Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia


{kind=link}
Abstract
:1. Introduction
2. Methods
3. Background
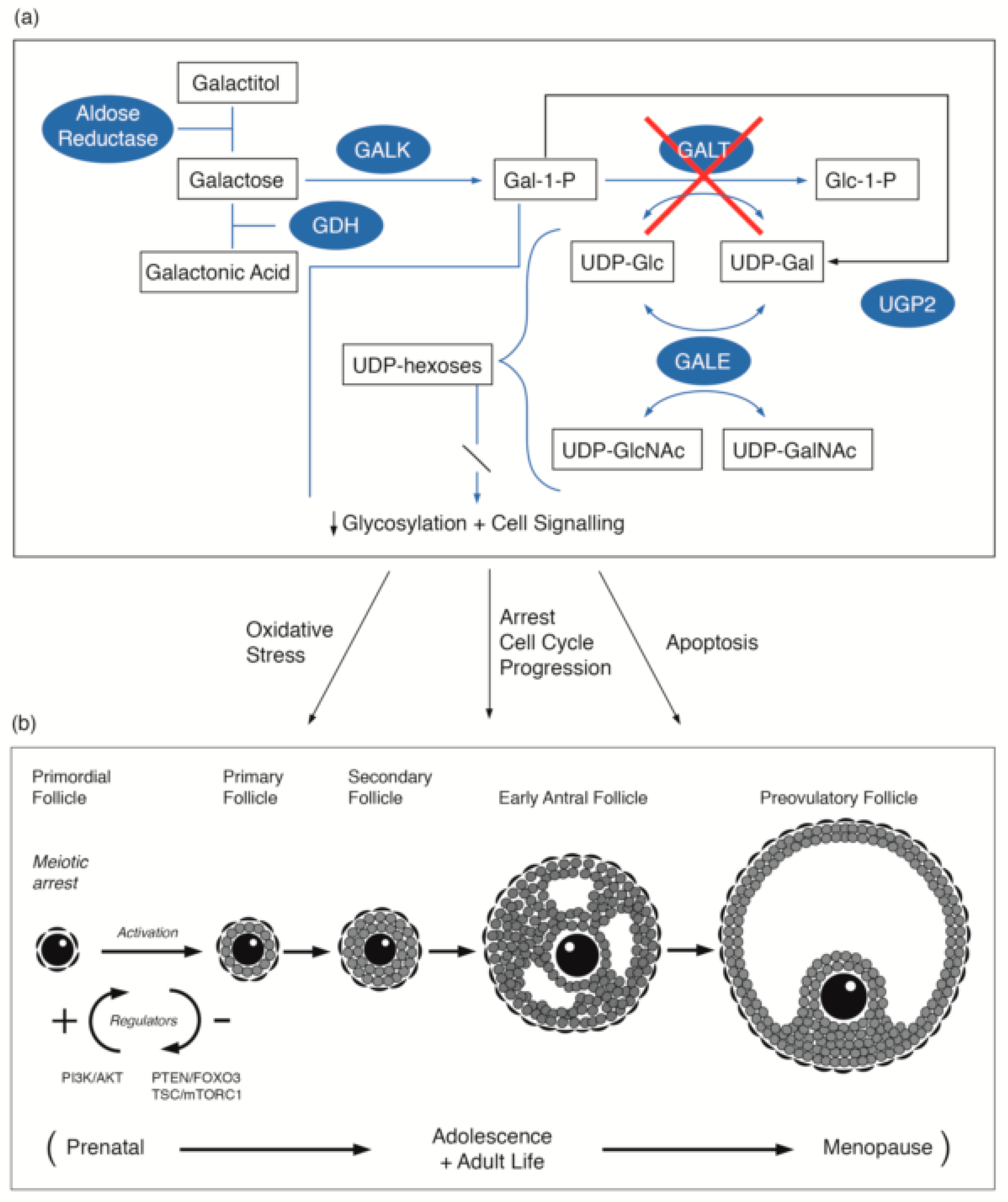
3.1. Galactosaemia and Galactose Metabolism
3.2. Primary Ovarian Insufficiency in Female Galactosaemia Patients
3.3. Link to Primordial Germ Cell Signalling
4. Approaches to Treatment
Fertility Preservation
5. New Horizons and Future Prospects for Fertility Treatments in Classical Galactosaemia
6. Conclusion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| AMH | Anti-Mullerian hormone |
| CG | Classical Galactosaemia |
| FOX03 | Forkhead box 03 gene |
| FSH | Follicle stimulating hormone |
| LH | Luteinizing hormone |
| Gal | Galactosaemia |
| Gal-1-P | Galactose-1-phosphate |
| GALT | Galactose-1-phosphate uridylytransferase |
| GALE | Galactose epimerase |
| GALK | Galactokinase |
| GALM | Galactose mutarotase |
| MTORC1 | Mammalian target of rapamycin complex 1 |
| P13K | Phosphatidylinositol 3-kinase |
| POI | Primary Ovarian Insufficiency |
| PTEN | Phosphatase and tensin homolog |
| UDP-Gal | Uridine diphosphate galactose |
| UGP | UDP-glucose pyrophosphorylase |
References
- Valle, D.; Antonarakis, S.; Ballabio, A.; Beaudet, A.; Mitchell, G.A. The Online Metabolic and Molecular Bases of Inherited Disease; The McGraw-Hill Companies: New York, NY, USA, 2008. [Google Scholar]
- Wada, Y.; Kikuchi, A.; Arai-Ichinoi, N.; Sakamoto, O.; Takezawa, Y.; Iwasawa, S.; Niihori, T.; Nyuzuki, H.; Nakajima, Y.; Ogawa, E.; et al. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet. Med. 2019, 21, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T. Classic Galactosemia and Clinical Variant Galactosemia. Gene Rev. 2017. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1518/ (accessed on 10 September 2019).
- Coss, K.P.; Doran, P.P.; Owoeye, C.; Codd, M.B.; Hamid, N.; Mayne, P.D.; Crushell, E.; Knerr, I.; Monavari, A.A.; Treacy, E.P. Classical galactosaemia in Ireland: Incidence, complications and outcomes of treatment. J. Inherit. Metab. Dis. 2013, 36, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, D.D.; Buist, N.R.; Donnell, G.N. Long-term prognosis in galactosaemia: Results of a survey of 350 cases. J. Inherit. Metab. Dis. 1990, 13, 802–818. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, S.; Shin, Y.; Jakobs, C.; Brodehl, J. Long-term outcome in 134 patients with galactosaemia. Eur. J. Pediatr. 1993, 152, 36–43. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Potter, N.L.; Gordon, C.M.; Green, R.C.; Greenstein, P.; Gubbels, C.S.; Rubio-Gozalbo, E.; Schomer, D.; Welt, C.; Anastasoaie, V.; et al. The adult galactosemic phenotype. J. Inherit. Metab. Dis. 2012, 35, 279–286. [Google Scholar] [CrossRef]
