Single-Channel Properties of the ROMK-Pore-Forming Subunit of the Mitochondrial ATP-Sensitive Potassium Channel

 , , ,
, , ,  and
and 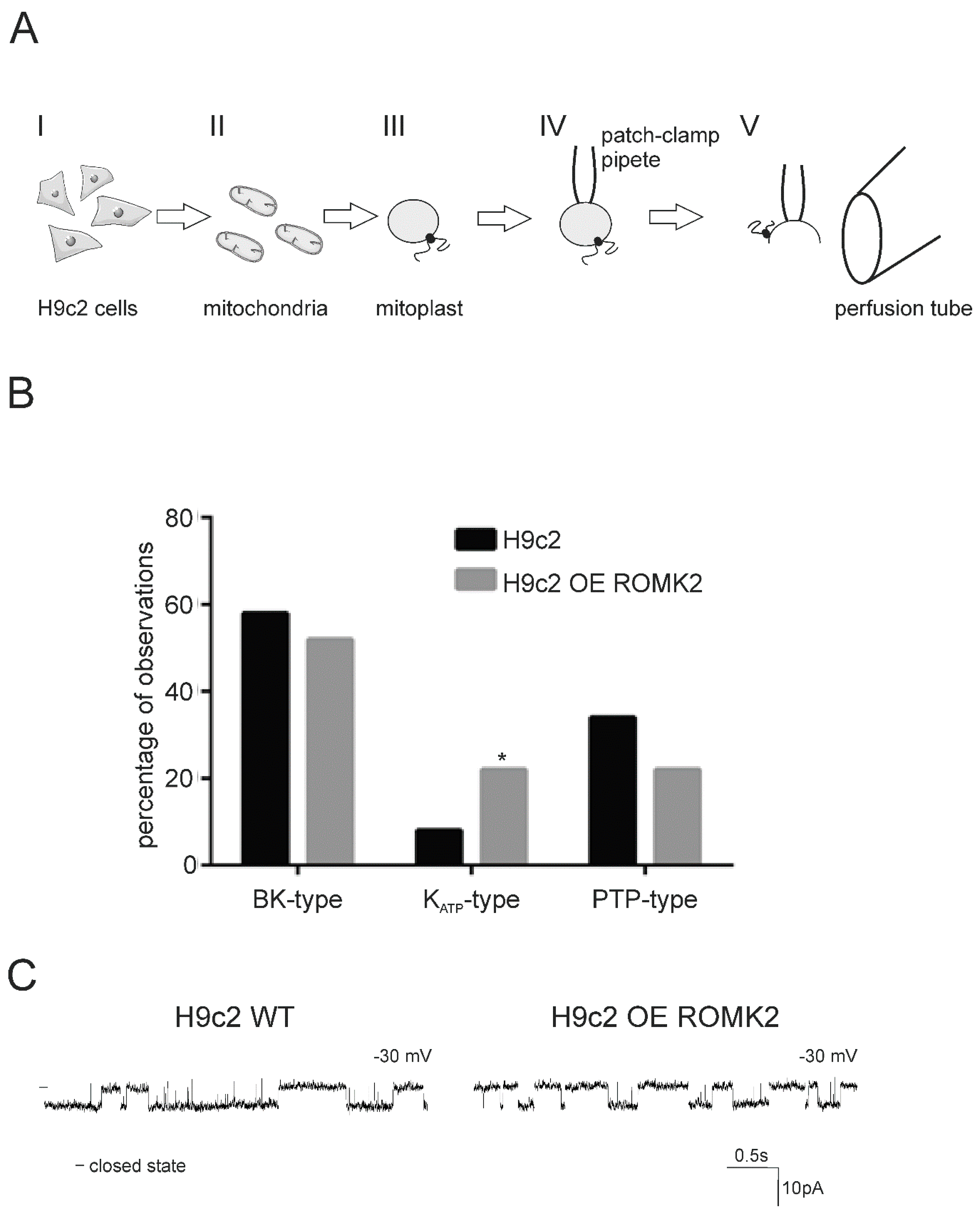{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Ion Channels Observed in H9c2 Mitochondria
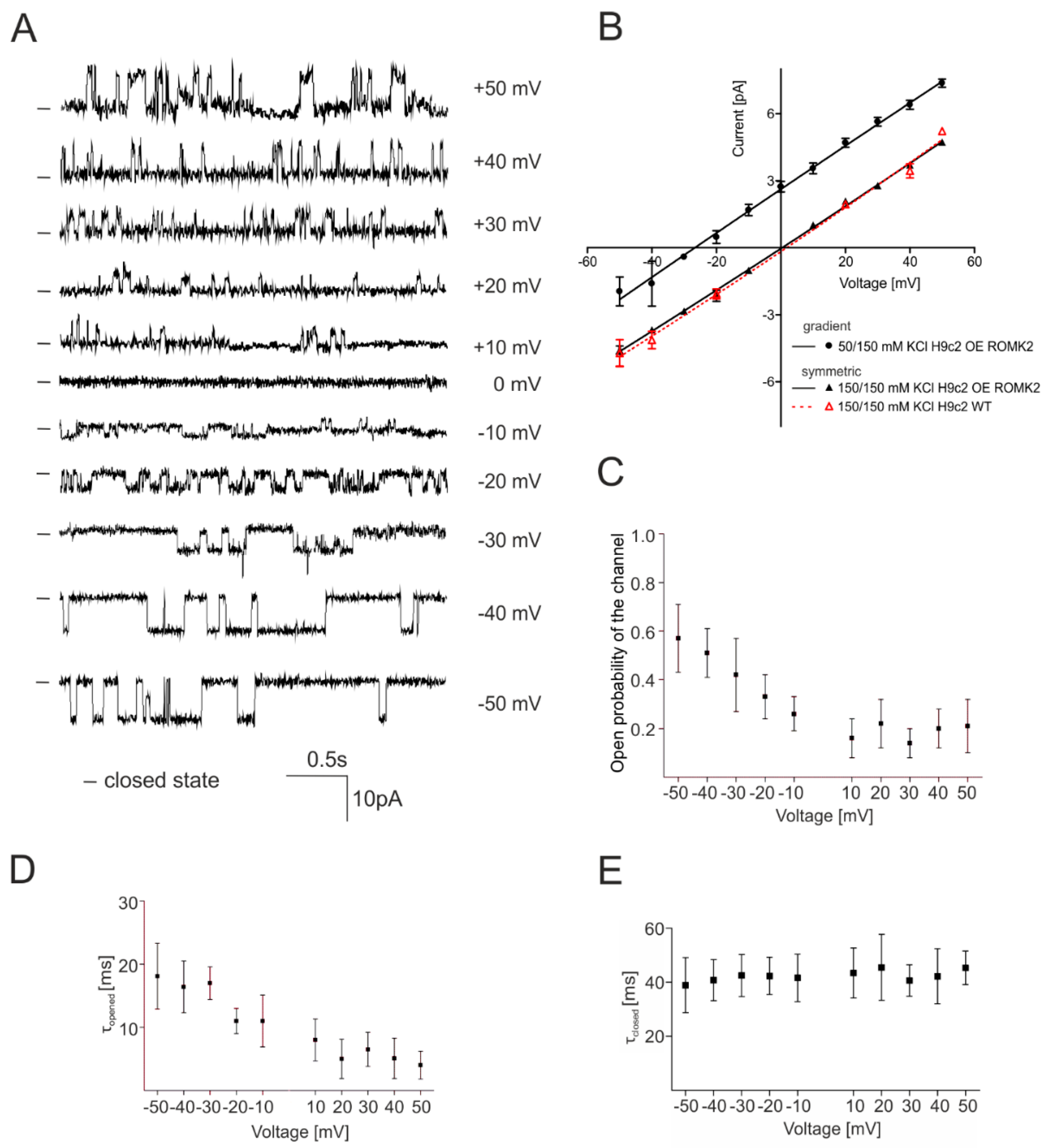
2.2. Identification of the mitoKATP Channel in Mitoplasts of ROMK Overexpressing H9c2
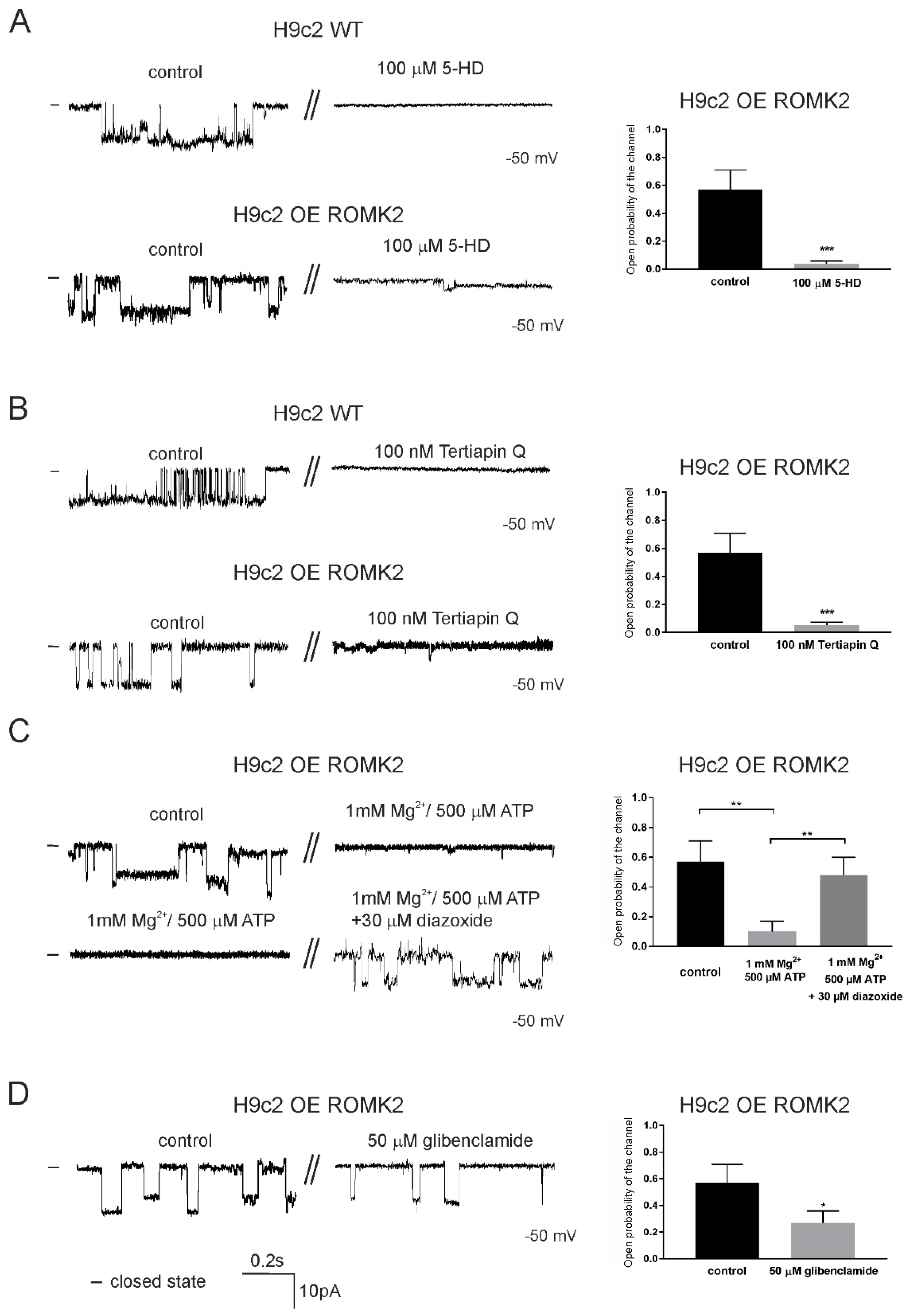
2.3. Specificity of the mitoKATP Channel Using the Patch-Clamp Technique
2.4. Pharmacological Properties of mitoKATP Using the Patch-Clamp Technique
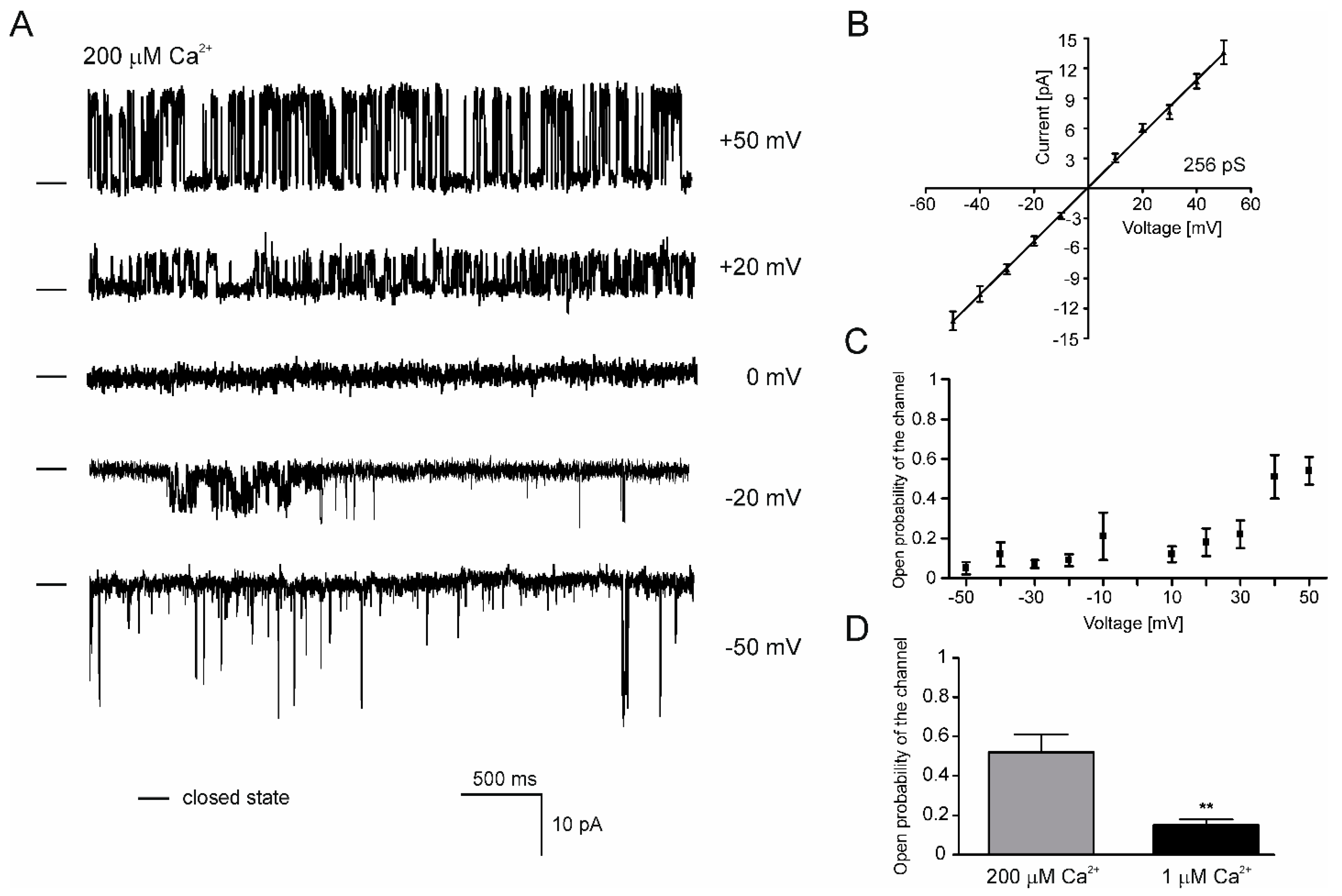
2.5. Properties of the mitoBKCa Channel from H9c2 OE ROMK2 Cells
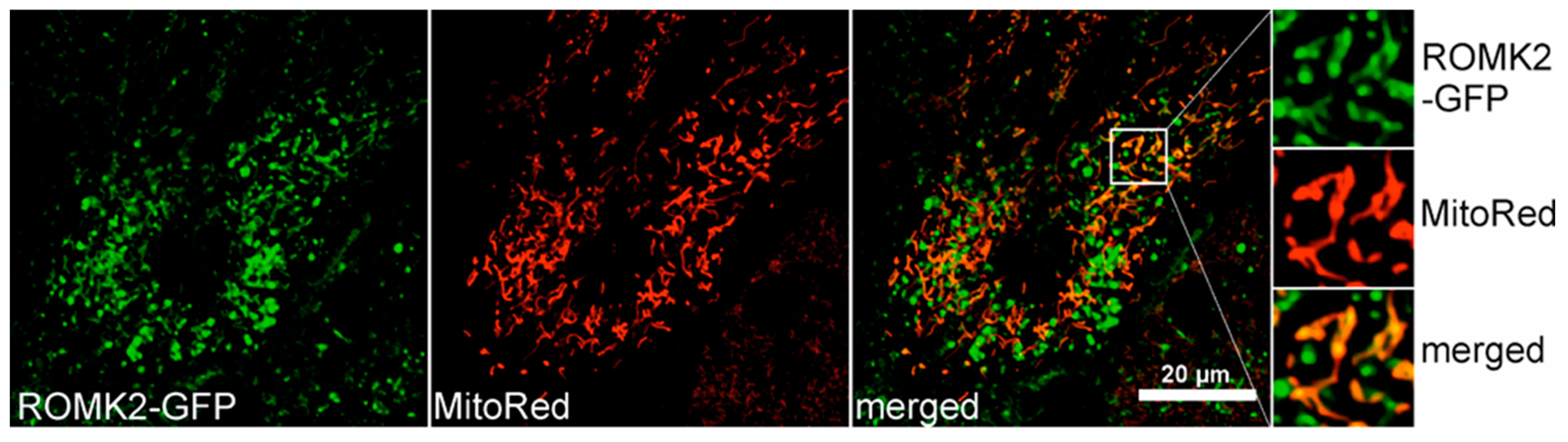
2.6. Localization of the ROMK protein after Transient Expression in H9c2 Cells
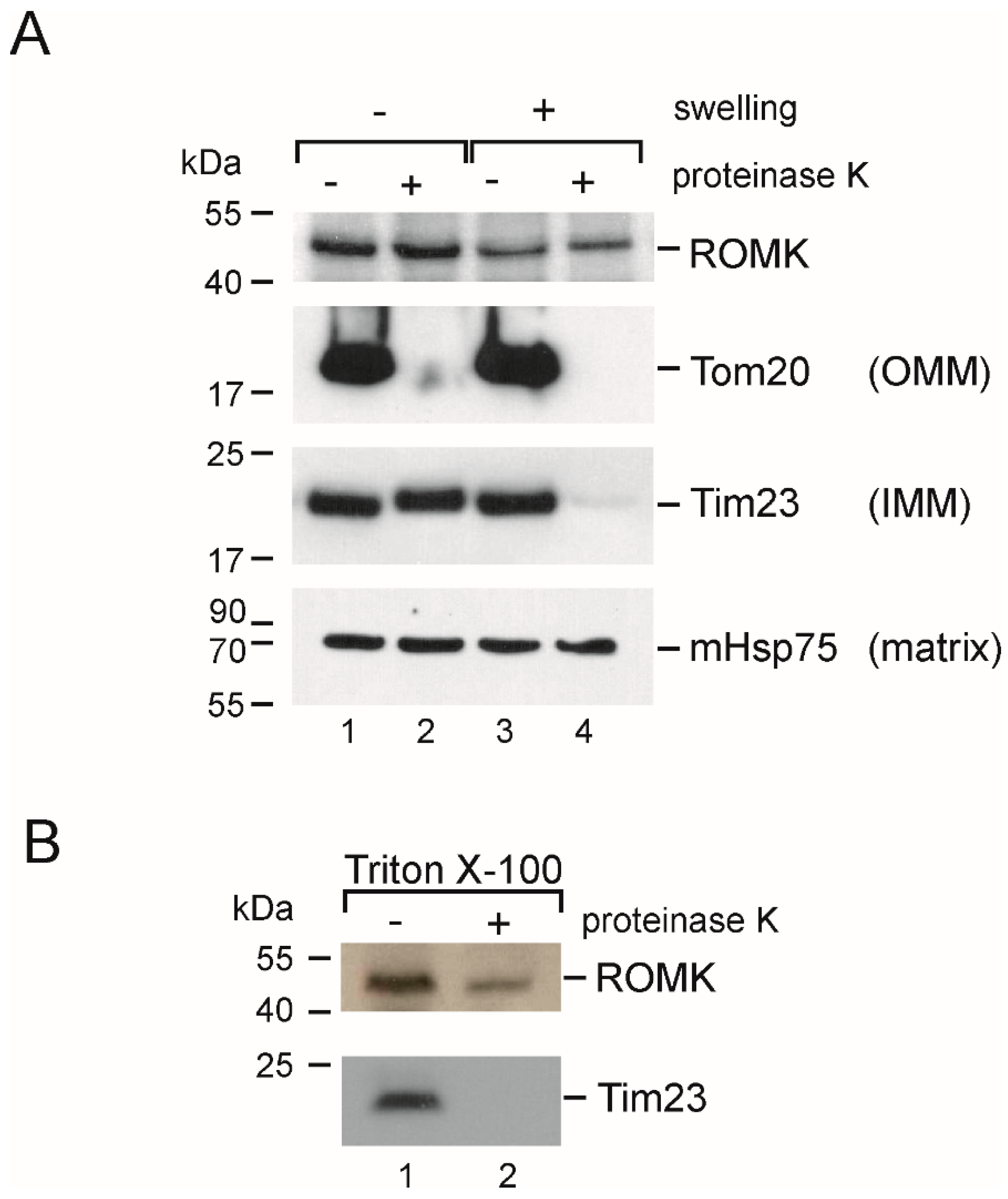
2.7. Localization in the Inner Mitochondrial Membrane and Possible Mitochondrial Topology of ROMK Protein in H9c2 OE ROMK2 Cells
3. Discussion
4. Materials and Methods
4.1. H9c2 WT and H9c2 OE ROMK2 Cells and Isolation of Mitochondria
4.2. Patch-Clamp Experiments
4.3. Transient Transfection and Fluorescence Staining of H9c2 Cells
4.4. Proteinase K and Western Blot Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-HD | 5-hydroxydecanoic acid |
| IMS | Mitochondrial intermembrane space |
| IPC | Ischemic preconditioning |
| MIM | Mitochondrial inner membrane |
| MOM | Mitochondrial outer membrane. |
| mitoKATP | Mitochondrial ATP-sensitive potassium channel |
| mitoBKCa | Mitochondrial large-conductance, calcium-regulated potassium channel |
| PK | Proteinase K |
| PTP | Permeability transition pore |
| ROMK | Renal outer medullary potassium channel |
| SUR | Sulfonylurea receptor |
References
- Perrelli, M.; Pagliaro, P.; Penna, C. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J. Cardiol. 2011, 3, 186–200. [Google Scholar] [CrossRef]
- Testai, L.; Rapposelli, S.; Martelli, A.; Breschi, M.; Calderone, V. Mitochondrial potassium channels as pharmacological target for cardioprotective drugs. Res. Rev. 2015, 35, 520–553. