The Landscape of Actionable Gene Fusions in Colorectal Cancer


 and
and
Abstract
1. Introduction
1.1. NTRK
1.2. ALK and ROS1
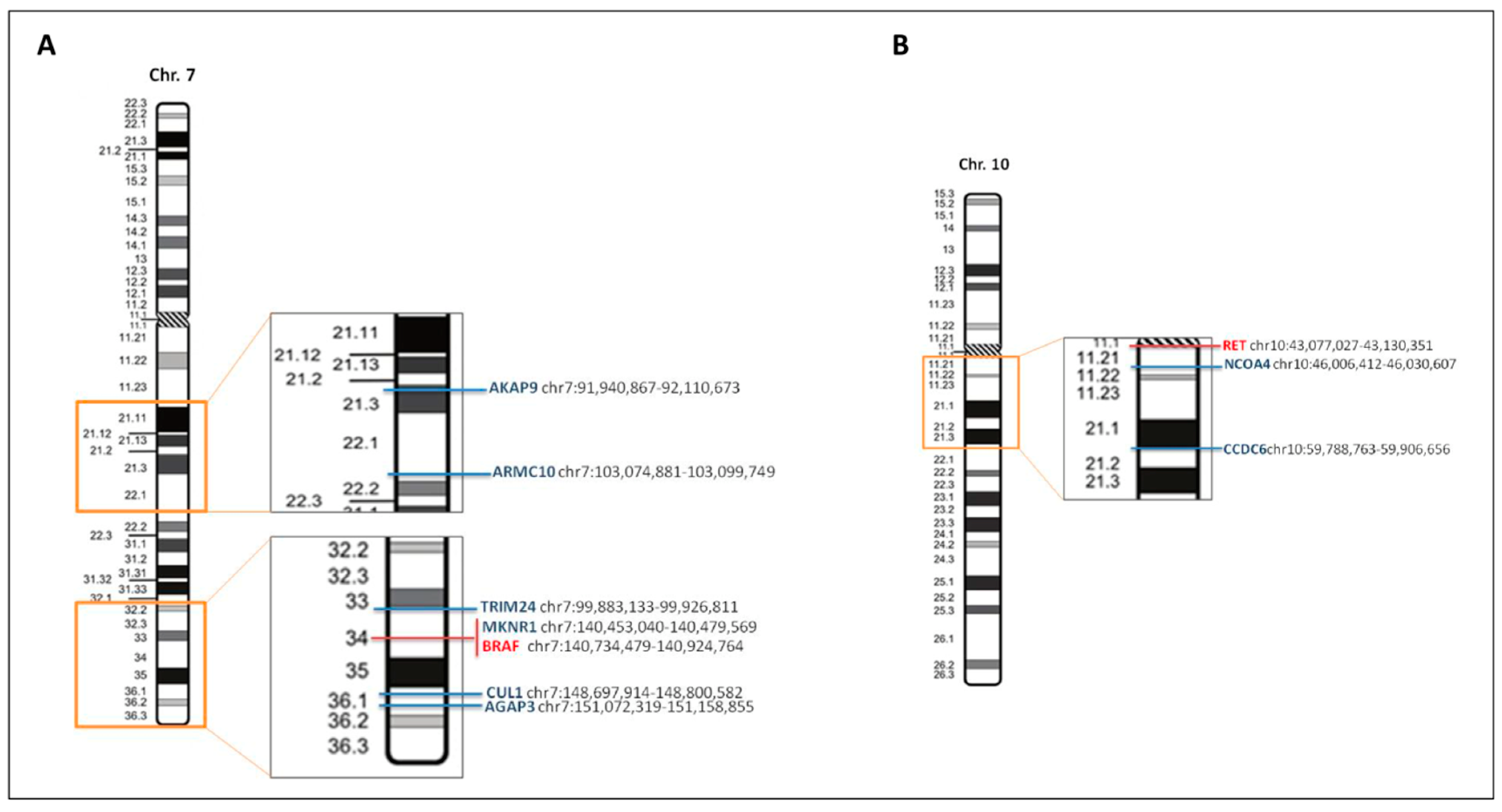
1.3. RET
1.4. BRAF
1.5. Other Gene Fusions: An Emerging Complexity
1.6. Methods to Detect Gene Fusions
2. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Stintzing, S.; Modest, D.P.; Rossius, L.; Lerch, M.M.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab for metastatic colorectal cancer (FIRE-3): A post-hoc analysis of tumour dynamics in the final RAS wild-type subgroup of this randomised open-label phase 3 trial. Lancet Oncol. 2016, 17, 1426–1434. [Google Scholar] [CrossRef]
- Loupakis, F.; Cremolini, C.; Masi, G.; Lonardi, S.; Zagonel, V.; Salvatore, L.; Cortesi, E.; Tomasello, G.; Ronzoni, M.; Spadi, R.; et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N. Engl. J. Med. 2014, 371, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Simkens, L.H.; van Tinteren, H.; May, A.; ten Tije, A.J.; Creemers, G.J.; Loosveld, O.J.; de Jongh, F.E.; Erdkamp, F.L.; Erjavec, Z.; van der Torren, A.M.; et al. Maintenance treatment with capecitabine and bevacizumab in metastatic colorectal cancer (CAIRO3): A phase 3 randomised controlled trial of the Dutch Colorectal Cancer Group. Lancet 2015, 385, 1843–1852. [Google Scholar] [CrossRef]
- Modest, D.P.; Pant, S.; Sartore-Bianchi, A. Treatment sequencing in metastatic colorectal cancer. Eur. J. Cancer 2019, 109, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Colon Cancer (version 1.2019). Available online: https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf (accessed on 15 June 2019).
