PD-L1/PD-1 Axis in Glioblastoma Multiforme

 ,
,
Abstract
1. Introduction
2. Central Nervous System Lymphatic System
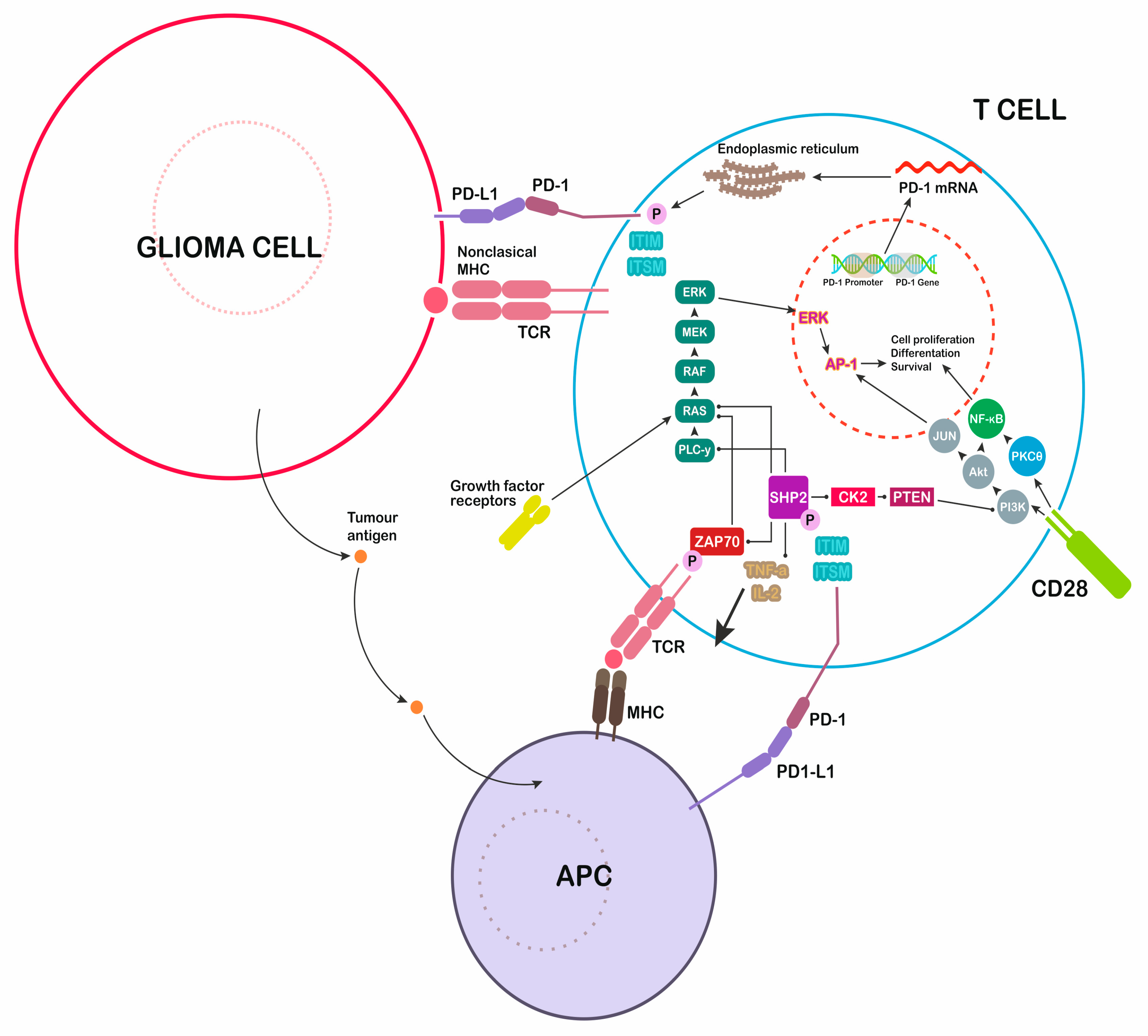
3. PD-L1 and PD-1 Structure
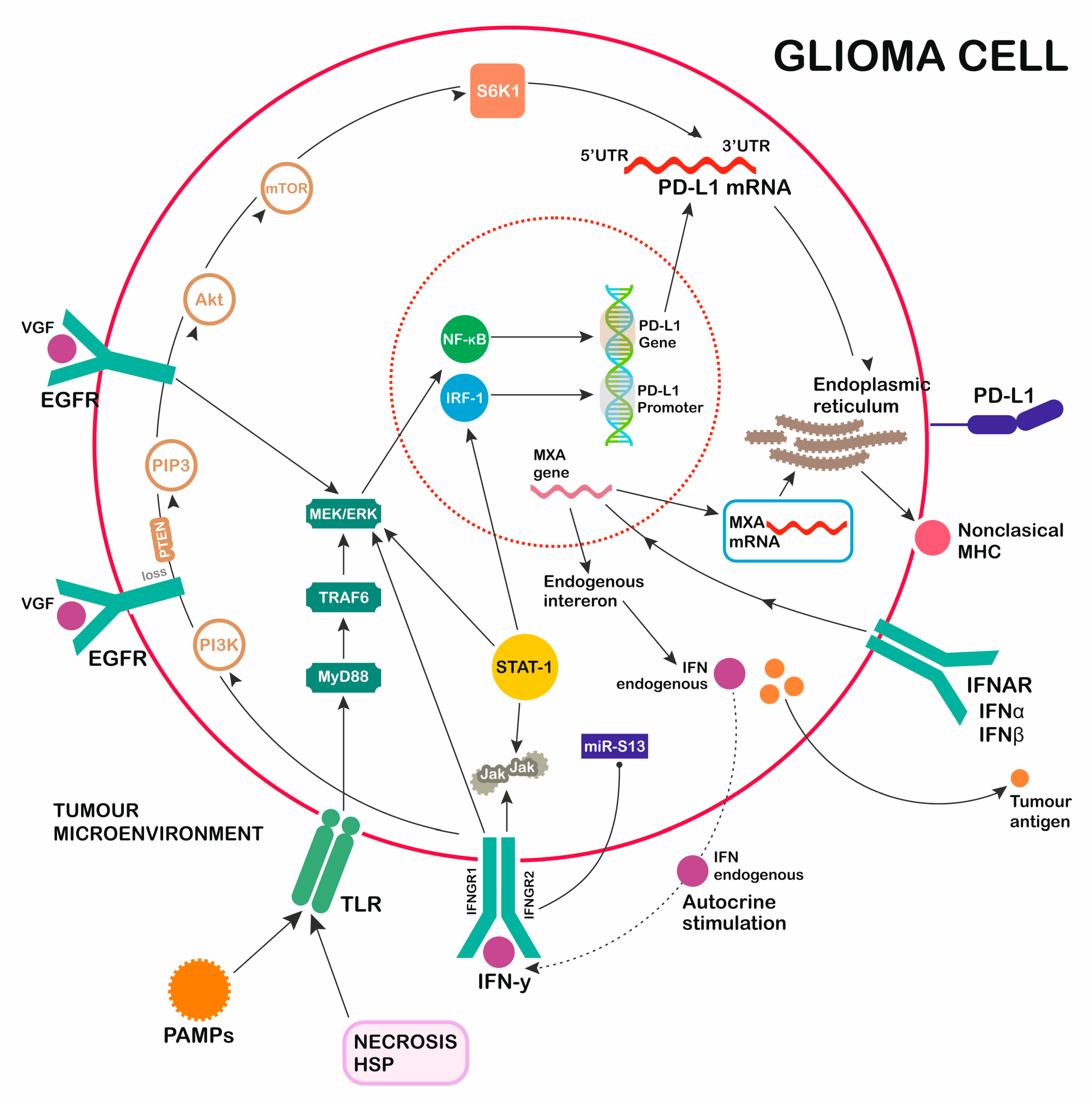
4. PD-1 Ligand Expression—Role of TLR Activation
5. EGFR Pathway
6. Angiogenesis in Glioblastomas
- Serine Proteases
- Matrix Metalloproteases (MMPs)
- Cysteine Proteases [72].
7. INF Receptor
8. Immunotherapy with Anti PD-1/PD-L1
9. Summary
Funding
Conflicts of Interest
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 3–9. [Google Scholar] [PubMed]
- Zarnett, O.J.; Sahgal, A.; Gosio, J.; Perry, J.; Berger, M.S.; Chang, S.; Das, S. Treatment of elderly patients with glioblastoma: A systematic evidence-based analysis. JAMA Neurol. 2015, 72, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Greer, L.; Pannullo, S.C.; Smith, A.W.; Taube, S.; Yondorf, M.Z.; Parashar, B.; Trichter, S.; Nedialkova, L.; Sabbas, A.; Christos, P.; et al. Accelerated hypofractionated radiotherapy in the era of concurrent temozolomide chemotherapy in elderly patients with glioblastoma multiforme. Cureus 2017, 9, e1388. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current standards of care in glioblastoma therapy. In Glioblastoma [Internet]; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2007; Chapter 11. [Google Scholar]
- Roy, S.; Lahiri, D.; Maji, T.; Biswas, J. Recurrent glioblastoma: Where we stand. South Asian J. Cancer 2015, 4, 163–173. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecularn subclasses of high-grade glioma predict prognosis, delineatea pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. 2013, 86, 343–349. [Google Scholar] [CrossRef]
- Freeman, G.J.; Hochrein, H.; O’Keeffe, M.; Luft, T.; Vandenabeele, S.; Grumont, R.J.; Maraskovsky, E.; Shortman, K. Engagement of the PD-1 immunoinhibitory receptor by a novel b7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Pachter, J.S.; de Vries, H.E.; Fabry, Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J. Neuropathol. Exp. Neurol. 2003, 62, 593–604. [Google Scholar] [CrossRef]
- Suter, T.; Biollaz, G.; Gatto, D.; Bernasconi, L.; Herren, T.; Reith, W.; Fontana, A. The brain as an immune privileged site: Dendritic cells of the central nervous system inhibit T cell activation. Eur. J. Immunol. 2003, 33, 2998–3006. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphaticvessels. Nature 2005, 523, 337–341. [Google Scholar] [CrossRef]
- Goldmann, J.; Kwidzinski, E.; Brandt, C.; Mahlo, J.; Richter, D.; Bechmann, I. T cells traffic frombrain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J. Leukoc. Biol. 2006, 80, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Noell, S.; Fallier-Becker, P.; Mack, A.F.; Wolburg-Buchholz, K. The disturbed blood-brain barrier in human glioblastoma. Mol Aspects Med. 2012, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Qin, Z.; Chen, Z.; Xie, L.; Wang, R.; Zhao, H. Tumor microenvironment in treatment of glioma. Open Med. 2017, 12, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Sengupta, S.; Han, Y.; Ulasov, I.V.; Lesniak, M.S. The presence of IL-17A and T helper 17 cells in experimental mouse brain tumors and human gliom. PLoS ONE 2010, 5, e15390. [Google Scholar] [CrossRef]
