Identification of 15 T Cell Restricted Genes Evaluates T Cell Infiltration of Human Healthy Tissues and Cancers and Shows Prognostic and Predictive Potential

 , , , and
, , , and 
Abstract
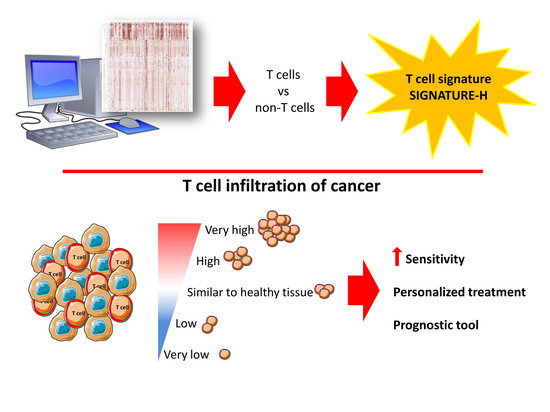
1. Introduction
2. Results
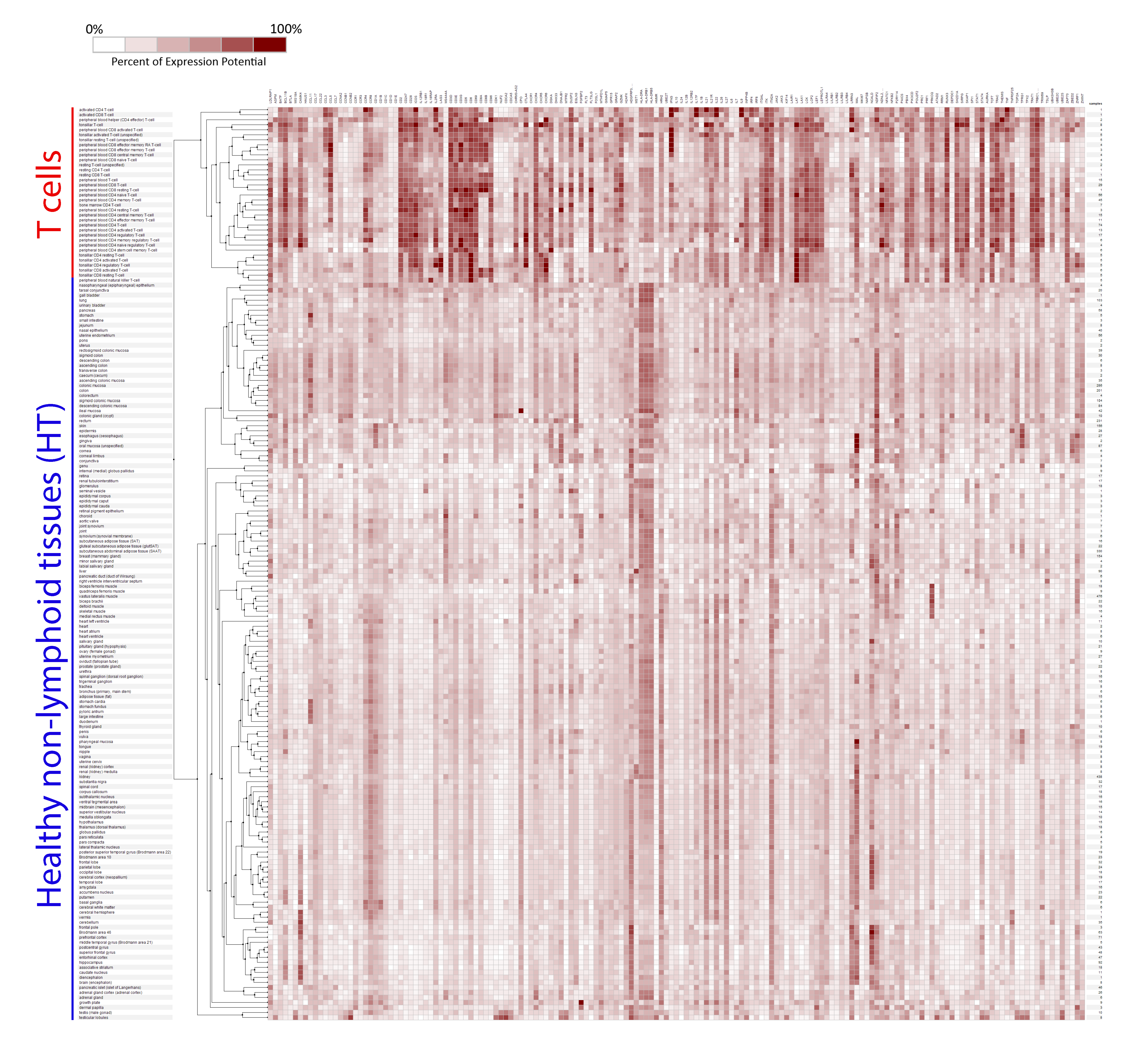
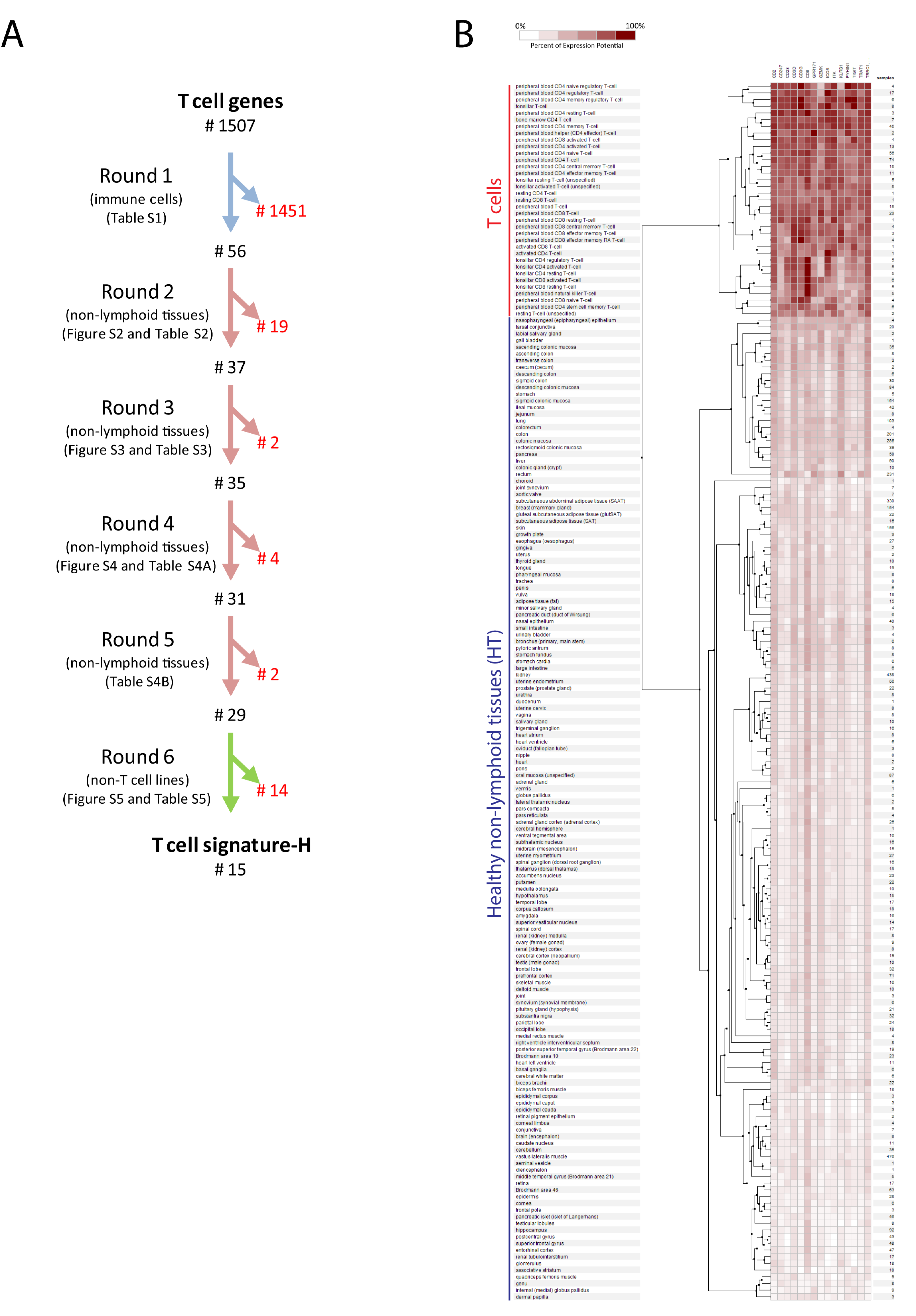
2.1. Selection of Genes for a New T Cell Signature (Signature-H)
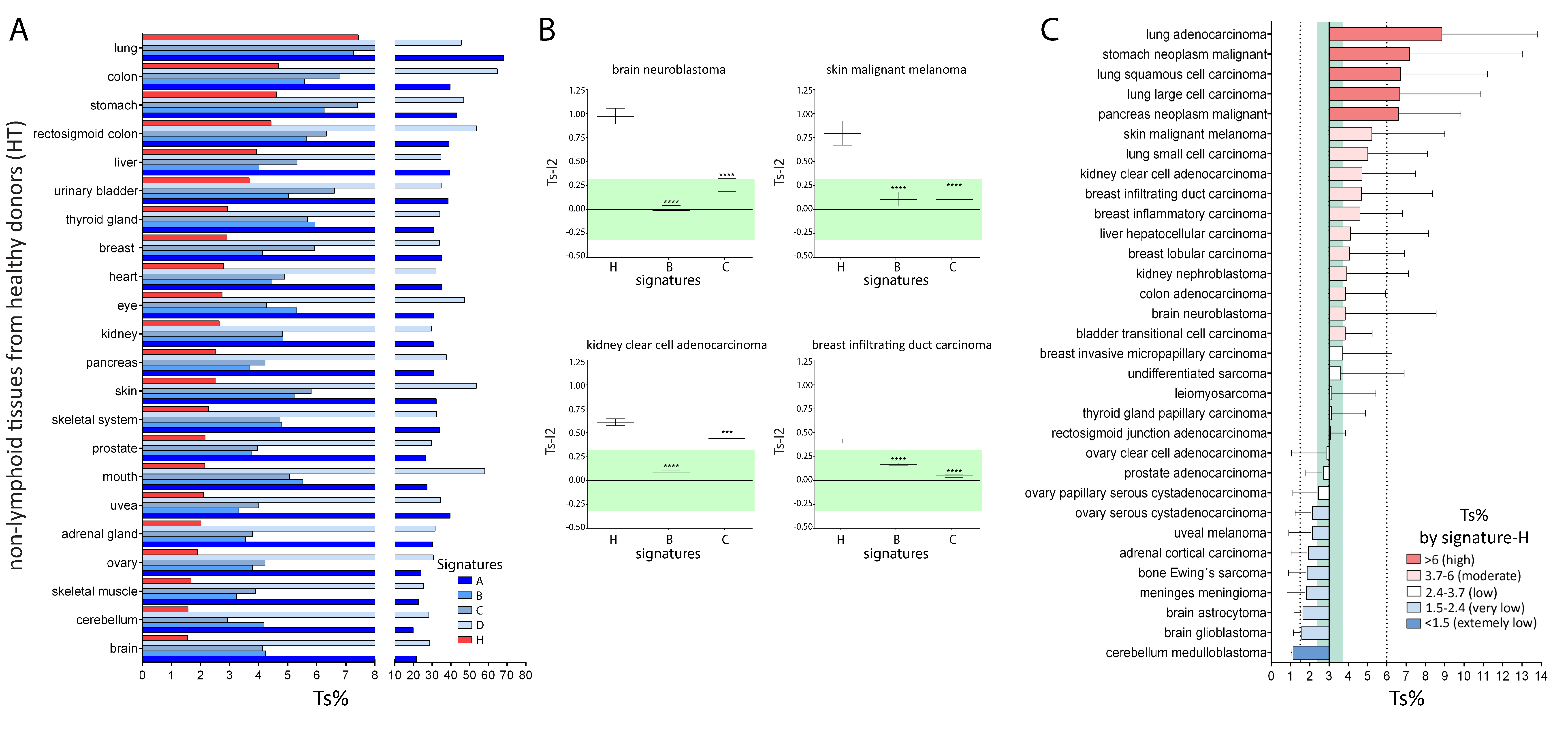
2.2. Comparison of Signature-H with Other T Cell Signatures for the Evaluation of Tci of Tissues from Healthy Donors
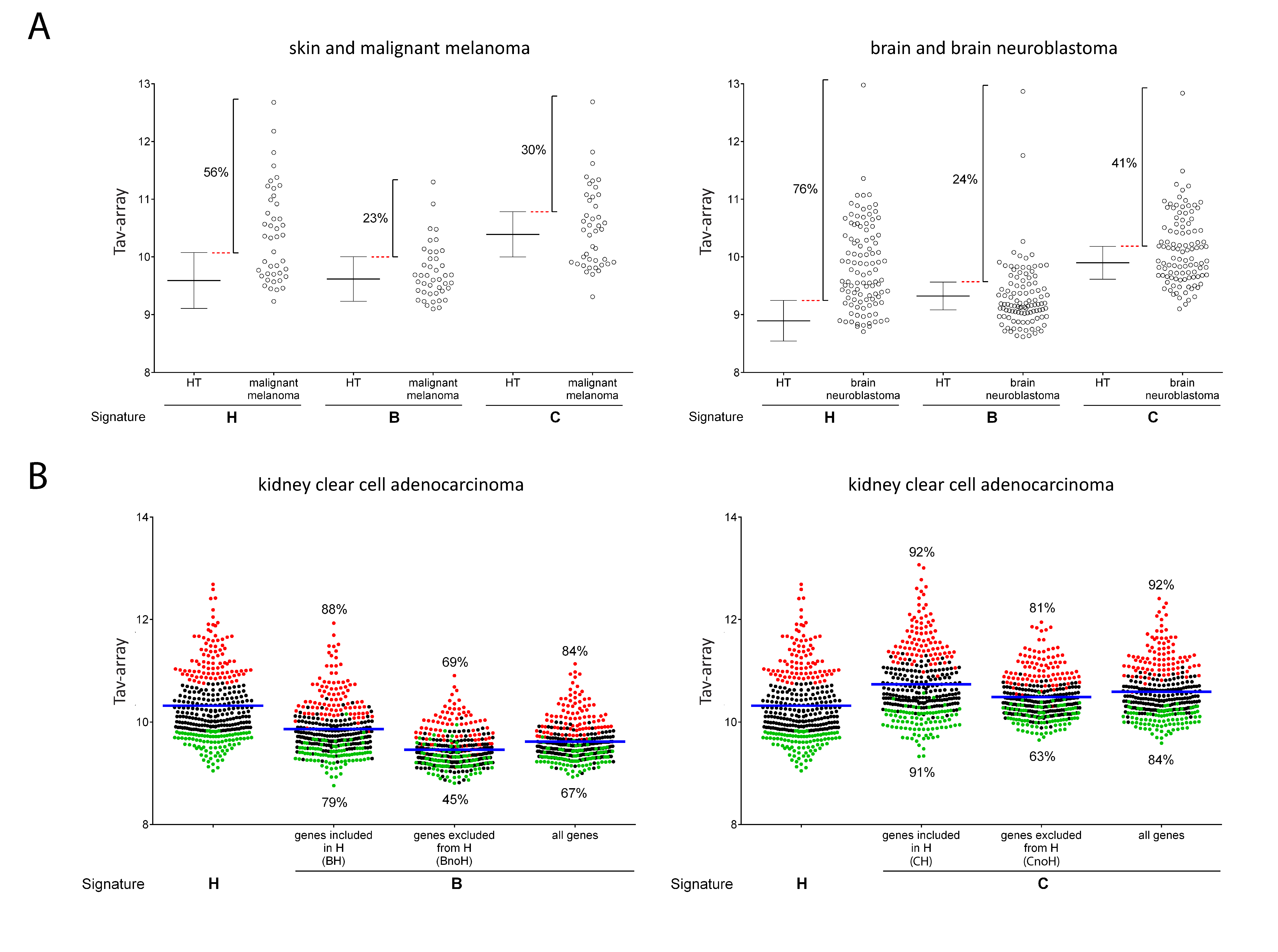
2.3. Comparison of Signature-H with Other T Cell Signatures for the Evaluation of Tci of Different Types of Cancers
2.4. Comparison of Signature-H with Other T Cell Signatures for the Evaluation of Tci of Cancer Specimens
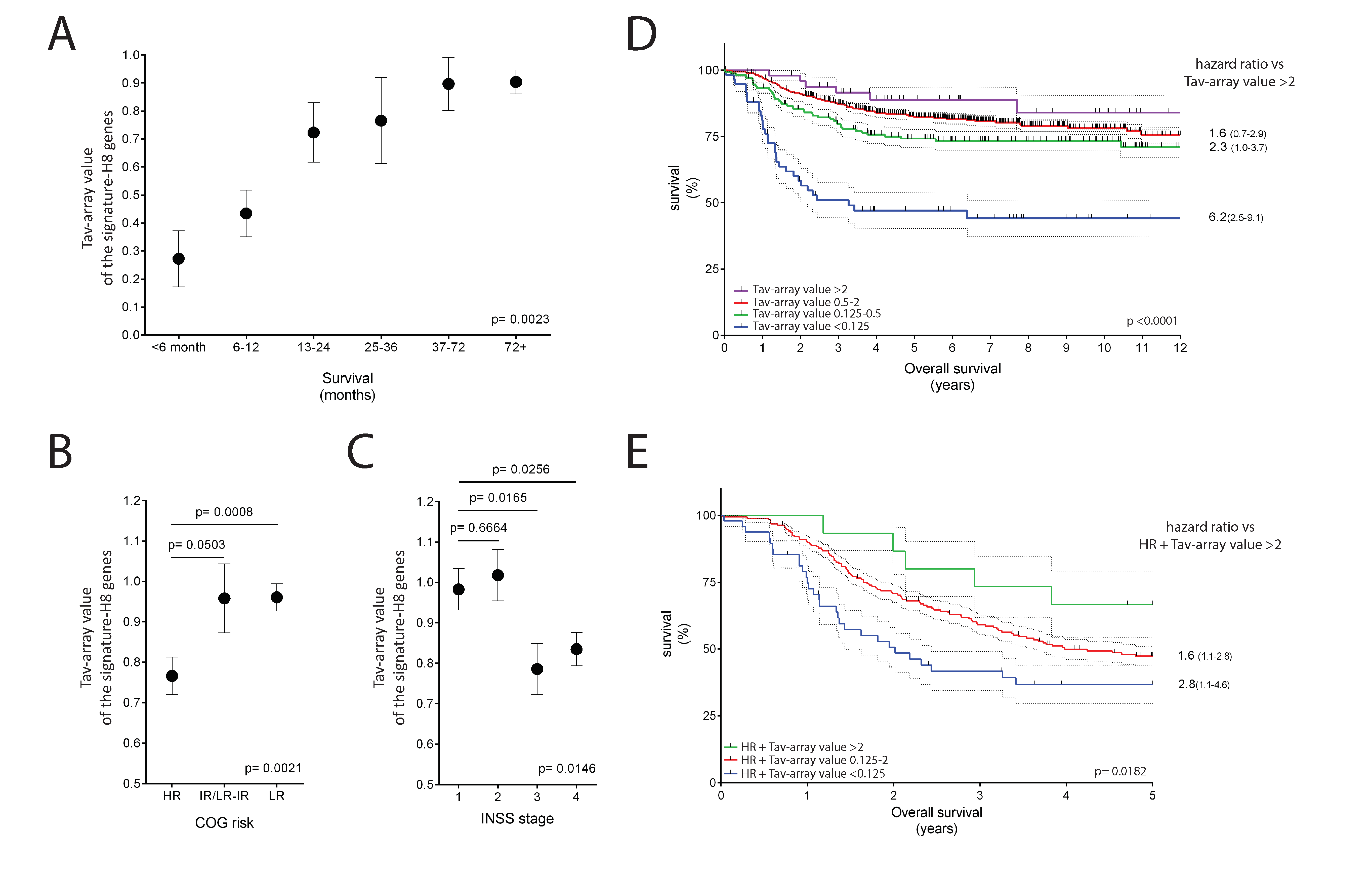
2.5. The Potential Use of Signature-H for Precision Medicine and Its Relevance for the Prognosis of Neuroblastoma Patients
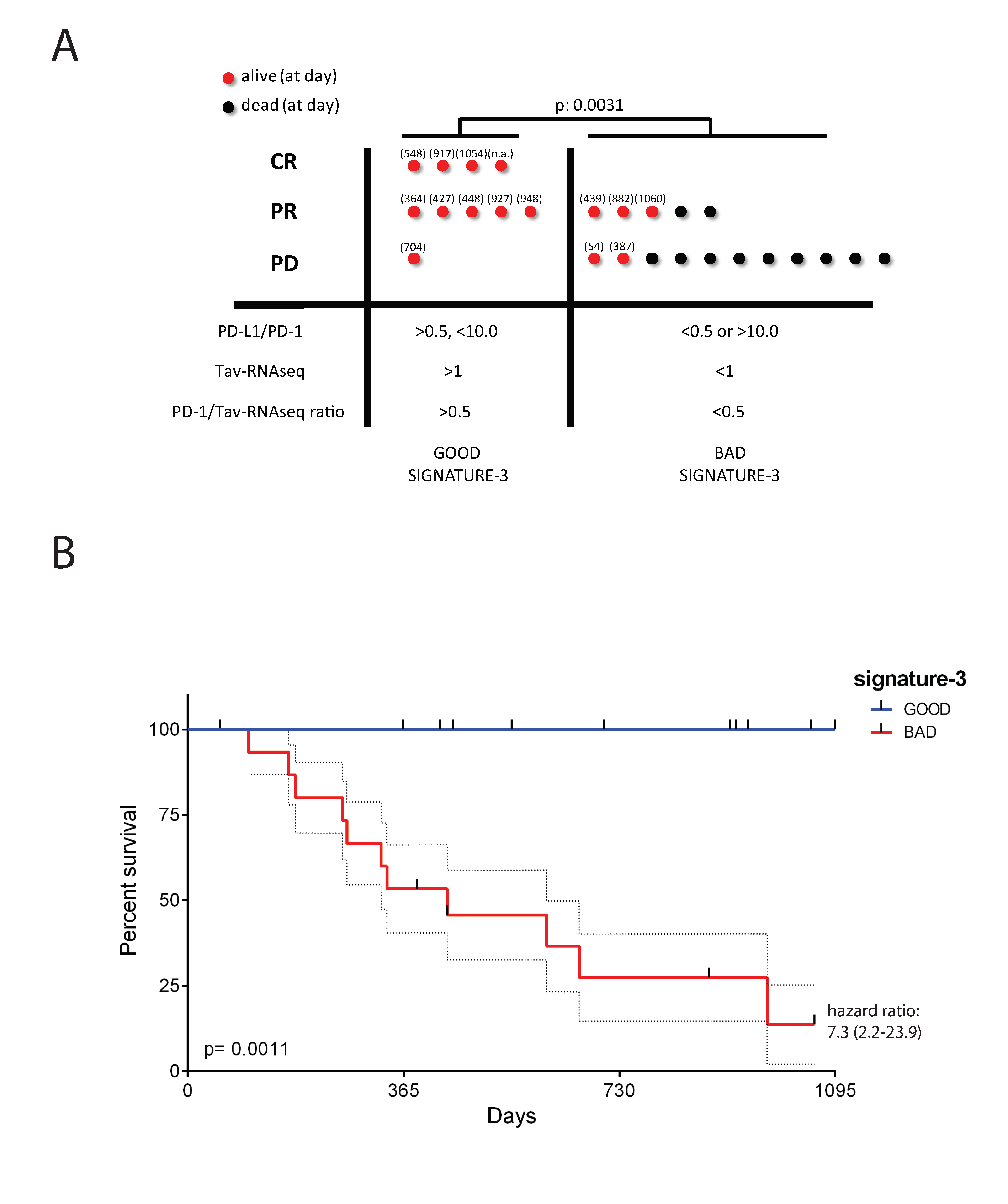
2.6. The Potential Use of Signature-H for Predicting Response to the Anti-PD-1 Antibody Nivolumab
3. Discussion
4. Materials and Methods
4.1. Data Source, Tumor Types, and Control Samples
4.2. T Cell Infiltration Scores Definition
4.3. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.-H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Rudensky, A.Y. Hallmarks of Tissue-Resident Lymphocytes. Cell 2016, 164, 1198–1211. [Google Scholar] [CrossRef]
