.; González-Díaz, H. ANN multiplexing model of drugs effect on macrophages; theoretical and flow cytometry study on the cytotoxicity of the anti-microbial drug G1 in spleen. Bioorganic Med. Chem. 2012, 20, 6181–6194. [Google Scholar] [CrossRef]
- Honório, K.M.; De Lima, E.F.; Quiles, M.G.; Romero, R.A.F.; Molfetta, F.A.; Da Silva, A.B.F.; Da Silva, A.B.F. Artificial Neural Networks and the Study of the Psychoactivity of Cannabinoid Compounds. Chem. Biol. Drug Des. 2010, 75, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Choi, S.H.; Lee, N.K.; Kang, S.K.; Choi, Y.J.; Shin, J.M.; Choi, K.; Jung, D.H. Machine learning study for the prediction of transdermal peptide. J. Comput. Mol. Des. 2011, 25, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Erol, R.; Ogulata, S.N.; Sahin, C.; Alparslan, Z.N.; Erol, R. A Radial Basis Function Neural Network (RBFNN) Approach for Structural Classification of Thyroid Diseases. J. Med Syst. 2008, 32, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Bezerianos, A.; Papadimitriou, S.; Alexopoulos, D. Radial basis function neural networks for the characterization of heart rate variability dynamics. Artif. Intell. Med. 1999, 15, 215–234. [Google Scholar] [CrossRef]
- Munteanu, C.-R.; Magalhaes, A.; Duardo-Sánchez, A.; Pazos, A.; González-Díaz, H. S2SNet: A Tool for Transforming Characters and Numeric Sequences into Star Network Topological Indices in Chemoinformatics, Bioinformatics, Biomedical, and Social-Legal Sciences. Curr. Bioinform. 2013, 8, 429–437. [Google Scholar] [CrossRef]
- Vazquez, J.; Aguiar, V.; Seoane, J.A.; Freire, A.; Serantes, J.; Dorado, J.; Pazos, A.; Munteanu, C.-R. Star Graphs of Protein Sequences and Proteome Mass Spectra in Cancer Prediction. Curr. Proteom. 2009, 6, 275–288. [Google Scholar] [CrossRef]
- Randić, M.; Zupan, J.; Vikic-Topic, D. On representation of proteins by star-like graphs. J. Mol. Graph. Model. 2007, 26, 290–305. [Google Scholar] [CrossRef]
- Fernández-Blanco, E.; Aguiar-Pulido, V.; Munteanu, C.R.; Dorado, J. Random Forest classification based on star graph topological indices for antioxidant proteins. J. Theor. Biol. 2013, 317, 331–337. [Google Scholar] [CrossRef]
- Fernandez-Lozano, C.; Cuiñas, R.F.; Seoane, J.A.; Fernández-Blanco, E.; Dorado, J.; Munteanu, C.-R. Classification of signaling proteins based on molecular star graph descriptors using Machine Learning models. J. Theor. Biol. 2015, 384, 50–58. [Google Scholar] [CrossRef]
- Munteanu, C.R.; González-Díaz, H.; Magalhães, A.L. Enzymes/non-enzymes classification model complexity based on composition, sequence, 3D and topological indices. J. Theor. Biol. 2008, 254, 476–482. [Google Scholar] [CrossRef]
- Wang, H.; Yan, L.; Huang, H.; Ding, C. From Protein Sequence to Protein Function via Multi-Label Linear Discriminant Analysis. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Hendryli, J.; Fanany, M.I. Classifying Abnormal Activities in Exam using Multi-Class Markov Chain LDA Based on MODEC Features. In Proceedings of the 4th International Conference on Information and Communication Technology (ICoICT), Bandung, Indonesia, 25–27 May 2016; pp. 1–6. [Google Scholar]