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.; Santos, R.; Perry, G.; Zhu, X.; Moreira, P.; Smith, M. Mitochondria: The missing link between preconditioning and neuroprotection. J. Alzheimer’s Dis. 2010, 20, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, P.; Dejean, L.; Kinnally, K. The therapeutic potential of mitochondrial channels in cancer, ischemia-reperfusion injury, and neurodegeneration. Mitochondrion 2012, 12, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Sasaki, N.; Seharaseyon, J.; O’Rourke, B.; Marban, E. Selective pharmacological agents implicate mitochondrial but not sarcolemmal K-ATP channels in ischemic cardioprotection. Circulation 2000, 101, 2418–2423. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Akao, M.; O’Rourke, B.; Marban, E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca2+ overload during simulated ischemia and reperfusion-Possible mechanism of cardioprotection. Circ. Res. 2001, 89, 891–898. [Google Scholar] [CrossRef]
- Laskowski, M.; Augustynek, B.; Kulawiak, B.; Koprowski, P.; Bednarczyk, P.; Jarmuszkiewicz, W.; Szewczyk, A. What do we not know about mitochondrial potassium channels? Biochim. Biophys. Acta 2016, 1857, 1247–1257. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef]
- Garlid, K.D.; Paucek, P.; YarovYarovoy, V.; Murray, H.N.; Darbenzio, R.B.; Dalonzo, A.J.; Lodge, N.J.; Smith, M.A.; Grover, G.J. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-Sensitive K+ channels - Possible mechanism of cardioprotection. Circ. Res. 1997, 81, 1072–1082. [Google Scholar] [CrossRef]
- Garlid, K.D.; Costa, A.D.; Quinlan, C.L.; Pierre, S.V.; Dos Santos, P. Cardioprotective signaling to mitochondria. J. Mol. Cell Cardiol. 2009, 46, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Downey, J.M.; Davis, A.M.; Cohen, M.V. Signaling pathways in ischemic preconditioning. Heart Fail. Rev. 2007, 12, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Nagase, H.; Kishi, K.; Higuti, T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 1991, 352, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk, P.; Kicińska, A.; Kominkova, V.; Ondrias, K.; Dołowy, K.; Szewczyk, A. Quinine inhibits mitochondrial ATP-regulated potassium channel from bovine heart. J. Membr. Biol. 2004, 199, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Paucek, P.; Mironova, G.; Mahdi, F.; Beavis, A.D.; Woldegiorgis, G.; Garlid, K.D. Reconstitution and partial-purification of the glibenclamide-sensitive, ATP-dependent K+-channel from rat-liver and beef-heart mitochondria. J. Biol. Chem. 1992, 267, 26062–26069. [Google Scholar] [PubMed]