- Gebhardt, T.; Palendira, U.; Tscharke, D.C.; Bedoui, S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol. Rev. 2018, 283, 54–76. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Kallies, A. Transcriptional Regulation of Tissue-Resident Lymphocytes. Trends Immunol. 2017, 38, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; Van Gisbergen, K.P.J.M.; Hombrink, P.; Van Lier, R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 2018, 19, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liao, W.; Li, Q.; Long, H.; Yin, H.; Zhao, M.; Chan, V.; Lau, C.-S.; Lu, Q. Pathogenic role of tissue-resident memory T cells in autoimmune diseases. Autoimmun. Rev. 2018, 17, 906–911. [Google Scholar] [CrossRef]
- Hendry, S.; Salgado, R.; Gevaert, T.; Russell, P.A.; John, T.; Thapa, B.; Christie, M.; van de Vijver, K.; Estrada, M.V.; Gonzalez-Ericsson, P.I.; et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors. Adv. Anat. Pathol. 2017, 24, 311–335. [Google Scholar] [CrossRef]
- Mina, M.; Boldrini, R.; Citti, A.; Romania, P.; D’Alicandro, V.; De Ioris, M.; Castellano, A.; Furlanello, C.; Locatelli, F.; Fruci, D. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology 2015, 4, 1–14. [Google Scholar] [CrossRef]