- Yi, Y.; Hsieh, I.-Y.; Huang, X.; Li, J.; Zhao, W. Glioblastoma Stem-Like Cells: Characteristics, Microenvironment, and Therapy. Front. Pharmacol. 2016, 7, 8884. [Google Scholar] [CrossRef]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Zhang, X.; Schwartz, J.C.; Guo, X.; Bhatia, S.; Cao, E.; Chen, L.; Zhang, Z.Y.; Edidin, M.A.; Nathenson, S.G.; Almo, S.C. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 2004, 20, 337–347. [Google Scholar] [CrossRef]
- Cheng, X.; Veverka, V.; Radhakrishnan, A.; Waters, L.C.; Muskett, F.W.; Morgan, S.H.; Huo, J.; Yu, C.; Evans, E.J.; Leslie, A.J.; et al. Structure and interactions of the human programmed cell death 1 receptor. J. Biol. Chem. 2013, 288, 11771–11785. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef]
- Baruch, K.; Deczkowska, A.; Rosenzweig, N.; Tsitsou-Kampeli, A.; Sharif, A.M.; Matcovitch-Natan, O.; Kertser, A.; David, E.; Amit, I.; Schwartz, M. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat. Med. 2016, 22, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Wintterle, S.; Schreiner, B.; Mitsdoerffer, M.; Schneider, D.; Chen, L.; Meyermann, R.; Weller, M.; Wiendl, H. Expression of the B7-related molecule B7-H1 by glioma cells: A potential mechanism of immune paralysis. Cancer Res. 2003, 63, 7462–7467. [Google Scholar]
- Wilmotte, R.; Burkhardt, K.; Kindler, V.; Belkouch, M.C.; Dussex, G.; Tribolet Nd Walker, P.R.; Dietrich, P.Y. B7-homolog 1 expression by human glioma: A new mechanism of immune evasion. Neuroreport 2005, 16, 1081–1085. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef]
- Asea, A. Novel signal transduction pathway utilized by extracellular hsprole of toll-like receptor (TLR) 2 and TLR4. J. Boil. Chem. 2002, 277, 15028–15034. [Google Scholar] [CrossRef]
- Verstrepen, L.; Bekaert, T.; Chau, T.-L.; Tavernier, J.; Chariot, A.; Beyaert, R. TLR-4, IL-1R and TNF-R signaling to NF-κB: Variations on a common theme. Cell. Mol. Life Sci. 2008, 65, 2964–2978. [Google Scholar] [CrossRef]
- Meng, Y.; Kujas, M.; Marie, Y.; Paris, S.; Thillet, J.; Delattre, J.-Y.; Carpentier, A.F. Expression of TLR9 within human glioblastoma. J. Neuro-Oncol. 2008, 88, 19–25. [Google Scholar] [CrossRef]
- Medvedev, A.E.; Sabroe, I.; Hasday, J.D.; Vogel, S.N. Invited review: Tolerance to microbial TLR ligands: Molecular mechanisms and relevance to disease. J. Endotoxin Res. 2006, 12, 133–150. [Google Scholar] [CrossRef]
- Aalaei-Andabili, S.H.; Rezaei, N. Toll like receptor (TLR)-induced differential expression of microRNAs (MiRs) and immune response against infection: A systematic review. J. Infect. 2013, 67, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Ampie, L.; Choy, W.; Lamano, J.B.; Fakurnejad, S.; Bloch, O.; Parsa, A.T. Heat shock protein vaccines against glioblastoma: From bench to bedside. J. Neuro-Oncol. 2015, 123, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Waziri, A. Glioblastoma-Derived Mechanisms of Systemic Immunosuppression. Neurosurg. Clin. N. Am. 2010, 21, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Ampie, L.; Woolf, E.C.; Dardis, C. Immunotherapeutic Advancements for Glioblastoma. Front. Oncol. 2015, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domaincontaining adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353. [Google Scholar] [CrossRef] [PubMed]