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef]
- Network, C.G.A. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Haan, J.C.; Labots, M.; Rausch, C.; Koopman, M.; Tol, J.; Mekenkamp, L.J.; van de Wiel, M.A.; Israeli, D.; van Essen, H.F.; van Grieken, N.C.; et al. Genomic landscape of metastatic colorectal cancer. Nat. Commun. 2014, 5, 5457. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef]
- Valeri, N. Streamlining detection of fusion genes in colorectal cancer: Having “faith” in precision oncology in the (tissue) “agnostic” era. Cancer Res. 2019, 79, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Sinha, C.; Kalyana-Sundaram, S.; Chinnaiyan, A.M. Landscape of gene fusions in epithelial cancers: Seq and ye shall find. Genome Med. 2015, 7, 129. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; van den Braak, R.R.J.C.; Pieterse, M.; van Roosmalen, M.J.; Sieuwerts, A.M.; Stangl, C.; Brunekreef, R.; Lalmahomed, Z.S.; Ooft, S.; van Galen, A.; et al. A systematic analysis of oncogenic gene fusions in primary colon cancer. Cancer Res. 2017, 77, 3814–3822. [Google Scholar] [CrossRef]
- Roche. Media Release. Available online: https://www.roche.com/media/releases/med-cor-2019-02-19b.htm (accessed on 15 June 2019).
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Cocco, E.; Benhamida, J.; Middha, S.; Zehir, A.; Mullaney, K.; Shia, J.; Yaeger, R.; Zhang, L.; Wong, D.; Villafania, L.; et al. Colorectal carcinomas containing hypermethylated MLH1 promoter and wild-type BRAF/KRAS are enriched for targetable kinase fusions. Cancer Res. 2019, 79, 1047–1053. [Google Scholar] [CrossRef]
- Medico, E.; Russo, M.; Picco, G.; Cancelliere, C.; Valtorta, E.; Corti, G.; Buscarino, M.; Isella, C.; Lamba, S.; Martinoglio, B.; et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 2015, 6, 7002. [Google Scholar] [CrossRef]
- Cremolini, C.; Morano, F.; Moretto, R.; Berenato, R.; Tamborini, E.; Perrone, F.; Rossini, D.; Gloghini, A.; Busico, A.; Zucchelli, G.; et al. Negative hyper-selection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: The PRESSING case-control study. Ann. Oncol. 2017, 28, 3009–3014. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Okamura, R.; Boichard, A.; Kato, S.; Sicklick, J.K.; Bazhenova, L.; Kurzrock, R. Analysis of NTRK alterations in pan-cancer adult and pediatric malignancies: Implications for NTRK-targeted therapeutics. JCO Precis. Oncol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Gatalica, Z.; Xiu, J.; Swensen, J.; Vranic, S. Molecular characterization of cancers with NTRK gene fusions. Mod Pathol. 2019, 32, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Milione, M.; Ardini, E.; Christiansen, J.; Valtorta, E.; Veronese, S.; Bosotti, R.; Pellegrinelli, A.; Testi, A.; Pietrantonio, F.; Fucà, G.; et al. Identification and characterization of a novel SCYL3-NTRK1 rearrangement in a colorectal cancer patient. Oncotarget 2017, 8, 55353–55360. [Google Scholar] [CrossRef] [PubMed]
- Hechtman, J.F.; Benayed, R.; Hyman, D.M.; Drilon, A.; Zehir, A.; Frosina, D.; Arcila, M.E.; Dogan, S.; Klimstra, D.S.; Ladanyi, M.; et al. Pan-trk immunohistochemistry is an efficient and reliable screen for the detection of NTRK fusions. Am. J. Surg. Pathol. 2017, 41, 1547–1551. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Tejpar, S.; Sartore-Bianchi, A.; Hechtman, J.F.; Christiansen, J.; Novara, L.; Tebbutt, N.; et al. ALK, ROS1, and NTRK rearrangements in metastatic colorectal cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. Gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef]