- Koch, M.; Beckhove, P.; op den Winkel, J.; Autenrieth, D.; Wagner, P.; Nummer, D.; Specht, S.; Antolovic, D.; Galindo, L.; Schmitz-Winnenthal, F.H.; et al. Tumor Infiltrating T Lymphocytes in Colorectal Cancer. Ann. Surg. 2006, 244, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Carbognin, L.; De Silva, P.; Criscitiello, C.; Lambertini, M. Tumor-infiltrating lymphocytes in breast cancer according to tumor subtype: Current state of the art. The Breast 2017, 35, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Sackstein, R.; Schatton, T.; Barthel, S.R. T-lymphocyte homing: an underappreciated yet critical hurdle for successful cancer immunotherapy. Lab. Investig. 2017, 97, 669–697. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C. Immunology and immunotherapy of neuroblastoma. Semin. Cancer Biol. 2011, 21, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; André, F.; Tesniere, A.; Kroemer, G. The anticancer immune response: indispensable for therapeutic success? J. Clin. Investig. 2008, 118, 1991–2001. [Google Scholar] [CrossRef] [PubMed]
- Turksma, A.W.; Coupe, V.M.H.; Shamier, M.C.; Lam, K.L.H.; de Weger, V.A.; Belien, J.A.M.; van den Eertwegh, A.J.; Meijer, G.A.; Meijer, C.J.L.M.; Hooijberg, E. Extent and Location of Tumor-Infiltrating Lymphocytes in Microsatellite-Stable Colon Cancer Predict Outcome to Adjuvant Active Specific Immunotherapy. Clin. Cancer Res. 2016, 22, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef]