- Echigo, R.; Sugimoto, N.; Yachie, A.; OhnoShosaku, T. Cannabinoids inhibit peptidoglycan-induced phosphorylation of NF-κB and cell growth in U87MG human malignant glioma cells. Oncol. Rep. 2012, 28, 1176–1180. [Google Scholar] [CrossRef]
- Wells, A. EGF receptor. Int. J. Biochem. Cell Biol. 1999, 31, 637–643. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Schmidt, U.; Unterberg, A.; Halatsch, M.E. Therapeutic inhibition of the epidermal growth factor receptor in high-grade gliomas: Where do we stand? Mol. Cancer Res. 2009, 7, 1000–1012. [Google Scholar] [CrossRef]
- Nagane, M.; Lin, H.; Cavenee, W.K.; Huang, H.-J. Aberrant receptor signaling in human malignant gliomas: Mechanisms and therapeutic implications. Cancer Lett. 2001, 162, S17–S21. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.-W.; Cao, X.; Zhu, H.; Ali-Osman, F. Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol. Cancer Res. 2010, 8, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Boil. 2005, 6, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.E.; Furnari, F.B.; Cavenee, W.K. Targeting EGFR for treatment of glioblastoma: Molecular basis to overcome resistance. Curr. Cancer Drug Targets 2012, 12, 197–209. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef]
- Zhu, H.; Acquaviva, J.; Ramachandran, P.; Boskovitz, A.; Woolfenden, S.; Pfannl, R.; Bronson, R.T.; Chen, J.W.; Weissleder, R.; Housman, D.E.; et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 2712–2716. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Hasselbalch, B.; Lassen, U.; Poulsen, H.S.; Stockhausen, M.-T. Cetuximab Insufficiently Inhibits Glioma Cell Growth Due to Persistent EGFR Downstream Signaling. Cancer Investig. 2010, 28, 775–787. [Google Scholar] [CrossRef]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef]
- Rich, J.N.; Rasheed, B.K.A.; Yan, H. EGFR mutations and sensitivity to gefitinib. N. Engl. J. Med. 2004, 351, 1260–1261. [Google Scholar] [PubMed]
- Ji, H.; Sharpless, N.E.; Wong, K.-K. EGFR Targeted Therapy: View from Biological Standpoint. Cell Cycle 2006, 5, 2072–2076. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krishnan, S.; Szabo, E.; Burghardt, I.; Frei, K.; Tabatabai, G.; Weller, M. Modulation of cerebral endothelial cell function by TGF-β in glioblastoma: VEGF-dependent angiogenesis versus endothelial mesenchymal transition. Oncotarget 2015, 6, 22480–22495. [Google Scholar] [CrossRef] [PubMed]
- Bullitt, E.; Reardon, D.A.; Smith, J.K. A review of micro- and macrovascular analyses in the assessment of tumor-associated vasculature as visualized by MR. NeuroImage 2007, 37 (Suppl. 1), S116–S119. [Google Scholar] [CrossRef]
- Plate, K.; Mennel, H. Vascular morphology and angiogenesis in glial tumors. Exp. Toxicol. Pathol. 1995, 47, 89–94. [Google Scholar] [CrossRef]
- Koh, J.; Go, H.; Shin, S.-J.; Jeon, Y.K.; Kim, P.-J.; Cho, Y.M.; Chung, D.H. Clinicopathologic Analysis of PD-L1 and PD-L2 Expression in Renal Cell Carcinoma: Association with Oncogenic Proteins Status. Ann. Surg. Oncol. 2015, 23, 694–702. [Google Scholar]
- Grochowski, C.; Staśkiewicz, G. Ultra high field TOF-MRA: A method to visualize small cerebral vessels. 7 T TOF-MRA sequence parameters on different MRI scanners—Literature review. Neurol. Neurochir. Pol. 2017, 51, 411–418. [Google Scholar] [CrossRef]
- Jain, R.K.; Di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef]