- Hsiao, S.J.; Zehir, A.; Sireci, A.N.; Aisner, D.L. Detection of tumor NTRK gene fusions to identify patients who may benefit from tyrosine kinase (TRK) inhibitor therapy. J. Mol. Diagn. 2019, 21, 553–571. [Google Scholar] [CrossRef]
- Hechtman, J.F.; Zehir, A.; Yaeger, R.; Wang, L.; Middha, S.; Zheng, T.; Hyman, D.M.; Solit, D.; Arcila, M.E.; Borsu, L.; et al. Identification of Targetable kinase alterations in patients with colorectal carcinoma that are preferentially associated with wild-type RAS/RAF. Mol. Cancer Res. 2016, 14, 296–301. [Google Scholar] [CrossRef]
- Sato, K.; Kawazu, M.; Yamamoto, Y.; Ueno, T.; Kojima, S.; Nagae, G.; Abe, H.; Soda, M.; Oga, T.; Kohsaka, S.; et al. Fusion kinases identified by genomic analyses of sporadic microsatellite instability-high colorectal cancers. Clin. Cancer Res. 2019, 25, 378–389. [Google Scholar] [CrossRef]
- Lipson, D.; Capelletti, M.; Yelensky, R.; Otto, G.; Parker, A.; Jarosz, M.; Curran, J.A.; Balasubramanian, S.; Bloom, T.; Brennan, K.W.; et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 2012, 18, 382–384. [Google Scholar] [CrossRef]
- Yakirevich, E.; Resnick, M.B.; Mangray, S.; Wheeler, M.; Jackson, C.L.; Lombardo, K.A.; Lee, J.; Kim, K.M.; Gill, A.J.; Wang, K.; et al. Oncogenic ALK fusion in rare and aggressive subtype of colorectal adenocarcinoma as a potential therapeutic target. Clin. Cancer Res. 2016, 22, 3831–3840. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.C.; Hong, J.Y.; Wang, K.; Kim, S.Y.; Jang, J.; Kim, S.T.; Park, J.O.; Lim, H.Y.; Kang, W.K.; et al. Detection of novel and potentially actionable anaplastic lymphoma kinase (ALK) rearrangement in colorectal adenocarcinoma by immunohistochemistry screening. Oncotarget 2015, 6, 24320–24332. [Google Scholar] [CrossRef]
- Selvam, P.; Kelly, K.; Hesse, A.N.; Spitzer, D.; Reddi, H.V. Evaluating gene fusions in solid tumors—Clinical experience using an RNA based 53 gene next-generation sequencing panel. Cancer Genet. 2019, 233–234, 32–42. [Google Scholar] [CrossRef]
- Choi, Y.; Kwon, C.H.; Lee, S.J.; Park, J.; Shin, J.Y.; Park, D.Y. Integrative analysis of oncogenic fusion genes and their functional impact in colorectal cancer. Br. J. Cancer 2018, 119, 230–240. [Google Scholar] [CrossRef]
- Jang, J.E.; Kim, H.P.; Han, S.W.; Jang, H.; Lee, S.H.; Song, S.H.; Bang, D.; Kim, T.Y. NFATC3-PLA2G15 fusion transcript identified by RNA sequencing promotes tumor invasion and proliferation in colorectal cancer cell lines. Cancer Res. Treat. 2019, 51, 391–401. [Google Scholar] [CrossRef]
- Ardini, E.; Menichincheri, M.; Banfi, P.; Bosotti, R.; De Ponti, C.; Pulci, R.; Ballinari, D.; Ciomei, M.; Texido, G.; Degrassi, A.; et al. Entrectinib, a Pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol. Cancer 2016, 15, 628–639. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Ou, S.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor entrectinib: Combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef]
- Basket Study of Entrectinib (RXDX-101) for the Treatment of Patients WITH Solid Tumors Harboring NTRK 1/2/3 (Trk A/B/C), ROS1, or ALK Gene Rearrangements (Fusions) (STARTRK-2). Available online: https://clinicaltrials.gov/ct2/show/NCT02568267 (accessed on 15 June 2019).
- Study of Entrectinib (Rxdx-101) in Children and Adolescents with no Curative First-Line Treatment Option, Recurrent or Refractory Solid Tumors and Primary Cns Tumors, with or without Trk, Ros1, or Alk Fusions. Available online: https://clinicaltrials.gov/ct2/show/NCT02650401 (accessed on 15 June 2019).