- Safo, S.E.; Ahn, J. General sparse multi-class linear discriminant analysis. Comput. Stat. Data Anal. 2016, 99, 81–90. [Google Scholar] [CrossRef]
- Beleites, C.; Salzer, R. Assessing and improving the stability of chemometric models in small sample size situations. Anal. Bioanal. Chem. 2008, 390, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Ion-Mărgineanu, A.; Kocevar, G.; Stamile, C.; Sima, D.M.; Durand-Dubief, F.; Van Huffel, S.; Sappey-Marinier, D. Machine Learning Approach for Classifying Multiple Sclerosis Courses by Combining Clinical Data with Lesion Loads and Magnetic Resonance Metabolic Features. Front. Mol. Neurosci. 2017, 11, 398. [Google Scholar] [CrossRef] [PubMed]
- Boughorbel, S.; Jarray, F.; El-Anbari, M. Optimal classifier for imbalanced data using Matthews Correlation Coefficient metric. PLoS ONE 2017, 12, 0177678. [Google Scholar] [CrossRef]

{kind=link}
| Training | Validation | Overall | |||||||
|---|---|---|---|---|---|---|---|---|---|
| All | −1 = Sn | 1 = Sp | All | −1 = Sn | 1 = Sp | All | −1 = Sn | 1 = Sp | |
| −1 | 98.13 | 40,781 | 778 | 98.27 | 13,613 | 240 | 98.16 | 54,394 | 1018 |
| 1 | 99.7 | 57 | 19,498 | 99.71 | 19 | 6498 | 99.71 | 76 | 25,996 |
| Total | 98.63 | 40,838 | 20,276 | 98.73 | 13,632 | 6738 | 98.66 | 54,470 | 27,014 |
| Eigenvalue | CanonicalR | Wilk’sLambda | Chi-Sqr. | df | p-value | MCC |
|---|---|---|---|---|---|---|
| 1.241879 | 0.744275 | 0.446054 | 49334.99 | 4.000000 | 0.00 | 0.97 |
| Obs. Sets a | Stat. Param. a | Pred. Stat. a | Predicted sets | ||
|---|---|---|---|---|---|
| 1 | −1 | nj | |||
| Training Series | |||||
| 1 | Sp a | 100 | 17,500 | 0 | 57,039 |
| −1 | Sn a | 100 | 0 | 39,539 | 0 |
| total | Ac a | 100 | 17,500 | 39,539 | 57,039 |
| Validation Series | |||||
| 1 | Sp a | 100 | 8572 | 0 | 24,445 |
| −1 | Sn a | 100 | 0 | 15,873 | 0 |
| total | Ac a | 100 | 8572 | 15,873 | 24,445 |
| Overall | |||||
| 1 | Sp a | 100 | 26,072 | 0 | 81,484 |
| −1 | Sn a | 100 | 0 | 55,412 | 0 |
| total | Ac a | 100 | 26,072 | 55,412 | 81,484 |
| Training | Validation | Overall | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Model | −1 = Sn | 1 = Sp | All | −1 = Sn | 1 = Sp | All | −1 = Sn | 1 = Sp | All | |
| BEST MLP: 4-9-2 | Total | 55,412 | 26,072 | 81,484 | 55,412 | 26,072 | 81,484 | 55,412 | 26,072 | 81,484 |
| Correct | 55,412 | 26,072 | 81,484 | 55,412 | 26,072 | 81,484 | 55,412 | 26,072 | 81,484 | |
| Incorrect | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Correct (%) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | |
| Incorrect (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| 1.MLP 4-7-2 | Total | 39,448 | 17,591 | 57,039 | 15,873 | 8572 | 24,445 | 55,412 | 26,072 | 81,484 |
| Correct | 39,448 | 17,567 | 57,015 | 15,873 | 8562 | 24,435 | 55,412 | 26,034 | 81,446 | |
| Incorrect | 0 | 24 | 24 | 0 | 10 | 10 | 0 | 38 | 38 | |
| Correct (%) | 100 | 99.86 | 99.96 | 100.00 | 99.88 | 99.96 | 100.00 | 99.85 | 99.95 | |
| Incorrect (%) | 0 | 0.14 | 0.04 | 0.00 | 0.12 | 0.04 | 0.00 | 0.15 | 0.05 | |
| 2.MLP 4-8-2 | Total | 39,448 | 17,591 | 57,039 | 15,873 | 8572 | 24,445 | 55,412 | 26,072 | 81,484 |
| Correct | 39,448 | 17,565 | 57,013 | 15,873 | 8563 | 24,436 | 55,412 | 26,037 | 81,449 | |