- Jumbo-Lucioni, P.P.; Garber, K.; Kiel, J.; Baric, I.; Berry, G.T.; Bosch, A.; Burlina, A.; Chiesa, A.; Pico, M.L.; Estrada, S.C.; et al. Diversity of approaches to classic galactosemia around the world: A comparison of diagnosis, intervention, and outcomes. J. Inherit. Metab. Dis. 2012, 35, 1037–1049. [Google Scholar] [CrossRef]
- Rubio-Gozalbo, M.E.; Haskovic, M.; Bosch, A.M.; Burnyte, B.; Coelho, A.I.; Cassiman, D.; Couce, M.L.; Dawson, C.; Demirbas, D.; Derks, T.; et al. The natural history of classic galactosaemia: Lessons from the GalNet registry. Orphanet J. Rare Dis. 2019, 14, 86. [Google Scholar] [CrossRef]
- Kaufman, F.; Kogut, M.D.; Donnell, G.N.; Koch, H.; Goebelsmann, U. Ovarian failure in galactosaemia. Lancet 1979, 314, 737–738. [Google Scholar] [CrossRef]
- Roth, S.; McGuire, E.J.; Roseman, S. Evidence for cell-surface glycosyltransferases: Their potential role in cellular recognition. J. Cell Biol. 1971, 51, 536. [Google Scholar] [CrossRef]
- Shin-Buehring, Y.S.; Beier, T.; Tan, A.; Osang, M.; Schaub, J. The activity of galactose-1-phosphate uridyltransferase and galactokinase in human fetal organs. Pediatr. Res. 1977, 11, 1045. [Google Scholar] [CrossRef]
- Heidenreich, R.A.; Mallee, J.; Rogers, S.; Segal, S. Developmental and tissue-specific modulation of rat galactose-1-phosphate uridyltransferase steady state messenger RNA and specific activity levels. Pediatr. Res. 1993, 34, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T.; Moate, P.J.; Reynolds, R.A.; Yager, C.T.; Ning, C.; Boston, R.C.; Segal, S. The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol. Genet. Metab. 2004, 81, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Schadewaldt, P.; Kamalanathan, L.; Hammen, H.W.; Wendel, U. Age dependence of endogenous galactose formation in Q188R homozygous galactosemic patients. Mol. Genet. Metab. 2004, 81, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Coman, D.J.; Murray, D.W.; Byrne, J.C.; Rudd, P.M.; Bagaglia, P.M.; Doran, P.D.; Treacy, E.P. Galactosemia, a single gene disorder with epigenetic consequences. Pediatr. Res. 2010, 67, 286–292. [Google Scholar] [CrossRef]
- Coss, K.P.; Treacy, E.P.; Cotter, E.J.; Knerr, I.; Murray, D.W.; Shin, Y.S.; Doran, P.P. Systemic gene dysregulation in classical Galactosaemia: Is there a central mechanism? Mol. Genet. Metab. 2014, 113, 177–187. [Google Scholar] [CrossRef]
- Balakrishnan, B.; Nicholas, C.; Siddiqi, A.; Chen, W.; Bales, E.; Feng, M.; Johnson, J.; Lai, K. Reversal of abberant PI3K/Akt signalling by Salubrinal in a GaltT-deficient mouse model. Biochem. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3286–3293. [Google Scholar] [CrossRef]