- Keller, S.; Schmidt, M.H.H. EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment. Int. J. Mol. Sci. 2017, 18, 1295. [Google Scholar] [CrossRef]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncology 2005, 7, 134–153. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Weathers, S.P.; de Groot, J. VEGF Manipulation in Glioblastoma. Oncology 2015, 29, 720–727. [Google Scholar] [PubMed]
- Budny, A.; Grochowski, C.; Kozłowski, P.; Kolak, A.; Kamińska, M.; Budny, B.; Abramiuk, M.; Burdan, F. Obesity as a tumour development triggering factor. Ann. Agric. Environ. Med. 2019, 26, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Hu, M.; Li, P.; Ma, J.; Xie, L.; Teng, F.; Zhu, Y.; Fan, B.; Mu, D.; Yu, J. Relationship between expression of PD-L1 and tumor angiogenesis, proliferation, and invasion in glioma. Oncotarget 2017, 8, 49702–49712. [Google Scholar] [CrossRef]
- Joseph, R.W.; Parasramka, M.; Eckel-Passow, J.E.; Serie, D.; Wu, K.; Jiang, L.; Kalari, K.; Thompson, R.H.; Ho, T.H.; Castle, E.P.; et al. Inverse association between programmed death ligand 1 and genes in the VEGF pathway in primary clear cell renal cell carcinoma. Cancer Immunol. Res. 2013, 1, 378–385. [Google Scholar] [CrossRef]
- Price, J.; Wilson, H.; Haites, N. Epidermal growth factor (EGF) Increases the in vitro invasion, motility and adhesion interactions of the primary renal carcinoma cell line, A704. Eur. J. Cancer 1996, 32, 1977–1982. [Google Scholar] [CrossRef]
- Shibata, T.; Kawano, T.; Nagayasu, H.; Okumura, K.; Arisue, M.; Hamada, J.-I.; Takeichi, N.; Hosokawa, M. Enhancing effects of epidermal growth factor on human squamous cell carcinoma motility and matrix degradation but not growth. Tumor Boil. 1996, 17, 168–175. [Google Scholar] [CrossRef]
- Huang, P.H.; Mukasa, A.; Bonavia, R.; Flynn, R.A.; Brewer, Z.E.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. USA 2007, 104, 12867–12872. [Google Scholar] [CrossRef]
- Bonavia, R.; Inda, M.; Vandenberg, S.; Cheng, S.; Nagane, M.; Hadwiger, P.; Tan, P.; Sah, D.; Cavenee, W.; Furnari, F. EGFRvIII promotes glioma angiogenesis and growth through the NF-κB, interleukin-8 pathway. Oncogene 2012, 31, 4054–4066. [Google Scholar] [CrossRef]
- Guillamo, J.-S.; Valable, S.; Marteau, L.; Leuraud, P.; Marie, Y.; Poupon, M.-F.; Parienti, J.-J.; Raymond, E.; Peschanski, M.; De Boüard, S. Molecular Mechanisms Underlying Effects of Epidermal Growth Factor Receptor Inhibition on Invasion, Proliferation, and Angiogenesis in Experimental Glioma. Clin. Cancer Res. 2009, 15, 3697–3704. [Google Scholar] [CrossRef]
- Kasza, A.; Kowanetz, M.; Po’slednik, K.; Witek, B.; Kordula, T.; Koj, A. Epidermal growth factor and pro-inflammatory cytokines regulate the expression of components of plasminogen activation system in U373-MG astrocytoma cells. Cytokine 2001, 16, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Abe, T.; Wakabayashi, Y.; Hikawa, T.; Matsuo, K.-I.; Yamada, Y.; Kuwano, M.; Hori, S. Up-regulation of Urokinase-type Plasminogen Activator and its Receptor Correlates with Enhanced Invasion Activity of Human Glioma Cells Mediated by Transforming Growth Factor-α or Basic Fibroblast Growth Factor. J. Neuro-Oncol. 2000, 46, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Muracciole, X.; Romain, S.; Dufour, H.; Palmari, J.; Chinot, O.; Ouafik, L.; Grisoli, F.; Branger, D.F.; Martin, P.M. PAI-1 and EGFR expression in adult glioma tumors: Toward a molecular prognostic classification. Int. J. Radiat. Oncol. Biol. Phys. 2002, 52, 592–598. [Google Scholar] [CrossRef]