| Incorrect | 0 | 26 | 26 | 0 | 9 | 9 | 0 | 35 | 35 | |
| Correct (%) | 100 | 99.85 | 99.95 | 100.00 | 99.90 | 99.96 | 100.00 | 99.87 | 99.96 | |
| Incorrect (%) | 0 | 0.15 | 0.05 | 0.00 | 0.10 | 0.04 | 0.00 | 0.13 | 0.04 | |
| 3.MLP 4-10-2 | Total | 39,448 | 17,591 | 57,039 | 15,873 | 8572 | 24,445 | 55,412 | 26,072 | 81,484 |
| Correct | 39,448 | 17,565 | 57,013 | 15,873 | 8563 | 24,436 | 55,412 | 26,037 | 81,449 | |
| Incorrect | 0 | 26 | 26 | 0 | 9 | 9 | 0 | 35 | 35 | |
| Correct (%) | 100 | 99.85 | 99.95 | 100.00 | 99.90 | 99.96 | 100.00 | 99.87 | 99.96 | |
| Incorrect (%) | 0 | 0.15 | 0.05 | 0.00 | 0.10 | 0.04 | 0.00 | 0.13 | 0.04 | |
| 4.MLP 4-11-2 | Total | 39,448 | 17,591 | 57,039 | 15,873 | 8572 | 24,445 | 55,412 | 26,072 | 81,484 |
| Correct | 39,448 | 17,566 | 57,014 | 15,873 | 8563 | 24,436 | 55,412 | 26,037 | 81,449 | |
| Incorrect | 0 | 25 | 25 | 0 | 9 | 9 | 0 | 35 | 35 | |
| Correct (%) | 100 | 99.86 | 99.96 | 100.00 | 99.90 | 99.96 | 100.00 | 99.87 | 99.96 | |
| Incorrect (%) | 0 | 0.14 | 0.04 | 0.00 | 0.10 | 0.04 | 0.00 | 0.13 | 0.04 | |
| 5.MLP 4-16-2 | Total | 39,448 | 17,591 | 57,039 | 15,873 | 8572 | 24,445 | 55,321 | 26,163 | 81,484 |
| Correct | 39,448 | 17,567 | 57,015 | 15,873 | 8572 | 24,445 | 55,321 | 26,139 | 81,460 | |
| Incorrect | 0 | 24 | 24 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Correct (%) | 100 | 99.86 | 99.96 | 100.00 | 100.00 | 100.00 | 100.00 | 99.91 | 99.97 | |
| Incorrect (%) | 0 | 0.14 | 0.04 | 0.00 | 0.00 | 0.00 | 0.00 | 0.09 | 0.03 | |
| 6.RBF 4-21-2 | Total | 39,539 | 17,500 | 57,039 | 15,873 | 8572 | 24,445 | 55,412 | 26,072 | 81,484 |
| Correct | 39,520 | 16,426 | 55,946 | 15,855 | 8059 | 23,914 | 55,375 | 24,485 | 79,860 | |
| Incorrect | 19 | 1074 | 1093 | 18 | 513 | 531 | 37 | 1587 | 1624 | |
| Correct (%) | 99.95 | 93.86 | 98.08 | 99.89 | 94.02 | 97.83 | 99.93 | 93.91 | 98.01 | |
| Incorrect (%) | 0.05 | 6.14 | 1.92 | 0.11 | 5.98 | 2.17 | 0.07 | 6.09 | 1.99 | |
| 7.RBF 4-29-2 | Total | 39,539 | 17,500 | 57,039 | 15,873 | 8572 | 24,445 | 55,412 | 26,072 | 81,484 |
| Correct | 39,165 | 17,475 | 56,640 | 15,714 | 8561 | 24,275 | 54,879 | 26,036 | 80,915 | |
| Incorrect | 374 | 25 | 399 | 159 | 11 | 170 | 533 | 36 | 569 | |
| Correct (%) | 99.05 | 99.86 | 99.3 | 99.00 | 99.87 | 99.30 | 99.04 | 99.86 | 99.30 | |
| Incorrect (%) | 0.95 | 0.14 | 0.7 | 1.00 | 0.13 | 0.70 | 0.96 | 0.14 | 0.70 | |
| 8.RBF 4-21-2 | Total | 39,539 | 17,500 | 57,039 | 15,873 | 8572 | 24,445 | 55,412 | 26,072 | 81,484 |
| Correct | 39,526 | 16,138 | 55,664 | 15,868 | 7873 | 23,741 | 55,394 | 24,011 | 79,405 | |
| Incorrect | 13 | 1362 | 1375 | 5 | 699 | 704 | 18 | 2061 | 2079 | |
| Correct (%) | 99.97 | 92.22 | 97.59 | 99.97 | 91.85 | 97.12 | 99.97 | 92.09 | 97.45 | |
| Incorrect (%) | 0.03 | 7.78 | 2.41 | 0.03 | 8.15 | 2.88 | 0.03 | 7.91 | 2.55 | |
| 9.RBF 4-28-2 | Total | 39,539 | 17,500 | 57,039 | 15,197 | 8571 | 23,768 | 53,008 | 26,060 | 81,484 |
| Correct | 39,489 | 16,000 | 23,489 | 15,197 | 8448 | 23,645 | 53,008 | 25,674 | 78,682 | |
| Incorrect | 50 | 1500 | 1,450 | 0 | 123 | 123 | 0 | 386 | 386 | |
| Correct (%) | 99.87 | 91.43 | 95.65 | 100.00 | 98.56 | 99.48 | 100.00 | 98.52 | 99.51 | |