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kim, H.R.; Ahn, M.J.; Yang, J.C.; Han, J.Y.; Lee, J.S.; Hochmair, M.J.; Li, J.Y.; Chang, G.C.; Lee, K.H.; et al. Brigatinib versus crizotinib in ALK-positive non-small-cell lung Cancer. N. Engl. J. Med. 2018, 379, 2027–2039. [Google Scholar] [CrossRef]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Solomon, B.J.; Besse, B.; Bauer, T.M.; Felip, E.; Soo, R.A.; Camidge, D.R.; Chiari, R.; Bearz, A.; Lin, C.C.; Gadgeel, S.M.; et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: Results from a global phase 2 study. Lancet Oncol. 2018, 19, 1654–1667. [Google Scholar] [CrossRef]
- Shaw, A.T.; Ou, S.H.; Bang, Y.J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Gainor, J.F.; Lee, D.H.; Curigliano, G.; Doebele, R.C.; Kim, D.-W.; Baik, C.S.; Tan, D.S.-W.; Lopes, G.; Gadgeel, S.M.; Cassier, P.A.; et al. Clinical activity and tolerability of BLU-667, a highly potent and selective RET inhibitor, in patients (pts) with advanced RET-fusion+ non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2019, 37 (Suppl. 15), 9008. [Google Scholar]
- Taylor, M.H.; Gainor, J.F.; Hu, M.I.-N.; Zhu, V.W.; Lopes, G.; Leboulleux, S.; Brose, M.S.; Schuler, M.H.; Bowles, D.W.; Kim, D.-W.; et al. Activity and tolerability of BLU-667, a highly potent and selective RET inhibitor, in patients with advanced RET-altered thyroid cancers. J. Clin. Oncol. 2019, 37 (Suppl. 15), 6018. [Google Scholar] [CrossRef]
- Drilon, A.E.; Subbiah, V.; Oxnard, G.R.; Bauer, T.M.; Velcheti, V.; Lakhani, N.J.; Besse, B.; Park, K.; Patel, J.D.; Cabanillas, M.E.; et al. A phase 1 study of LOXO-292, a potent and highly selective RET inhibitor, in patients with RET-altered cancers. J. Clin. Oncol. 2018, 36 (Suppl. 15), 102. [Google Scholar] [CrossRef]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol 2016, 27 (Suppl. 3), iii4–iii15. [Google Scholar] [CrossRef]
- Bergethon, K.; Shaw, A.T.; Ou, S.H.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef]
- Sasaki, T.; Rodig, S.J.; Chirieac, L.R.; Jänne, P.A. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur. J. Cancer 2010, 46, 1773–1780. [Google Scholar] [CrossRef]
- Alese, O.B.; El-Rayes, B.F.; Sica, G.; Zhang, G.; Alexis, D.; La Rosa, F.G.; Varella-Garcia, M.; Chen, Z.; Rossi, M.R.; Adsay, N.V.; et al. Anaplastic lymphoma kinase (ALK) gene alteration in signet ring cell carcinoma of the gastrointestinal tract. Adv. Med. Oncol. 2015, 7, 56–62. [Google Scholar] [CrossRef]
- Lin, E.; Li, L.; Guan, Y.; Soriano, R.; Rivers, C.S.; Mohan, S.; Pandita, A.; Tang, J.; Modrusan, Z. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol. Cancer Res. 2009, 7, 1466–1476. [Google Scholar] [CrossRef]
- Bavi, P.; Jehan, Z.; Bu, R.; Prabhakaran, S.; Al-Sanea, N.; Al-Dayel, F.; Al-Assiri, M.; Al-Halouly, T.; Sairafi, R.; Uddin, S.; et al. ALK gene amplification is associated with poor prognosis in colorectal carcinoma. Br. J. Cancer 2013, 109, 2735–2743. [Google Scholar] [CrossRef]
- Amatu, A.; Somaschini, A.; Cerea, G.; Bosotti, R.; Valtorta, E.; Buonandi, P.; Marrapese, G.; Veronese, S.; Luo, D.; Hornby, Z.; et al. Novel CAD-ALK gene rearrangement is drugable by entrectinib in colorectal cancer. Br. J. Cancer 2015, 113, 1730–1734. [Google Scholar] [CrossRef]