- Garlid, K.D.; Paucek, P.; YarovYarovoy, V.; Sun, X.C.; Schindler, P.A. The mitochondrial K-ATP channel as a receptor for potassium channel openers. J. Biol. Chem. 1996, 271, 8796–8799. [Google Scholar] [CrossRef] [PubMed]
- Bajgar, R.; Seetharaman, S.; Kowaltowski, A.J.; Garlid, K.D.; Paucek, P. Identification and properties of a novel intracellular (mitochondrial) ATP-sensitive potassium channel in brain. J. Biol. Chem. 2001, 276, 33369–33374. [Google Scholar] [CrossRef] [PubMed]
- Debska, G.; May, R.; Kicinska, A.; Szewczyk, A.; Elger, C.E.; Kunz, W.S. Potassium channel openers depolarize hippocampal mitochondria. Brain Res. 2001, 892, 42–50. [Google Scholar] [CrossRef]
- Debska, G.; Kicinska, A.; Skalska, J.; Szewczyk, A.; May, R.; Elger, C.; Kunz, W. Opening of potassium channels modulates mitochondrial function in rat skeletal muscle. Biochim. Biophys. Acta 2002, 1556, 97–105. [Google Scholar] [CrossRef]
- Bednarczyk, P.; Kicinska, A.; Laskowski, M.; Kulawiak, B.; Kampa, R.; Walewska, A.; Krajewska, M.; Jarmuszkiewicz, W. Evidence for a mitochondrial ATP-regulated potassium channel in human dermal fibroblasts. Biochim. Et Biophys. Acta Bioenerg. 2018, 1859, 309–318. [Google Scholar] [CrossRef]
- Cancherini, D.; Trabuco, L.; Reboucas, N.; Kowaltowski, A. ATP-sensitive K+ channels in renal mitochondria. Am. J. Physiol. Renal. Physiol. 2003, 1291–1296. [Google Scholar] [CrossRef]
- Costa, A.; Krieger, M. Evidence for an ATP-sensitive K+ channel in mitoplasts isolated from Trypanosoma cruzi and Crithidia fasciculata. Int. J. Parasitol. 2009, 39, 955–961. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wojtovich, A.; Burwell, L.; Sherman, T.; Nehrke, K.; Brookes, P.; The, C. elegans mitochondrial K+(ATP) channel: A potential target for preconditioning. Biochem. Biophys. Rescommun. 2008, 376, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Kicinska, A.; Swida, A.; Bednarczyk, P.; Dolowy, K.; Jarmuszkiewicz, W.; Szewczyk, A. ATP-sensitive potassium channel in mitochondria of the eukaryotic microorganism, Acanthamoeba castellanii. J. Biol. Chem. 2007, 282, 17433–17441. [Google Scholar] [CrossRef] [PubMed]