- Colhoun, H.O.; Rubio-Gozalbo, E.M.; Bosch, A.M.; Knerr, I.; Dawson, C.; Brady, J.; Galligan, M.; Stepien, K.; O’Flaherty, R.; Catherine Moss, C.; et al. Fertility in classical galactosaemia, a study of n-glycan, hormonal and inflammatory gene interactions. Orphanet. J. Rare Dis. 2018, 13, 164. [Google Scholar] [CrossRef]
- Sanchez, F.; Smitz, J. Molecular control of oogenesis. Biochim. Biophys. Acta 2012, 1822, 1896–1912. [Google Scholar] [CrossRef] [Green Version]
- Calderon, F.R.O.; Phansalkar, A.R.; Crockett, D.K.; Miller, M.; Mao, R. Mutation database for the galactose-1-phosphate uridylytransferase (GALT) gene. Hum. Mutat. 2007, 28, 939–943. [Google Scholar] [CrossRef]
- Lai, K.; Willis, A.C.; Elsas, L.J. The biochemical role of glutamine 188 in human galactose-1-phosphate uridyltransferase. J. Biol. Chem. 1999, 274, 6559–6566. [Google Scholar] [CrossRef]
- Lai, K.; Langley, S.; Singh, R.H.; Dembure, P.P.; Hjelm, L.; Elsas, L.J. A prevalent mutation for galactosaemia among black Americans. J. Pediatr. 1996, 128, 89–95. [Google Scholar] [CrossRef]
- Nelson, L.M. Clinical Practice. Primary ovarian insufficiency. N. Engl. J. Med. 2009, 360, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, F.R.; Kogut, M.D.; Donnell, G.N.; Goebelsmann, U.; March, C.; Koch, R. Hypergonadotropic hypogonadism in female patients with galactosemia. N. Engl. J. Med. 1981, 304, 994–998. [Google Scholar] [CrossRef]
- Gubbels, C.S.; Land, J.A.; Rubio-Gozalbo, M.E. Fertility and impact of pregnancies on the mother and child in classic galactosemia. Obstet. Gynecol. Surv. 2008, 63, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Fridovich-Keil, J.L.; Gubbles, C.S.; Spencer, J.B.; Sanders, R.D.; Land, J.A.; Rubio-Gozalbo, E. Ovarian function in girls and women with GALT-deficiency galactosemia. J. Inherit. Metab. Dis. 2011, 34, 357–366. [Google Scholar] [CrossRef]
- Guerrero, N.V.; Singh, R.H.; Manatunga, A.; Berry, G.T.; Steiner, R.D.; Elsas, L.J.I. Risk factors for premature ovarian failure in females with galactosemia. J. Pediatr. 2000, 137, 833–841. [Google Scholar] [CrossRef]
- Sanders, R.D.; Spencer, J.B.; Epstein, M.P.; Pollak, S.V.; Vardhana, P.A.; Lustbader, J.W.; Fridovich-Keil, J.L. Biomarkers of ovarian function in girls and women with classic galactosaemia. Fertil. Steril. 2009, 92, 344–351. [Google Scholar] [CrossRef]
- Spencer, J.B.; Badik, J.R.; Ryan, E.L.; Gleason, T.J.; Broadaway, K.A.; Epstein, M.P.; Fridovich-Keil, J.L. Modifiers of ovarian function in girls and women with classic galactosaemia. J. Clin. Endocrinol. Metab. 2013, 98, E1257–1265. [Google Scholar] [CrossRef]
- Kaufman, F.R.; Donnell, G.N.; Roe, T.F.; Kogut, M.D. Gonadal function in patients with galactosaemia. J. Inherit. Metab. Dis. 1986, 9, 140–146. [Google Scholar] [CrossRef]