- Houang, M.; Toon, C.W.; Clarkson, A.; Sioson, L.; de Silva, K.; Watson, N.; Singh, N.R.; Chou, A.; Gill, A.J. ALK and ROS1 overexpression is very rare in colorectal adenocarcinoma. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Lin, C.; Wu, J.; Guo, L.; Qiu, T.; Ling, Y.; Shan, L.; Zhou, H.; Zhao, D.; Wang, J.; et al. Anaplastic lymphoma kinase rearrangement in digestive tract cancer: Implication for targeted therapy in Chinese population. PLoS ONE 2015, 10, e0144731. [Google Scholar] [CrossRef] [PubMed]
- Aisner, D.L.; Nguyen, T.T.; Paskulin, D.D.; Le, A.T.; Haney, J.; Schulte, N.; Chionh, F.; Hardingham, J.; Mariadason, J.; Tebbutt, N.; et al. ROS1 and ALK fusions in colorectal cancer, with evidence of intratumoral heterogeneity for molecular drivers. Mol. Cancer Res. 2014, 12, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, L.M. RET revisited: Expanding the oncogenic portfolio. Nat. Rev. Cancer 2014, 14, 173. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Hu, Z.I.; Lai, G.G.Y.; Tan, D.S.W. Targeting RET-driven cancers: Lessons from evolving preclinical and clinical landscapes. Nat. Rev. Clin. Oncol. 2017, 15, 151. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Elisei, R. RET/PTC Translocations and clinico-pathological features in human papillary thyroid carcinoma. Front. Endocrinol. 2012, 3, 54. [Google Scholar] [CrossRef]
- Kohno, T.; Ichikawa, H.; Totoki, Y.; Yasuda, K.; Hiramoto, M.; Nammo, T.; Sakamoto, H.; Tsuta, K.; Furuta, K.; Shimada, Y.; et al. KIF5B-RET fusions in lung adenocarcinoma. Nat. Med. 2012, 18, 375–377. [Google Scholar] [CrossRef]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378. [Google Scholar] [CrossRef]
- Le Rolle, A.-F.; Klempner, S.J.; Garrett, C.R.; Seery, T.; Sanford, E.M.; Balasubramanian, S.; Ross, J.S.; Stephens, P.J.; Miller, V.A.; Ali, S.M.; et al. Identification and characterization of RET fusions in advanced colorectal cancer. Oncotarget 2015, 6, 28929–28937. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Morano, F.; Fucà, G.; Nikolinakos, P.; Drilon, A.; Hechtman, J.F.; Christiansen, J.; et al. RET fusions in a small subset of advanced colorectal cancers at risk of being neglected. Ann. Oncol. 2018, 29, 1394–1401. [Google Scholar] [CrossRef]
- Roskoski, R.; Sadeghi-Nejad, A. Role of RET protein-tyrosine kinase inhibitors in the treatment RET-driven thyroid and lung cancers. Pharmacol. Res. 2018, 128, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Elisei, R.; Müller, S.; Schöffski, P.; Brose, M.; Shah, M.; Licitra, L.; Krajewska, J.; Kreissl, M.C.; Niederle, B.; et al. Overall survival analysis of EXAM, a phase III trial of cabozantinib in patients with radiographically progressive medullary thyroid carcinoma. Ann. Oncol. 2017, 28, 2813–2819. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W.; Cao, Q.; Sheets, M.P.; Wilson, D.; et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov. 2018, 8, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Velcheti, V.; Tuch, B.B.; Ebata, K.; Busaidy, N.L.; Cabanillas, M.E.; Wirth, L.J.; Stock, S.; Smith, S.; Lauriault, V.; et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann. Oncol. 2018, 29, 1869–1876. [Google Scholar] [CrossRef]
- Phase 1/2 Study of the Highly-selective RET Inhibitor, Pralsetinib (BLU-667), in Patients with Thyroid Cancer, Non-Small Cell Lung Cancer, and Other Advanced Solid Tumors (ARROW). Available online: https://clinicaltrials.gov/ct2/show/NCT03037385 (accessed on 16 June 2019).
- Phase 1/2 Study of LOXO-292 in Patients with Advanced Solid Tumors, RET Fusion-Positive Solid Tumors, and Medullary Thyroid Cancer (LIBRETTO-001). Available online: https://clinicaltrials.gov/ct2/show/NCT03157128 (accessed on 16 June 2019).