- Pastore, D.; Stopelli, M.; Di Fonzo, N.; Passarella, S. The existence of the K+ channel in plant mitochondria. J. Biol. Chem. 1999, 274, 26683–26690. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kotake, K.; Fujikura, K.; Inagaki, N.; Suzuki, T.; Gonoi, T.; Seino, S.; Takata, K. Kir6.1: A possible subunit of ATP-sensitive K+ channels in mitochondria. Biochem. Biophys. Res. Commun. 1998, 241, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.; Rucker, J.; Marban, E. Is Kir6.1 a subunit of mitoKATP? Biochem. Biophys. Res. Commun. 2008, 366, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Wojtovich, A.P.; Urciuoli, W.R.; Chatterjee, S.; Fisher, A.B.; Nehrke, K.; Brookes, P.S. Kir6.2 is not the mitochondrial K-ATP channel but is required for cardioprotection by ischemic preconditioning. American J. Physiol. Heart Circ. Physiol. 2013, 304, H1439–H1445. [Google Scholar] [CrossRef]
- Foster, D.; Ho, A.; Rucker, J.; Garlid, A.; Sidor, A.; Garlid, K.; O’Rourke, B. Mitochondrial ROMK channel is a molecular component of mitoKATP. Circ. Res. 2012, 111, 446–454. [Google Scholar] [CrossRef]
- Fretwell, L.; Dickenson, J.M. Role of large-conductance Ca2+-activated potassium channels in adenosine A (1) receptor-mediated pharmacological preconditioning in H9c2 cells. Eur. J. Pharmacol. 2009, 618, 37–44. [Google Scholar] [CrossRef]
- Choma, K.; Bednarczyk, P.; Koszela-Piotrowska, I.; Kulawiak, B.; Kunz, W.; Dołowy, K.; Szewczyk, A. Single channel studies of the ATP-regulated potassium channel in brain mitochondria. J. Bioenerg Biomembr. 2009, 41, 323–334. [Google Scholar] [CrossRef]
- Bednarczyk, P.; Dołowy, K.; Szewczyk, A. Matrix Mg2+ regulates mitochondrial ATP-dependent potassium channel from heart. FEBS Lett. 2005, 579, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Mironova, G.D.; Skarga, Y.Y.; Grigoriev, S.M.; Negoda, A.E.; Kolomytkin, O.V.; Marinov, B.S. Reconstitution of the mitochondrial ATP-dependent potassium channel into bilayer lipid membrane. J. Bioenerg. Biomembr. 1999, 31, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Dahlem, Y.A.; Horn, T.F.W.; Buntinas, L.; Gonoi, T.; Wolf, G.; Siemen, D. The human mitochondrial KATP channel is modulated by calcium and nitric oxide: A patch-clamp approach. Biochim. Et Biophys. Acta 2004, 1656, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk, P.; Kowalczyk, J.; Beresewicz, M.; Dolowy, K.; Szewczyk, A.; Zablocka, B. Identification of a voltage-gated potassium channel in gerbil hippocampal mitochondria. Biochem. Biophys. Res. Commun. 2010, 397, 614–620. [Google Scholar] [CrossRef]