- Grochowski, C.; Radzikowska, E.; Maciejewski, R. Neural stem cell therapy—Brief review. Clin. Neurol. Neurosurg. 2018, 173, 8–14. [Google Scholar] [CrossRef]
- Choe, G.; Park, J.K.; Jouben-Steele, L.; Kremen, T.J.; Liau, L.M.; Vinters, H.V.; Cloughesy, T.F.; Mischel, P.S. Active matrix metalloproteinase 9 expression is associated with primary glioblastoma subtype. Clin. Cancer Res. 2002, 8, 2894–2901. [Google Scholar]
- Ellerbroek, S.M.; Halbleib, J.M.; Benavidez, M.; Warmka, J.K.; Wattenberg, E.V.; Stack, M.S.; Hudson, L.G. Phosphatidylinositol 3-kinase activity in epidermal growth factor-stimulated matrix metalloproteinase-9 production and cell surface association. Cancer Res. 2001, 61, 1855–1861. [Google Scholar]
- Anand, M.; Van Meter, T.E.; Fillmore, H.L. Epidermal growth factor induces matrix metalloproteinase-1 (MMP-1) expression and invasion in glioma cell lines via the MAPK pathway. J. Neuro-Oncol. 2011, 104, 679–687. [Google Scholar] [CrossRef]
- Kesanakurti, D.; Chetty, C.; Maddirela, D.R.; Gujrati, M.; Rao, J.S. Functional cooperativity by direct interaction between PAK4 and MMP-2 in the regulation of anoikis resistance, migration and invasion in glioma. Cell Death Dis. 2012, 3, e445. [Google Scholar] [CrossRef]
- Gole, B.; Huszthy, P.C.; Popovi’c, M.; Jeruc, J.; Ardebili, Y.S.; Bjerkvig, R.; Lah, T.T. The regulation of cysteine cathepsins and cystatins in human gliomas. Int. J. Cancer 2012, 131, 1779–1789. [Google Scholar] [CrossRef]
- Rempel, S.A.; Rosenblum, M.L.; Mikkelsen, T.; Yan, P.S.; Ellis, K.D.; Golembieski, W.A.; Sameni, M.; Rozhin, J.; Ziegler, G.; Sloane, B.F. Cathepsin B expression and localization in glioma progression and invasion. Cancer Res. 1994, 54, 6027–6031. [Google Scholar]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.O.; Westphal, M.; Hagel, C.; Ergün, S.; Stavrou, D.; Rosen, E.M.; Lamszus, K. Levels of vascular endothelial growth factor, hepatocyte growth factor/scatter factor and basic fibroblast growth factor in human gliomas and their relation to angiogenesis. Int. J. Cancer 1999, 84, 10–18. [Google Scholar] [CrossRef]
- Brat, D.J.; Bellail, A.C.; Van Meir, E.G. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-Oncology 2005, 7, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [PubMed]
- Domanski, P.; Colamonici, O.R. The type-I interferon receptor. The long and short of it. Cytokine Growth Factor Rev. 1996, 7, 143–151. [Google Scholar] [CrossRef]
- Novick, D.; Cohen, B.; Rubinstein, M. The human interferon a/b receptor: Characterization and molecular cloning. Cell 1994, 77, 391–400. [Google Scholar] [CrossRef]
- Darnell, J.; Kerr, I.; Stark, G. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Smith, P.L.; Lombardi, G.; Foster, G.R. Type I interferons and the innate immune response—More than just antiviral cytokines. Mol. Immunol. 2005, 42, 869–877. [Google Scholar] [CrossRef]
- Friedman, R.L.; Stark, G.R. α-Interferon-induced transcription of HLA and metallothionein genes containing homologous upstream sequences. Nature 1985, 314, 637–639. [Google Scholar] [CrossRef]