- Peeters, M.; Oliner, K.S.; Parker, A.; Siena, S.; Van Cutsem, E.; Huang, J.; Humblet, Y.; Van Laethem, J.L.; André, T.; Wiezorek, J.; et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin. Cancer Res. 2013, 19, 1902–1912. [Google Scholar] [CrossRef]
- Loupakis, F.; Moretto, R.; Aprile, G.; Muntoni, M.; Cremolini, C.; Iacono, D.; Casagrande, M.; Ferrari, L.; Salvatore, L.; Schirripa, M.; et al. Clinico-pathological nomogram for predicting BRAF mutational status of metastatic colorectal cancer. Br. J. Cancer 2016, 114, 30–36. [Google Scholar] [CrossRef]
- Jones, D.T.; Kocialkowski, S.; Liu, L.; Pearson, D.M.; Bäcklund, L.M.; Ichimura, K.; Collins, V.P. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008, 68, 8673–8677. [Google Scholar] [CrossRef]
- Cordioli, M.I.; Moraes, L.; Carvalheira, G.; Sisdelli, L.; Alves, M.T.; Delcelo, R.; Monte, O.; Longui, C.A.; Cury, A.N.; Cerutti, J.M. AGK-BRAF gene fusion is a recurrent event in sporadic pediatric thyroid carcinoma. Cancer Med. 2016, 5, 1535–1541. [Google Scholar] [CrossRef]
- Kulkarni, A.; Al-Hraishawi, H.; Simhadri, S.; Hirshfield, K.M.; Chen, S.; Pine, S.; Jeyamohan, C.; Sokol, L.; Ali, S.; Teo, M.L.; et al. BRAF fusion as a novel mechanism of acquired resistance to vemurafenib in BRAF V600E mutant melanoma. Clin. Cancer Res. 2017, 23, 5631–5638. [Google Scholar] [CrossRef]
- Hutchinson, K.E.; Lipson, D.; Stephens, P.J.; Otto, G.; Lehmann, B.D.; Lyle, P.L.; Vnencak-Jones, C.L.; Ross, J.S.; Pietenpol, J.A.; Sosman, J.A.; et al. BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin. Cancer Res. 2013, 19, 6696–6702. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Chmielecki, J.; Gay, L.; Johnson, A.; Chudnovsky, J.; Yelensky, R.; Lipson, D.; Ali, S.M.; Elvin, J.A.; et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int. J. Cancer 2016, 138, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- A Study of LGK974 in Patients with Malignancies Dependent on Wnt Ligands. Available online: https://clinicaltrials.gov/ct2/show/NCT01351103 (accessed on 6 August 2019).
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Sheehan, C.E.; Boguniewicz, A.B.; Otto, G.; Downing, S.R.; Sun, J.; He, J.; Curran, J.A.; Ali, S.; et al. Relapsed classic E-cadherin (CDH1) mutated invasive lobular breast cancer demonstrates a high frequency of HER2 (ERBB2) gene mutations. Clin. Cancer Res. 2013, 19, 2668. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Gay, L.M.; Al-Rohil, R.N.; Nazeer, T.; Sheehan, C.E.; Jennings, T.A.; Otto, G.A.; Donahue, A.; He, J.; et al. A high frequency of activating extracellular domain ERBB2 (HER2) mutation in micropapillary urothelial carcinoma. Clin. Cancer Res. 2014, 20, 68. [Google Scholar] [CrossRef]
- Chmielecki, J.; Ross, J.S.; Wang, K.; Frampton, G.M.; Palmer, G.A.; Ali, S.M.; Palma, N.; Morosini, D.; Miller, V.A.; Yelensky, R.; et al. Oncogenic alterations in ERBB2/HER2 represent potential therapeutic targets across tumors from diverse anatomic sites of origin. Oncologist 2015, 20, 7–12. [Google Scholar] [CrossRef]
- Connell, C.M.; Doherty, G.J. Activating HER2 mutations as emerging targets in multiple solid cancers. ESMO Open 2017, 2, e000279. [Google Scholar] [CrossRef]
- Laurent-Puig, P.; Balogoun, R.; Cayre, A.; Le Malicot, K.; Tabernero, J.; Mini, E.; Folprecht, G.; van Laethem, J.L.; Thaler, J.; Petersen, L.N.; et al. Taieb ERBB2 alterations a new prognostic biomarker in stage III colon cancer from a FOLFOX based adjuvant trial (PETACC8). Ann. Oncol. 2016, 27, 149–206. [Google Scholar] [CrossRef]