- Jiang, M.; Ljubkovic, M.; Nakae, Y.; Shi, Y.; Kwok, W.; Stowe, D.; Bosnjak, Z. Characterization of human cardiac mitochondrial ATP-sensitive potassium channel and its regulation by phorbol ester in vitro. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, 1770–1776. [Google Scholar] [CrossRef]
- Welling, P.; Ho, K. A comprehensive guide to the ROMK potassium channel: Form and function in health and disease. Am. J. Physiol. Ren. Physiol. 2009, 297, 849–863. [Google Scholar] [CrossRef]
- Wang, W. Two types of K+ channel in thick ascending limb of rat kidney. Am. J. Physiol. Ren. Fluid. Electrolyte Physiol. 1994, 267, 599–605. [Google Scholar] [CrossRef]
- Balderas, E.; Zhang, J.; Stefani, E.; Toro, L. Mitochondrial. BKCa channel. Front. Physiol. 2015, 6, 104. [Google Scholar] [CrossRef]
- Sato, T.; Li, Y.L.; Saito, T.; Nakaya, H. Minoxidil opens mitochondrial K-ATP channels and confers cardioprotection. Br. J. Pharmacol. 2004, 141, 360–366. [Google Scholar] [CrossRef]
- Jaburek, M.; Yarov-Yarovoy, V.; Paucek, P.; Garlid, K.D. State-dependent inhibition of the mitochondrial K-ATP channel by glyburide and 5-hydroxydecanoate. J. Biol. Chem. 1998, 273, 13578–13582. [Google Scholar]
- Jin, W.; Klem, A.; Lewis, J.; Lu, Z. Mechanisms of inward-rectifier K+ channel inhibition by tertiapin-Q. Biochemistry 1999, 38, 14294–14301. [Google Scholar] [CrossRef] [PubMed]
- Hilder, T.A.; Chung, S.H. Conduction and Block of Inward Rectifier K+ Channels: Predicted Structure of a Potent Blocker of Kir2.1. Biochemistry 2013, 52, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Doupnik, C.A. Venom-derived peptides inhibiting Kir channels: Past, present, and future. Neuropharmacology 2017, 127, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Kanjhan, R.; Coulson, E.J.; Adams, D.J.; Bellingham, M.C. Tertiapin-Q blocks recombinant and native large conductance K+ channels in a use-dependent manner. J. Pharmacol. Exp. Ther. 2005, 314, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, Y.; Wang, S.; McDonald, T.; Van Eyk, J.E.; Sidor, A.; O’Rourke, B. Cytoprotective role of Ca2+- activated K+ channels in the cardiac inner mitochondrial membrane. Science 2002, 298, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Frankenreiter, S.; Bednarczyk, P.; Kniess, A.; Bork, N.I.; Straubinger, J.; Koprowski, P.; Wrzosek, A.; Mohr, E.; Logan, A.; Murphy, M.P.; et al. cGMP-Elevating Compounds and Ischemic Conditioning Provide Cardioprotection Against Ischemia and Reperfusion Injury via Cardiomyocyte-Specific BK Channels. Circulation 2017, 136, 2337–2355. [Google Scholar] [CrossRef]