- Hamada, T.; Soong, T.R.; Masugi, Y.; Kosumi, K.; Nowak, J.A.; da Silva, A.; Mu, X.J.; Twombly, T.S.; Koh, H.; Yang, J.; et al. TIME (Tumor Immunity in the MicroEnvironment) classification based on tumor CD274 (PD-L1) expression status and tumor-infiltrating lymphocytes in colorectal carcinomas. Oncoimmunology 2018, 7, e1442999. [Google Scholar] [CrossRef]
- Burugu, S.; Asleh-Aburaya, K.; Nielsen, T.O. Immune infiltrates in the breast cancer microenvironment: detection, characterization and clinical implication. Breast Cancer 2017, 24, 3–15. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef]
- Malissen, N.; Grob, J.-J. Metastatic Melanoma: Recent Therapeutic Progress and Future Perspectives. Drugs 2018, 78, 1197–1209. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Perea, F.; Sánchez-Palencia, A.; Gómez-Morales, M.; Bernal, M.; Concha, Á.; García, M.M.; González-Ramírez, A.R.; Kerick, M.; Martin, J.; Garrido, F.; et al. HLA class I loss and PD-L1 expression in lung cancer: impact on T-cell infiltration and immune escape. Oncotarget 2018, 9, 4120–4133. [Google Scholar] [PubMed]
- Angelova, M.; Charoentong, P.; Hackl, H.; Fischer, M.L.; Snajder, R.; Krogsdam, A.M.; Waldner, M.J.; Bindea, G.; Mlecnik, B.; Galon, J.; et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wan, Y.-W.; Pang, K.; Chow, L.M.L.; Liu, Z. Digital sorting of complex tissues for cell type-specific gene expression profiles. BMC Bioinform. 2013, 14, 89. [Google Scholar] [CrossRef] [PubMed]