| Incorrect (%) | 0.03 | 7.78 | 4.35 | 0.00 | 1.44 | 0.52 | 0.00 | 1.48 | 0.49 | |
| 10.RBF 4-26-2 | Total | 39,539 | 17,500 | 57,039 | 15,873 | 8572 | 24,445 | 55,412 | 26,072 | 81,484 |
| Correct | 11,880 | 6629 | 18,509 | 4748 | 3170 | 7918 | 16,628 | 9799 | 26,427 | |
| Incorrect | 27659 | 10871 | 38530 | 11125 | 5402 | 16527 | 38784 | 16273 | 55057 | |
| Correct (%) | 30.05 | 37.88 | 32.45 | 29.91 | 36.98 | 32.39 | 30.01 | 37.58 | 32.43 | |
| Incorrect (%) | 69.95 | 62.12 | 67.55 | 70.09 | 63.02 | 67.61 | 69.99 | 62.42 | 67.57 | |
| Model | Class | Fail | Total Class |
|---|---|---|---|
| 1. MLP 4-7-2 | 6.4 | 1 | 104 |
| 6.5 | 34 | 36 | |
| 2. MLP 4-8-2 | 1.6 | 3 | 4 |
| 6.4 | 1 | 104 | |
| 6.5 | 34 | 36 | |
| 3. MLP 4-10-2 | 1.6 | 3 | 4 |
| 6.4 | 1 | 104 | |
| 6.5 | 33 | 36 | |
| 4. MLP 4-11-2 | 1.6 | 3 | 4 |
| 6.4 | 1 | 104 | |
| 6.5 | 32 | 36 | |
| 5. MLP 4-16-2 | 6.4 | 1 | 104 |
| 6.5 | 33 | infer 36 |
| Input Variable | Variable Sensitivity | Variable Name/Details |
|---|---|---|
| <Tr5(srn)> | 15,896,991 | Expected value of Trace of order 5 of the srn for the sequence |
| D Tr5(srn) | 1,288,626 | Deviation of Trace of order 5 of the srn with respect to the mean value of the class |
| <Tr3(srn)> | 591,331.9 | Expected value of Trace of order 3 of the srn for the sequence |
| D Tr3(srn) | 108.7591 | Deviation of Trace of order 3 of the srn with respect to the mean value of the class |
| EC Subclass | Number of Sequences | EC Subclass | Number of Sequences | EC Subclass | Number of Sequences |
|---|---|---|---|---|---|
| 1.1 | 555 | 2.3 | 722 | 4.6 | 120 |
| 1.2 | 250 | 2.4 | 424 | 4.99 | 95 |
| 1.3 | 172 | 2.5 | 291 | 5.1 | 176 |
| 1.4 | 108 | 2.6 | 19 | 5.2 | 74 |
| 1.5 | 5 | 2.7 | 3112 | 5.3 | 247 |
| 1.6 | 4 | 2.8 | 71 | 5.4 | 160 |
| 1.7 | 91 | 2.9 | 10 | 5.5 | 115 |
| 1.8 | 165 | 3.1 | 1559 | 5.6 | 159 |
| 1.9 | 73 | 3.11 | 7 | 5.99 | 3 |
| 1.10 | 555 | 3.13 | 3 | 6.1 | 277 |
| 1.11 | 136 | 3.2 | 700 | 6.2 | 38 |
| 1.12 | 32 | 3.3 | 164 | 6.3 | 291 |
| 1.13 | 123 | 3.4 | 1481 | 6.4 | 104 |
| 1.14 | 244 | 3.5 | 561 | 6.5 | 36 |
| 1.15 | 162 | 3.6 | 417 | 7.1 | 8827 |
| 1.16 | 173 | 3.7 | 69 | 7.2 | 927 |
| 1.17 | 121 | 3.8 | 77 | 7.4 | 189 |
| 1.18 | 45 | 3.9 | 3 | 7.5 | 187 |
| 1.20 | 250 | 4.1 | 486 | 7.6 | 197 |
| 1.21 | 28 | 4.2 | 460 | ||
| 1.23 | 3 | 4.3 | 97 | ||
| 2.1 | 522 | 4.4 | 39 | ||
| 2.2 | 107 | 4.5 | 25 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Concu, R.; Cordeiro, M.N.D.S. Alignment-Free Method to Predict Enzyme Classes and Subclasses. Int. J. Mol. Sci. 2019, 20, 5389. https://doi.org/10.3390/ijms20215389
Concu R, Cordeiro MNDS. Alignment-Free Method to Predict Enzyme Classes and Subclasses. International Journal of Molecular Sciences. 2019; 20(21):5389. https://doi.org/10.3390/ijms20215389
Chicago/Turabian StyleConcu, Riccardo, and M. Natália D. S. Cordeiro. 2019. "Alignment-Free Method to Predict Enzyme Classes and Subclasses" International Journal of Molecular Sciences 20, no. 21: 5389. https://doi.org/10.3390/ijms20215389
APA StyleConcu, R., & Cordeiro, M. N. D. S. (2019). Alignment-Free Method to Predict Enzyme Classes and Subclasses. International Journal of Molecular Sciences, 20(21), 5389. https://doi.org/10.3390/ijms20215389