- Singh, H.; Lu, R.; Bopassa, J.C.; Meredith, A.L.; Stefani, E.; Toro, L. mitoBK(Ca) is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location. Proc. Natl. Acad. Sci. USA 2013, 110, 10836–10841. [Google Scholar] [CrossRef]
- Ramu, Y.; Klem, A.M.; Lu, Z. Short variable sequence acquired in evolution enables selective inhibition of various inward-rectifier K+ channels. Biochemistry 2004, 43, 10701–10709. [Google Scholar] [CrossRef]
- Han, J.H.; Kang, D.W.; Kim, D. Properties and modulation of the G protein-coupled K+ channel in rat cerebellar granule neurons: ATP versus phosphatidylinositol 4,5-bisphosphate. J. Physiol. Lond. 2003, 550, 693–706. [Google Scholar] [CrossRef]
- Pleumsamran, A.; Wolak, M.L.; Kim, D. Inhibition of ATP-induced increase in muscarinic K+ current by trypsin, alkaline pH, and anions. Am. J. Physiol. Heart Circ. Physiol. 1998, 275, H751–H759. [Google Scholar] [CrossRef]
- Giebisch, G. Renal potassium channels: Function, regulation, and structure. Kidney Int. 2001, 60, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Talanov, E.Y.; Pavlik, L.L.; Mikheeva, I.B.; Murzaeva, S.V.; Ivanov, A.N.; Mironova, G.D. Ultrastructural Localization of the ROMK Potassium Channel in Rat Liver and Heart. Biochem. Mosc. Suppl. Ser. A 2016, 10, 195–198. [Google Scholar] [CrossRef]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabo, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 2019, 572, 609. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, A.; Czyż, A.; Nałęcz, M. ATP-regulated potassium channel blocker, glibenclamide, uncouples mitochondria. Pol. J. Pharm. 1997, 49, 49–52. [Google Scholar]
- Tanemoto, M.; Vanoye, C.; Dong, K.; Welch, R.; Abe, T.; Hebert, S.; Xu, J. Rat homolog of sulfonylurea receptor 2B determines glibenclamide sensitivity of ROMK2 in Xenopus laevis oocyte. Am. J. Physiol. 2000, 278, 659–666. [Google Scholar]
- Ye, B.; Kroboth, S.; Pu, J.; Sims, J.; Aggarwal, N.; McNally, E.; Makielski, J.; Shi, N. Molecular identification and functional characterization of a mitochondrial sulfonylurea receptor 2 splice variant generated by intraexonic splicing. Circ. Res. 2009, 105, 1083–1093. [Google Scholar] [CrossRef]