- Cull, V.S.; Tilbrook, P.A.; Bartlett, E.J.; Brekalo, N.L.; James, C.M. Type I interferon differential therapy for erythroleukemia: Specificity of STAT activation. Blood 2003, 101, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Kochs, G. Human MxA Protein: An Interferon-Induced Dynamin-Like GTPase with Broad Antiviral Activity. J. Interferon Cytokine Res. 2011, 31, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Grochowski, C.; Blicharska, E.; Baj, J.; Mierzwińska, A.; Brzozowska, K.; Forma, A.; Maciejewski, R. Serum iron, Magnesium, Copper, and Manganese Levels in Alcoholism: A Systematic Review. Molecules 2019, 24, 1361. [Google Scholar] [CrossRef] [PubMed]
- Silginer, M.; Nagy, S.; Happold, C.; Schneider, H.; Weller, M.; Roth, P. Autocrine activation of the IFN signaling pathway may promote immune escape in glioblastoma. Neuro-Oncology 2017, 19, 1338–1349. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’tknow JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Kim, J.; Myers, A.C.; Chen, L.; Pardoll, D.M.; Truong-Tran, Q.-A.; Lane, A.P.; McDyer, J.F.; Fortuno, L.; Schleimer, R.P. Constitutive and Inducible Expression of B7 Family of Ligands by Human Airway Epithelial Cells. Am. J. Respir. Cell Mol. Boil. 2005, 33, 280–289. [Google Scholar] [CrossRef]
- Tseng, S.-Y.; Otsuji, M.; Gorski, K.; Huang, X.; Slansky, J.E.; Pai, S.I.; Shalabi, A.; Shin, T.; Pardoll, D.M.; Tsuchiya, H. B7-Dc, a New Dendritic Cell Molecule with Potent Costimulatory Properties for T Cells. J. Exp. Med. 2001, 193, 839–846. [Google Scholar] [CrossRef]
- Lee, S.-J.; Jang, B.-C.; Lee, S.-W.; Yang, Y.-I.; Suh, S.-I.; Park, Y.-M.; Oh, S.; Shin, J.-G.; Yao, S.; Chen, L.; et al. Interferon regulatory factor- 1 is prerequisite to the constitutive expression and IFN-γ-induced upregulation of B7-H1 (CD274). FEBS Lett. 2006, 580, 755–762. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef]
- Redman, J.; Hill, E.; Aldeghaither, D.; Weiner, L. Mechanisms of action of therapeutic antibodies for cancer. Mol. Immunol. 2015, 67, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lee, H.T.; Shin, W.; Chae, J.; Choi, J.; Kim, S.H.; Lim, H.; Heo, T.W.; Park, K.Y.; Lee, Y.J.; et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Commun. 2016, 7, 13354. [Google Scholar] [CrossRef] [PubMed]
- Mayor, M.; Yang, N.; Sterman, D.; Jones, D.R.; Adusumilli, P.S. Immunotherapy for non-small cell lung cancer: Current concepts and clinical trials. Eur. J. Cardio-Thorac. Surg. 2016, 49, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Jimeno, A. Atezolizumab: A novel PD-L1 inhibitor in cancer therapy with a focus in bladder and non-small cell lung cancers. Drugs Today 2017, 53, 217–237. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2018. MAbs 2018, 10, 183–203. [Google Scholar] [CrossRef]
- Kimiz-Gebologlu, I.; Gulce-Iz, S.; Biray-Avci, C. Monoclonal antibodies in cancer immunotherapy. Mol. Biol. Rep. 2018, 45, 2935–2940. [Google Scholar] [CrossRef]
- Topalian, S.L.; Weiner, G.J.; Pardoll, D.M. Cancer immunotherapy comes of age. J. Clin. Oncol. 2011, 29, 4828–4836. [Google Scholar] [CrossRef]
- Huang, B.Y.; Zhan, Y.P.; Zong, W.J.; Yu, C.J.; Li, J.F.; Qu, Y.M.; Han, S. The PD-1/B7-H1 Pathway Modulates the Natural Killer Cells versus Mouse Glioma Stem Cells. PLoS ONE 2015, 10, e0134715. [Google Scholar] [CrossRef]