- Valtorta, E.; Martino, C.; Sartore-Bianchi, A.; Penaullt-Llorca, F.; Viale, G.; Risio, M.; Rugge, M.; Grigioni, W.; Bencardino, K.; Lonardi, S.; et al. Assessment of a HER2 scoring system for colorectal cancer: Results from a validation study. Mod Pathol. 2015, 28, 1481. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.D.; Southward, K.; Chambers, P.; Cross, D.; Barrett, J.; Hemmings, G.; Taylor, M.; Wood, H.; Hutchins, G.; Foster, J.M.; et al. HER2 overexpression and amplification as a potential therapeutic target in colorectal cancer: Analysis of 3256 patients enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trials. J. Pathol. 2016, 238, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Sartore-Bianchi, A.; Marsoni, S.; Hurwitz, H.I.; McCall, S.J.; Penault-Llorca, F.; Srock, S.; Bardelli, A.; Trusolino, L. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann. Oncol 2018, 29, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.A.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: Results from MyPathway, an open-label, phase IIa multiple basket study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef]
- Vaughn, C.P.; Costa, J.L.; Feilotter, H.E.; Petraroli, R.; Bagai, V.; Rachiglio, A.M.; Marino, F.Z.; Tops, B.; Kurth, H.M.; Sakai, K.; et al. Simultaneous detection of lung fusions using a multiplex RT-PCR next generation sequencing-based approach: A multi-institutional research study. BMC Cancer 2018, 18, 828. [Google Scholar] [CrossRef]
- Heyer, E.E.; Deveson, I.W.; Wooi, D.; Selinger, C.I.; Lyons, R.J.; Hayes, V.M.; O’Toole, S.A.; Ballinger, M.L.; Gill, D.; Thomas, D.M.; et al. Diagnosis of fusion genes using targeted RNA sequencing. Nat. Commun. 2019, 10, 1388. [Google Scholar] [CrossRef]
- Pfarr, N.; Stenzinger, A.; Penzel, R.; Warth, A.; Dienemann, H.; Schirmacher, P.; Weichert, W.; Endris, V. High-throughput diagnostic profiling of clinically actionable gene fusions in lung cancer. Genes Chromosom. Cancer 2016, 55, 30–44. [Google Scholar] [CrossRef]
- Wallander, M.L.; Geiersbach, K.B.; Tripp, S.R.; Layfield, L.J. Comparison of reverse transcription-polymerase chain reaction, immunohistochemistry, and fluorescence in situ hybridization methodologies for detection of echinoderm microtubule-associated proteinlike 4-anaplastic lymphoma kinase fusion-positive non-small cell lung carcinoma: Implications for optimal clinical testing. Arch. Pathol. Lab. Med. 2012, 136, 796–803. [Google Scholar] [CrossRef]
- Carter, T.C.; He, M.M. Challenges of identifying clinically actionable genetic variants for precision medicine. J. Healthc. Eng. 2016. [Google Scholar] [CrossRef]
- Rogers, T.M.; Arnau, G.M.; Ryland, G.L.; Huang, S.; Lira, M.E.; Emmanuel, Y.; Perez, O.D.; Irwin, D.; Fellowes, A.P.; Wong, S.Q.; et al. Multiplexed transcriptome analysis to detect ALK, ROS1 and RET rearrangements in lung cancer. Sci. Rep. 2017, 7, 42259. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Schirripa, M.; Antoniotti, C.; Moretto, R.; Salvatore, L.; Masi, G.; Falcone, A.; Loupakis, F. First-line chemotherapy for mCRC—A review and evidence-based algorithm. Nat. Rev. Clin. Oncol. 2015, 12, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Morano, F.; Corallo, S.; Miceli, R.; Lonardi, S.; Raimondi, A.; Cremolini, C.; Rimassa, L.; Bergamo, F.; Sartore-Bianchi, A.; et al. Maintenance therapy with panitumumab alone vs panitumumab plus fluorouracil-leucovorin in patients with RAS wild-type metastatic colorectal cancer: A phase 2 randomized clinical trial. JAMA Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Huijberts, S.; Grothey, A.; Yaeger, R.; Cuyle, P.J.; Elez, E.; Fakih, M.; Montagut, C.; Peeters, M.; Yoshino, T.; et al. Binimetinib, encorafenib, and cetuximab triplet therapy for patients with BRAF V600E-mutant metastatic colorectal cancer: Safety lead-in results from the phase III BEACON colorectal cancer study. J. Clin. Oncol. 2019, 37, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Kopets, S.; Grothey, A.; Van Cutsem, A.; Yaeger, R.; Wasa, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Gollerkeri, A.; Maharry, K.; et al. BEACON CRC: A randomized, 3-Arm, phase 3 study of encorafenib and cetuximab with or without binimetinib vs. choice of either irinotecan or FOLFIRI plus cetuximab in BRAF V600E-mutant metastatic colorectal cancer. Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Morano, F.; Corallo, S.; Lonardi, S.; Raimondi, A.; Cremolini, C.; Rimassa, L.; Murialdo, R.; Zaniboni, A.; Sartore-Bianchi, A.; Tomasello, G.; et al. Negative hyperselection of patients with RAS and BRAF-wild-type metastatic colorectal cancer who received panitumumab-based maintenance therapy. J. Clin. Oncol. 2019, in press. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Lenz, H.-J.J.; Van Cutsem, E.; Limon, M.L.; Wong, K.Y.; Hendlisz, A.; Aglietta, M.; Garcia-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.; et al. Durable clinical benefit with nivolumab (NIVO) plus low-dose ililimumab (IPI) as first-line therapy in micro satellite instability-high/mismatch repair deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC). Ann. Oncol. 2019, 29, 714. [Google Scholar]
- Pelster, M.S.; Amaria, R.N. Combined targeted therapy and immunotherapy in melanoma: A review of the impact on the tumor microenvironment and outcomes of early clinical trials. Adv. Med. Oncol. 2019, 11. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Fusion Partners | NTRK1- | NTRK2- | NTRK3- | ALK- | ROS1- | RET1- | BRAF- | FGFR2- | FGFR3- | ERBB2- |
| LMNA [29] | DAB2IP [29] | ETV6 [29] | SPTBN1 [19] | GOPC [27] | CCDC6 [27] | AGAP3 [19] | MYH15 [19] | STAB1 [19] | GRB7 [30] | |
| TPM3 [29] | EML4 [16] | CAD [27] | SLC34A2 [27] | GEMIN5 [19] | TRIM24 [19] | SPDYE4 [19] | ||||
| SCYL3 [29] | KANK1 [31] | EML4 [27] | NCOA4 [19] | CUL1 [19] | ||||||
| PLEKHA6 [29] | CENPF [27] | RUFY1 [31] | MKRN1 [19] | |||||||
| PRKAR1B [27] | TNIP1 [27] | ARMC10 [31] | ||||||||
| MAPRE3 [27] | SNRNP70 [27] | AKAP9 [31] | ||||||||
| STRN [27] | ||||||||||
| C2orf44 [32] | ||||||||||
| PPP1R21 [33] | ||||||||||
| SMEK2 [34] |
| Gene Fusion | Drug | Disease | References |
|---|---|---|---|
| NTRK1,2,3- | Entrectinib | Solid tumors | [39,40,41] |
| Larotrectinib | Solid tumors | [18] | |
| ALK- | Crizotinib | NSCLC | [42] |
| Entrectinib | NSCLC | [39,40,41] | |
| Alectinib | NSCLC | [43] | |
| Brigatinib | NSCLC | [44] | |
| Ceritinib | NSCLC | [45] | |
| Lorlatinib | NSCLC | [46] | |
| ROS1- | Crizotinib | NSCLC | [47] |
| Lorlatinib | NSCLC | [46] | |
| Entrectinib | NSCLC | [39,40,41] | |
| RET- | Cabozantinib | RCC | [44] |
| Vandetanib | Thyroid cancer | [48] | |
| Lenvatinib | RCC, Thyroid cancer | [49,50] | |
| Sorafenib | RCC, HCC | [51,52] | |
| Alectinib | NSCLC | [43] | |
| Regorafenib | CRC | [53] | |
| BLU-667 * | Solid tumors | [54,55] | |
| LOXO-292 + | Solid tumors | [56] | |
| FGFR2,3- | Erdafitinib | Urothelial carcinoma | [57] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagani, F.; Randon, G.; Guarini, V.; Raimondi, A.; Prisciandaro, M.; Lobefaro, R.; Di Bartolomeo, M.; Sozzi, G.; de Braud, F.; Gasparini, P.; et al. The Landscape of Actionable Gene Fusions in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 5319. https://doi.org/10.3390/ijms20215319
Pagani F, Randon G, Guarini V, Raimondi A, Prisciandaro M, Lobefaro R, Di Bartolomeo M, Sozzi G, de Braud F, Gasparini P, et al. The Landscape of Actionable Gene Fusions in Colorectal Cancer. International Journal of Molecular Sciences. 2019; 20(21):5319. https://doi.org/10.3390/ijms20215319
Chicago/Turabian StylePagani, Filippo, Giovanni Randon, Vincenzo Guarini, Alessandra Raimondi, Michele Prisciandaro, Riccardo Lobefaro, Maria Di Bartolomeo, Gabriella Sozzi, Filippo de Braud, Patrizia Gasparini, and et al. 2019. "The Landscape of Actionable Gene Fusions in Colorectal Cancer" International Journal of Molecular Sciences 20, no. 21: 5319. https://doi.org/10.3390/ijms20215319
APA StylePagani, F., Randon, G., Guarini, V., Raimondi, A., Prisciandaro, M., Lobefaro, R., Di Bartolomeo, M., Sozzi, G., de Braud, F., Gasparini, P., & Pietrantonio, F. (2019). The Landscape of Actionable Gene Fusions in Colorectal Cancer. International Journal of Molecular Sciences, 20(21), 5319. https://doi.org/10.3390/ijms20215319