- Chen, Y.T.; Mattison, D.R.; Feigenbaum, L.; Fukui, H.; Schulman, J.D. Reduction in oocyte number following prenatal exposure to a diet high in galactose. Science 1981, 214, 1145–1147. [Google Scholar] [CrossRef]
- Hoefnagel, D.; Wurster-Hill, D.; Child, E.L. Ovarian failure in galactosaemia. Lancet 1979, 314, 1197. [Google Scholar] [CrossRef]
- Fraser, I.S.; Russell, P.; Greco, S.; Robertson, D.M. Resistant ovary syndrome and premature ovarian failure in young women with galactosaemia. Clin. Reprod. Fertil. 1986, 4, 133–138. [Google Scholar] [PubMed]
- Morrow, R.J.; Atkinson, A.B.; Carson, D.J.; Carson, N.A.; Sloan, J.M.; Traub, A.I. Ovarian failure in a young woman with galactosaemia. Ulster Med. J. 1985, 54, 218. [Google Scholar] [PubMed]
- Robinson, A.C.; Dockeray, C.J.; Cullen, M.J.; Sweeney, E.C. Hypergonadotrophic hypogonadism in classical galactosaemia: Evidence for defective oogenesis. Case report. Br. J. Obstet. Gynaecol. 1984, 91, 199–200. [Google Scholar] [CrossRef]
- Levy, H.L.; Driscoll, S.G.; Porensky, R.S.; Wender, D.F. Ovarian failure in galactosemia (letter). N. Eng. J. Med. 1984, 310, 50. [Google Scholar]
- Rubio-Gozalbo, M.E.; Gubbels, C.S.; Bakker, J.A.; Menheere, P.P.; Wodzig, W.K.; Land, J.A. Gonadal function in male and female patients with classic galactosemia. Hum. Reprod. Update 2010, 16, 177–188. [Google Scholar] [CrossRef]
- Mamsen, L.S.; Kelsey, T.; Ernst, E.; Macklon, K.T.; Lund, A.M.; Andersen, C.Y. Cryopreservation of ovarian tissue may be considered in young girls with galactosaemia. J. Assis. Reprod. Genet. 2018, 2018 35, 1209–1217. [Google Scholar] [CrossRef]
- Van Erven, B.; Berry, G.T.; Cassiman, D.; Connolly, G.; Forga, M.; Gautschi, M.; Gubbels, C.S.; Hollak, C.E.M.; Janssen, M.C.; Knerr, I.; et al. Fertility in adult women with classic galactosaemia and primary ovarian insufficiency. Fertil. Steril. 2017, 108, 168–174. [Google Scholar] [CrossRef]
- Thakur, M.; Feldman, G.; Puscheck, E.E. Primary ovarian insufficiency in classic galactosaemia: Current understanding and future research opportunities. J. Assist. Reprod. Genet. 2018, 35, 3–16. [Google Scholar] [CrossRef]
- Gibson, J.B. Gonadal function in galactosemics and in galactose-intoxicated animals. Eur. J. Pediatr. 1995, 154, S14. [Google Scholar] [CrossRef]
- Meyer, W.R.; Doyle, M.B.; Grifo, J.A.; Lipetz, K.J.; Oates, P.J.; DeCherney, A.H.; Diamond, M.P. Aldose reductase inhibition prevents galactose-induced ovarian dysfunction in the Sprague-Dawley rat. Am. J. Obstet. Gynecol. 1992, 167, 1837–1843. [Google Scholar] [CrossRef]
- Rubio-Gozalbo, M.E.; Coelho, A.I.; Haskovic, M.; Van Scherpenzeel, M.; Lindhout, M.; Zijlstra, F.; Vanoevelen, J.M.; Bierau, J.; lefeber, D.J. Nucleotide sugar profile in the galactosaemia zebrafish model reveals new pathogenic mechanisms and potential readouts. J. Inherit. Metab. Dis. 2019, 42, S1. [Google Scholar]