- Cari, L.; Nocentini, G.; Migliorati, G.; Riccardi, C. Potential effect of tumor-specific Treg-targeted antibodies in the treatment of human cancers: A bioinformatics analysis. Oncoimmunology 2018, 7, e1387705. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef]
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. Genevestigator V3: A Reference Expression Database for the Meta-Analysis of Transcriptomes. Adv. Bioinform. 2008, 2008, 1–5. [Google Scholar] [CrossRef]
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update—simplifying data submissions. Nucleic Acids Res. 2015, 43, D1113–D1116. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M. Recent Advances in Neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Zahoor, H.; Pavicic, P.G.; Przybycin, C.; Ko, J.; Stephens, L.; Radivoyevitch, T.; Jia, X.; Diaz-Montero, C.M.; Finke, J.; Rayman, P.A.; et al. Evaluation of T cell infiltration in matched biopsy and nephrectomy samples in renal cell carcinoma. Medicine 2018, 97, e12344. [Google Scholar] [CrossRef] [PubMed]
- Lizotte, P.H.; Ivanova, E.V.; Awad, M.M.; Jones, R.E.; Keogh, L.; Liu, H.; Dries, R.; Almonte, C.; Herter-Sprie, G.S.; Santos, A.; et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight 2016, 1, e89014. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Busund, L.