- Konstas, A.; Dabrowski, M.; Korbmacher, C.; Tucker, S. Intrinsic sensitivity of Kir1.1 (ROMK) to glibenclamide in the absence of SUR2B. Implications for the identity of the renal ATP-regulated secretory K+ channel. J. Biol. Chem. 2002, 277, 21346–21351. [Google Scholar] [CrossRef]
- Ruknudin, A.; Schulze, D.; Sullivan, S.; Lederer, W.; Welling, P. Novel subunit composition of a renal epithelial KATP channel. J. Biol. Chem. 1998, 273, 14165–14171. [Google Scholar] [CrossRef]
- Yarov-Yarovoy, V.; Paucek, P.; Jaburek, M.; Garlid, K. The nucleotide regulatory sites on the mitochondrial KATP channel face the cytosol. Biochim. Biophys. Acta 1997, 1321, 128–136. [Google Scholar] [CrossRef]
- McKee, E.E.; Bentley, A.T.; Smith, R.M.; Ciaccio, C.E. Origin of guanine nucleotides in isolated heart mitochondria. Biochem. Biophys. Res. Commun. 1999, 257, 466–472. [Google Scholar] [CrossRef]
- Vozza, A.; Blanco, E.; Palmieri, L.; Palmieri, F. Identification of the mitochondrial GTP/GDP transporter in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 20850–20857. [Google Scholar] [CrossRef] [PubMed]
- Olszewska, A.; Bednarczyk, P.; Siemen, D.; Szewczyk, A. Modulation of the mitochondrial large-conductance calcium-regulated potassium channel by polyunsaturated fatty acids. Biochim. Biophys. Acta 2014, 1837, 1602–1610. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bednarczyk, P.; Wieckowski, M.R.; Broszkiewicz, M.; Skowronek, K.; Siemen, D.; Szewczyk, A. Putative Structural and Functional Coupling of the Mitochondrial BKCa Channel to the Respiratory Chain. PLoS ONE 2013, 8, e68125. [Google Scholar] [CrossRef] [PubMed]
- Walewska, A.; Kulawiak, B.; Szewczyk, A.; Koprowski, P. Mechanosensitivity of mitochondrial large-conductance calcium-activated potassium channels. Biochim. Biophys. Acta 2018, 1859, 797–805. [Google Scholar] [CrossRef]
- Kajma, A.; Szewczyk, A. A new pH-sensitive rectifying potassium channel in mitochondria from the embryonic rat hippocampus. Biochim. Et Biophys. Acta Bioenerg. 2012, 1817, 1867–1878. [Google Scholar] [CrossRef]
- Chacinska, A.; Koehler, C.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef]