- Reardon, D.A.; Dietrich, J.; Kaley, T.J.; Gan, H.K.; Dunn, G.P.; Cloughesy, T.F.; Lim, M.; Clarke, J.L.; Park, A.J.; Pan, L.S.; et al. Phase II study to evaluate the clinical efficacy and safety of MEDI4736 in patients with glioblastoma (GBM). J. Clin. Oncol. 2015, 33, TPS2077. [Google Scholar] [CrossRef]
- Reardon, D.A.; De Groot, J.F.; Colman, H.; Jordan, J.T.; Daras, M.; Clarke, J.L.; Nghiemphu, P.L.; Gaffey, S.C.; Peters, K.B. Safety of pembrolizumab in combination with bevacizumab in recurrent glioblastoma (rGBM). J. Clin. Oncol. 2016, 34, 2010. [Google Scholar] [CrossRef]
- Sahebjam, S.; Johnstone, P.A.; Forsyth, P.; Arrington, J.; Jaglal, M.; Tran, N.D.; Vrionis, F.D.; Etame, A.B.; Wicklund, M.; Elie, A.L.; et al. Atim-a phase i trial of hypofractionated stereotactic irradiation (hfsrt) with pembrolizumab and bevacizumab in patients with recurrent high grade gliomas. Neuro-Oncology 2016, 18, vi21. [Google Scholar] [CrossRef]
- Sahebjam, S.; Johnstone, P.A.; Forsyth, P.A.; Arrington, J.; Vrionis, F.D.; Etame, A.B.; Tran, N.; Dalvi, P.; Kim, S.; Macaulay, R.; et al. Safety and antitumor activity of hypofractionated stereotactic irradiation (HFSRT) with pembrolizumab (Pembro) and bevacizumab (Bev) in patients (pts) with recurrent high grade gliomas: Preliminary results from phase I study. J. Clin. Oncol. 2016, 34, 2041. [Google Scholar] [CrossRef]
- Brahmer, J.; Rasco, D.; Chen, M.; Masteller, E.; Qazi, I.; Rogers, S.; Sankar, N.; Sikorski, R.; Hambleton, J.; Hodi, F.S. Abstract B143: A phase 1a/1b study of FPA008 in combination with nivolumab in patients with selected advanced cancers. Cancer Immunol. Res. 2016, 4, B143. [Google Scholar]
- Sahebjam, S.; Forsyth, P.A.; Arrington, J.; Tran, N.D.; Jaglal, M.V.; Mokhtari, S.; Long, W.; Macaulay, R.J.; Wicklund, M.; Drury-Sibiga, A.; et al. Nivolumab combined with hypofractionated stereotactic irradiation (HFSRT) for patients with recurrent high grade gliomas: A phase I trial (NCT02829931). J. Clin. Oncol. 2017, 35, TPS2084. [Google Scholar] [CrossRef]
- Sanborn, R.E.; Pishvaian, M.J.; Kluger, H.M.; Callahan, M.K.; Weise, A.M.; Lutzky, J.; Yellin, M.J.; Rawls, T.; Vitale, L.; Halim, A.; et al. Clinical results with combination of anti-CD27 agonist antibody, varlilumab, with anti-PD1 antibody nivolumab in advanced cancer patients. J. Clin. Oncol. 2017, 35, 3007. [Google Scholar] [CrossRef]
- Perez, R.P.; Riese, M.J.; Lewis, K.D.; Saleh, M.N.; Daud, A.; Berlin, J.; Lee, J.J.; Mukhopadhyay, S.; Zhou, L.; Serbest, G.; et al. Epacadostat plus nivolumab in patients with advanced solid tumors: Preliminary phase I/II results of ECHO-204. J. Clin. Oncol. 2017, 35, 3003. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kaley, T.J.; Dietrich, J.; Clarke, J.L.; Dunn, G.P.; Lim, M.; Cloughesy, T.F.; Gan, H.K.; Park, A.J.; Schwarzenberger, P.; et al. Phase 2 study to evaluate safety and efficacy of MEDI4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: An update. J. Clin. Oncol. 2017, 35, 2042. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; Donovan, L. OS09.5 Synergistic effect of reirradiation and PD-1 inhibitors in recurrent high-grade gliomas. Neuro-Oncology 2017, 19, iii19. [Google Scholar] [CrossRef][Green Version]
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; De Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: CheckMate 143. Neuro-Oncology 2017, 19, iii21–iii22. [Google Scholar] [CrossRef]