-T.; Kilvær, T.L.; Andersen, S.; Richardsen, E.; Paulsen, E.E.; Hald, S.; Khanehkenari, M.R.; Cooper, W.A.; Kao, S.C.; et al. The Role of Tumor-Infiltrating Lymphocytes in Development, Progression, and Prognosis of Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2016, 11, 789–800. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Oberthuer, A.; Juraeva, D.; Hero, B.; Volland, R.; Sterz, C.; Schmidt, R.; Faldum, A.; Kahlert, Y.; Engesser, A.; Asgharzadeh, S.; et al. Revised Risk Estimation and Treatment Stratification of Low- and Intermediate-Risk Neuroblastoma Patients by Integrating Clinical and Molecular Prognostic Markers. Clin. Cancer Res. 2015, 21, 1904–1915. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef]
- Tomioka, N.; Azuma, M.; Ikarashi, M.; Yamamoto, M.; Sato, M.; Watanabe, K.; Yamashiro, K.; Takahashi, M. The therapeutic candidate for immune checkpoint inhibitors elucidated by the status of tumor-infiltrating lymphocytes (TILs) and programmed death ligand 1 (PD-L1) expression in triple negative breast cancer (TNBC). Breast Cancer 2018, 25, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Bagatell, R. Mechanisms of neuroblastoma regression. Nat. Rev. Clin. Oncol. 