- Kulawiak, B.; Hopker, J.; Gebert, M.; Guiard, B.; Wiedemann, N.; Gebert, N. The mitochondrial protein import machinery has multiple connections to the respiratory chain. Biochim. Biophys. Acta 2013, 1827, 612–626. [Google Scholar] [CrossRef]
- Dolga, A.M.; Netter, M.F.; Perocchi, F.; Doti, N.; Meissner, L.; Tobaben, S.; Grohm, J.; Zischka, H.; Plesnila, N.; Decher, N.; et al. Mitochondrial Small Conductance SK2 Channels Prevent Glutamate-induced Oxytosis and Mitochondrial Dysfunction. J. Biol. Chem. 2013, 288, 10792–10804. [Google Scholar] [CrossRef]
- Hu, X.L.; Xu, X.; Huang, Y.M.; Fassett, J.; Flagg, T.P.; Zhang, Y.; Nichols, C.G.; Bache, R.J.; Chen, Y.J. Disruption of Sarcolemmal ATP-Sensitive Potassium Channel Activity Impairs the Cardiac Response to Systolic Overload. Circ. Res. 2008, 103, 1009–1017. [Google Scholar] [CrossRef]
- Glukhov, A.V.; Flagg, T.P.; Fedorov, V.V.; Efimov, I.R.; Nichols, C.G. Differential K-ATP channel pharmacology in intact mouse heart. J. Mol. Cell. Cardiol. 2010, 48, 152–160. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laskowski, M.; Augustynek, B.; Bednarczyk, P.; Żochowska, M.; Kalisz, J.; O’Rourke, B.; Szewczyk, A.; Kulawiak, B. Single-Channel Properties of the ROMK-Pore-Forming Subunit of the Mitochondrial ATP-Sensitive Potassium Channel. Int. J. Mol. Sci. 2019, 20, 5323. https://doi.org/10.3390/ijms20215323
Laskowski M, Augustynek B, Bednarczyk P, Żochowska M, Kalisz J, O’Rourke B, Szewczyk A, Kulawiak B. Single-Channel Properties of the ROMK-Pore-Forming Subunit of the Mitochondrial ATP-Sensitive Potassium Channel. International Journal of Molecular Sciences. 2019; 20(21):5323. https://doi.org/10.3390/ijms20215323
Chicago/Turabian StyleLaskowski, Michał, Bartłomiej Augustynek, Piotr Bednarczyk, Monika Żochowska, Justyna Kalisz, Brian O’Rourke, Adam Szewczyk, and Bogusz Kulawiak. 2019. "Single-Channel Properties of the ROMK-Pore-Forming Subunit of the Mitochondrial ATP-Sensitive Potassium Channel" International Journal of Molecular Sciences 20, no. 21: 5323. https://doi.org/10.3390/ijms20215323
APA StyleLaskowski, M., Augustynek, B., Bednarczyk, P., Żochowska, M., Kalisz, J., O’Rourke, B., Szewczyk, A., & Kulawiak, B. (2019). Single-Channel Properties of the ROMK-Pore-Forming Subunit of the Mitochondrial ATP-Sensitive Potassium Channel. International Journal of Molecular Sciences, 20(21), 5323. https://doi.org/10.3390/ijms20215323