- Deken, M.A.; Gadiot, J.; Jordanova, E.S.; Lacroix, R.; Van Gool, M.; Kroon, P.; Pineda, C.; Foppen, M.H.G.; Scolyer, R.; Song, J.-Y.; et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. OncoImmunology 2016, 5, e1238557. [Google Scholar] [CrossRef] [PubMed]
- Ebert, P.J.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology 2016, 18, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Berghoff, A.S.; Wick, W.; Weller, M. Clinical Neuropathology mini-review 6-2015: PD-L1: Emerging biomarker in glioblastoma? Clin. Neuropathol. 2015, 34, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced squamous-cell nonesmall-cell lung cancer. N. Engl. J Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody incancer. N. Engl. J Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Mazières, J.; Planchard, D.; Stinchcombe, T.E.; Dy, G.K.; Antonia, S.J.; Horn, L.; Lena, H.; Minenza, E.; Mennecier, B.; et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer. Lancet Oncol. 2015, 16, 257–265. [Google Scholar] [CrossRef]
- Festino, L.; Botti, G.; Lorigan, P.; Masucci, G.V.; Hipp, J.D.; Horak, C.E.; Melero, I.; Ascierto, P.A. Cancer Treatment with Anti-PD-1/PD-L1 Agents: Is PD-L1 Expression a Biomarker for Patient Selection? Drugs 2016, 76, 925–945. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical trial Identification Number | Study Phase | Number of Patients | Monoclonal Antibody |
|---|---|---|---|
| NCT02829931 | I | 26 | Nivolumab |
| NCT02313272 | I | 46 | Pembrolizumab, bevacizumab |
| NCT02529072 | I | 66 | Nivolumab |
| NCT02658981 | I | 68 | Anti-LAG-3, urelumab, nivolumab |
| NCT02526017 | I | 280 | nivolumab |
| NCT03233152 | I | 6 | Ipilimumab, nivolumab |
| NCT02937844 | I | 20 | Anti-PD-L1 CSR T cells |
| NCT03058289 | I/II | 60 | anti-PD-1 antibody |
| NCT02327078 | I/II | 291 | Nivolumab, epacadostat |
| NCT02311582 | I/II | 52 | MK-3475, MRI-guided laser ablation |
| NCT02866747 | I/II | 62 | Durvalumab |
| NCT02798406 | II | 48 | pembrolizumab |
| NCT02335918 | II | 205 | Varlilumab, nivolumab |
| NCT02968940 | II | 43 | Avelumab |
| NCT02794883 | II | 36 | Durvalumab, tremelimumab |
| NCT02336165 | II | 159 | bevacizumab |
| NCT02337491 | II | 82 | Pembrolizumab, bevacizumab |
| NCT03014804 | II | 30 | Nivolumab |
| NCT02550249 | II | 29 | Nivolumab |
| NCT02852655 | Pilot | 30 | Pembrolizumab |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litak, J.; Mazurek, M.; Grochowski, C.; Kamieniak, P.; Roliński, J. PD-L1/PD-1 Axis in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5347. https://doi.org/10.3390/ijms20215347
Litak J, Mazurek M, Grochowski C, Kamieniak P, Roliński J. PD-L1/PD-1 Axis in Glioblastoma Multiforme. International Journal of Molecular Sciences. 2019; 20(21):5347. https://doi.org/10.3390/ijms20215347
Chicago/Turabian StyleLitak, Jakub, Marek Mazurek, Cezary Grochowski, Piotr Kamieniak, and Jacek Roliński. 2019. "PD-L1/PD-1 Axis in Glioblastoma Multiforme" International Journal of Molecular Sciences 20, no. 21: 5347. https://doi.org/10.3390/ijms20215347
APA StyleLitak, J., Mazurek, M., Grochowski, C., Kamieniak, P., & Roliński, J. (2019). PD-L1/PD-1 Axis in Glioblastoma Multiforme. International Journal of Molecular Sciences, 20(21), 5347. https://doi.org/10.3390/ijms20215347