2014, 11, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Lauder, I.; Aherne, W. The Significance of Lymphocytic Infiltration in Neuroblastoma. Br. J. Cancer 1972, 26, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, G.; Cari, L.; Ronchetti, S.; Riccardi, C. Modulation of tumor immunity: a patent evaluation of WO2015026684A1. Expert. Opin. Ther. Pat. 2016, 26, 417–425. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Signature in the Manuscript | Signature-A | Signature-B | Signature-C | Signature-D | / | / | / | Signature-H | Number of Signatures in Which the Gene is Overexpressed (Signature-H Excluded) | |
| References | [22] | [23] | [24] | [25] | [27] | [28] | [29] | the present study | ||
| Populations Investigated by the Study | T cells | T cells | T cells | T cells | T cells subpopulations | T cells subpopulations | T cells subpopulations | T cells | ||
| Number of the Genes Included in the Signature | 86 | 17 | 19 | 76 | 341 | 105 | 1002 | 15 | ||
| Gene | CD2 | X | X | X | 2 | |||||
| CD247 | X | X | X | 2 | ||||||
| CD28 | X | X | X | X | X | X | 5 | |||
| CD3D | X | X | X | X | X | X | X | X | 7 | |
| CD3G | X | X | X | X | X | X | X | X | 7 | |
| CD6 | X | X | X | X | X | X | X | 6 | ||
| GPR171 | X | X | 1 | |||||||
| GZMK | X | X | X | X | 3 | |||||
| ICOS | X | X | X | X | X | 4 | ||||
| ITK | X | X | X | X | X | 4 | ||||
| KLRB1 | X | X | 1 | |||||||
| PYHIN1 | X | X | 1 | |||||||
| TIGIT | X | X | 1 | |||||||
| TRAT1 | X | X | X | X | X | X | 5 | |||