- Davis, D.; Liu, X.; Segaloff, D.L. Identification of the sites of N-linked glycosylation on the follicle-stimulating hormone (FSH) receptor and assessment of their role in FSH receptor function. Mol. Endocrinol. 1995, 9, 159–170. [Google Scholar] [PubMed]
- Gubbels, C.S.; Land, J.A.; Evers, J.L.; Bierau, J.; Menheere, P.P.; Robben, S.G.; Rubio-Gozalbo, M.E. Primary ovarian insufficiency in classic galactosemia: Role of FSH dysfunction and timing of the lesion. J. Inherit. Metab. Dis. 2013, 36, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Coss, K.P.; Byrne, J.C.; Coman, D.J.; Adamczyk, B.; Abrahams, J.L.; Saldova, R.; Brown, A.Y.; Walsh, O.; Hendroff, U.; CArolan, C.; et al. IgG N-glycans as potential biomarkers for determining galactose tolerance in Classical Galactosaemia. Mol. Genet. Metab. 2012, 105, 212–220. [Google Scholar] [CrossRef]
- Coss, K.P.; Hawkes, C.P.; Crushell, E.; Adamczyk, B.; Saldova, R.; Knerr, I.; Monavari, A.A.; Rudd, P.M.; Treacy, E.P. IgG N-glycan abnormalities in children with Galactosaemia. J. Proteome Res. 2014, 13, 385–394. [Google Scholar] [CrossRef]
- Maratha, A.; Stockmann, H.; Coss, K.P.; Rubio-Gozalbo, E.M.; Knerr, I.; Fitzgibbon, M.; McVeigh, T.P.; Foley, P.; Moss, C.; Colhoun, H.-E.; et al. Classical galactosaemia: Novel insights in IgG n-glycosylation and n-glycan biosynthesis. Eur. J. Hum. Genet. 2016, 24, 976–984. [Google Scholar] [CrossRef]
- Aittomäki, K.; Lucena, J.L.; Pakarinen, P.; Sistonen, P.; Tapanainen, J.; Gromoll, J.; Kaskikari, R.; Sankila, E.M.; Lehväslaiho, H.; Engel, A.R.; et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell 1995, 82, 959–968. [Google Scholar] [CrossRef] [Green Version]
- Swartz, W.J.; Mattison, D.R. Galactose inhibition of ovulation in mice. Fertil. Steril. 1988, 49, 522–526. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Chakrabarti, J.; Banerjee, S.; Pal, A.K.; Goswami, S.K.; Chakravarty, B.N.; Kabir, S.N. Galactose toxicity in the rat as a model for premature ovarian failure: An experimental approach readdressed. Hum. Reprod. 2003, 18, 2031–2038. [Google Scholar] [CrossRef]
- Vanoevelen, J.M.; van Erven, B.; Bierau, J.; Huang, X.; Berry, G.T.; Vos, R.; Coelo, A.I.; Rubio-Gozalbo, M.E. Impaired fertility and motor function in a zebrafish model for classic galactosaemia. J. Inherit. Metab. Dis. 2018, 41, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Gubbels, C.S.; Welt, C.K.; Dumoulin, J.C.; Robben, S.G.; Gordon, C.M.; Dunselman, G.A.; Rubio-Gozalbo, M.E.; Berry, G.T. The male reproductive system in classic galactosaemia: Cyptorchidism and low semen volume. J. Inherit. Metab. Dis. 2013, 36, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Siddiqi, A.; Witt, B.; Yuzyuk, T.; Johnson, B.; Fraser, N.; Chen, W.; Rascon, R.; Yin, X.; Goli, H.; et al. Subfertility and growth restriction in a new galactose-1-phosphate uridylytransferase (GALT)-deficient mouse model. Eur. J. Hum. Genet. 