| TRBC1 | X | X | 1 | |||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cari, L.; De Rosa, F.; Petrillo, M.G.; Migliorati, G.; Nocentini, G.; Riccardi, C. Identification of 15 T Cell Restricted Genes Evaluates T Cell Infiltration of Human Healthy Tissues and Cancers and Shows Prognostic and Predictive Potential. Int. J. Mol. Sci. 2019, 20, 5242. https://doi.org/10.3390/ijms20205242
Cari L, De Rosa F, Petrillo MG, Migliorati G, Nocentini G, Riccardi C. Identification of 15 T Cell Restricted Genes Evaluates T Cell Infiltration of Human Healthy Tissues and Cancers and Shows Prognostic and Predictive Potential. International Journal of Molecular Sciences. 2019; 20(20):5242. https://doi.org/10.3390/ijms20205242
Chicago/Turabian StyleCari, Luigi, Francesca De Rosa, Maria Grazia Petrillo, Graziella Migliorati, Giuseppe Nocentini, and Carlo Riccardi. 2019. "Identification of 15 T Cell Restricted Genes Evaluates T Cell Infiltration of Human Healthy Tissues and Cancers and Shows Prognostic and Predictive Potential" International Journal of Molecular Sciences 20, no. 20: 5242. https://doi.org/10.3390/ijms20205242
APA StyleCari, L., De Rosa, F., Petrillo, M. G., Migliorati, G., Nocentini, G., & Riccardi, C. (2019). Identification of 15 T Cell Restricted Genes Evaluates T Cell Infiltration of Human Healthy Tissues and Cancers and Shows Prognostic and Predictive Potential. International Journal of Molecular Sciences, 20(20), 5242. https://doi.org/10.3390/ijms20205242