2014, 22, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.L. Reproductive effects of maternal metabolic disorders: Implications for pediatrics and obstetrics. Turk. J. Pediatr. 1996, 38, 335–344. [Google Scholar]
- Forges, T.; Monnier-Barbarino, P.; Leheup, B.; Jouvet, P. Pathophysiology of impaired ovarian function in galactosaemia. Hum. Reprod. Update 2006, 12, 537–584. [Google Scholar] [CrossRef]
- Menezo, Y.J.; Lescaille, M.; Nicollet, B.; Servy, E.J. Pregnancy and delivery after stimulation with rFSH of a galatosemia patient suffering hypergonadotropic hypogonadism: Case report. J. Assist. Reprod. Genet. 2004, 21, 89–90. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, K. Cellular and molecular regulation of the activation of mammalian primordial follicles: Somatic cells initiate follicle activation in adulthood. Hum. Reprod. Update 2015, 21, 779–786. [Google Scholar] [CrossRef]
- Kim, S.Y.; Ebbert, K.; Cordeiro, M.H.; Romero, M.; Zhu, J.; Serna, V.A.; Whelan, K.A.; Woodruff, T.K.; Kurita, T. Cell autonomous phosphoinositide 3-kinase activation on oocytes disrupts normal ovarian function through promoting survival and overgrowth of ovarian follicles. Endocrinology 2015, 156, 1464–1476. [Google Scholar] [CrossRef]
- Li, J.; Kawamura, K.; Cheung, Y.; Liu, S.; Klein, C.; Liu, S.; Duan, E.K.; Hsueh, A.J. Activation of dormant ovarian follicles to generate mature eggs. Proc. Natl. Acad. Sci. 2010, 107, 10280–10284. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Kim, J.; Li, X.X.; Hsueh, A.J. Promotion of ovarian follicle growth following mTOR activation: Synergistic effects of AKT stimulators. PLoS ONE 2015, 10, e0117769. [Google Scholar] [CrossRef]
- Sun, X.; Su, Y.; He, Y.; Zhang, J.; Liu, W.; Zhang, H.; Hou, Z.; Liu, J.; Li, J. New strategy for in vitro activation of primordial follicles with mTOR and PI3K stimulators. Cell Cycle 2015, 14, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welling, L.; Bernstein, L.E.; Berry, G.T.; Burlina, A.B.; Eyskens, F.; Gautschi, M.; Grünewald, S.; Gubbels, C.S.; Knerr, I.; Labrune, P.; et al. Galactosemia Network (GalNet). International clinical guideline for the management of classical galactosaemia: Diagnosis, treatment and follow up. J. Inherit. Metab. Dis. 2017, 40, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Roness, H.; Meirow, D. Follicle reserve loss in ovarian tissue transplantation. Reproduction 2019. [Google Scholar] [CrossRef] [PubMed]
- Shea, L.D.; Woodruff, T.K.; Shikanov, A. Bioengineering the ovarian follicle microenvironment. Annu. Rev. Biomed. Eng. 2014, 16, 29–52. [Google Scholar] [CrossRef] [PubMed]
- Skory, R.M.; Xu, Y.; Shea, L.D.; Woodruff, T.K. Engineering the ovarian cycle using in vitro follicle culture. Hum. Reprod. 2015, 30, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- van Erven, B.; Gubbels, C.S.; van Golde, R.J.; Dunselman, G.A.; Derhaag, J.G.; de Wert, G.; Geraedts, J.P.; Bosch, A.M.; Treacy, E.P.; Welt, C.K.; et al. Fertility preservation in female classic galactosemia patients. Orphanet, J. Rare Dis. 2013, 8, 107. [Google Scholar] [CrossRef]
- Zhai, J.; Yao, G.; Dong, F.; Bu, Z.; Cheng, Y.; Sato, Y.; Hu, L.; Zhang, Y.; Wang, J.; Dai, S.; et al. In vitro activation of follicles and fresh tissue autotransplantation in primary ovarian insufficiency patients. J. Clin. Endocrinol. Metab. 2016, 101, 4405–4412. [Google Scholar] [CrossRef]
- Kawamura, K.; Kawamura, N.; Hsueh, A.J. Activation of dormant follicles: A new treatment for premature ovarian failure? Curr. Opin. Obstet. Gynecol. 2016, 28, 217–222. [Google Scholar] [CrossRef]
- Tang, M.; Odejinmi, S.I.; Vankayalapati, H.; Wierenga, K.J.; Lai, K. Innovative therapy for classic galactosemia – tale of two HTS. Mol. Genet. Metab. 2012, 105, 44–55. [Google Scholar] [CrossRef]
- Martini, P.G.V.; Guey, L.T. A new era for rare genetic diseases: messenger RNA therapy. Hum. Gene Ther. 2019, 30, 1180–1189. [Google Scholar] [CrossRef]
- Truman, A.M.; Tilly, J.L.; Woods, D.C. Ovarian regeneration: The potential for stem cell contribution in the postnatal ovary to sustained endocrine function. Mol. Cell Endocrinol. 2017, 445, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, T.K. Oncofertility: A grand collaboration between reproductive medicine and oncology. Reproduction 2015, 150, S1–S10. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abidin, Z.; Treacy, E.P. Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia. Int. J. Mol. Sci. 2019, 20, 5236. https://doi.org/10.3390/ijms20205236
Abidin Z, Treacy EP. Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia. International Journal of Molecular Sciences. 2019; 20(20):5236. https://doi.org/10.3390/ijms20205236
Chicago/Turabian StyleAbidin, Zaza, and Eileen P. Treacy. 2019. "Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia" International Journal of Molecular Sciences 20, no. 20: 5236. https://doi.org/10.3390/ijms20205236
APA StyleAbidin, Z., & Treacy, E. P. (2019). Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia. International Journal of Molecular Sciences, 20(20), 5236. https://doi.org/10.3390/ijms20205236